 Review
Review

Affiliation:
1Experimental Hepatology and Drug Targeting (HEVEPHARM), University of Salamanca, Institute for Biomedical Research of Salamanca (IBSAL), 37007 Salamanca, Spain
ORCID: https://orcid.org/0000-0002-4811-8230
Affiliation:
1Experimental Hepatology and Drug Targeting (HEVEPHARM), University of Salamanca, Institute for Biomedical Research of Salamanca (IBSAL), 37007 Salamanca, Spain
2Center for the Study of Liver and Gastrointestinal Diseases (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
ORCID: https://orcid.org/0000-0001-9271-3320
Affiliation:
1Experimental Hepatology and Drug Targeting (HEVEPHARM), University of Salamanca, Institute for Biomedical Research of Salamanca (IBSAL), 37007 Salamanca, Spain
ORCID: https://orcid.org/0000-0003-1577-2110
Affiliation:
1Experimental Hepatology and Drug Targeting (HEVEPHARM), University of Salamanca, Institute for Biomedical Research of Salamanca (IBSAL), 37007 Salamanca, Spain
2Center for the Study of Liver and Gastrointestinal Diseases (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
ORCID: https://orcid.org/0000-0003-2649-8778
Affiliation:
1Experimental Hepatology and Drug Targeting (HEVEPHARM), University of Salamanca, Institute for Biomedical Research of Salamanca (IBSAL), 37007 Salamanca, Spain
2Center for the Study of Liver and Gastrointestinal Diseases (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
ORCID: https://orcid.org/0000-0003-1186-6849
Affiliation:
1Experimental Hepatology and Drug Targeting (HEVEPHARM), University of Salamanca, Institute for Biomedical Research of Salamanca (IBSAL), 37007 Salamanca, Spain
2Center for the Study of Liver and Gastrointestinal Diseases (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
Email: mjmonte@usal.es
ORCID: https://orcid.org/0000-0003-1844-7428
Explor Dig Dis. 2022;1:137–153 DOI: https://doi.org/10.37349/edd.2022.00010
Received: June 22, 2022 Accepted: September 12, 2022 Published: December 7, 2022
Academic Editor: Han Moshage, University of Groningen, The Netherlands
The article belongs to the special issue CHOLESTASIS
Several metabolic pathways are involved in the biotransformation of C27 neutral cholesterol to C24 primary bile acids (BAs), mainly cholic acid (CA) and chenodeoxycholic acid (CDCA), which are then conjugated with glycine or taurine. This process can start with the modification of the steroid ring or the shortening of the side chain and involves enzymes present in different subcellular compartments. Inborn errors affecting the biogenesis of organelles, such as peroxisomes, or the expression or function of specific enzymes of these convergent routes result in: i) the lack of mature C24-BAs, with the subsequent impairment in digestion and absorption of dietary fat and liposoluble vitamins, such as vitamin K, which may account for a deficient hepatic synthesis of several coagulation factors; ii) the accumulation of intermediate metabolites, which may affect hepatocyte physiology, causing cholestasis as a commonly shared alteration besides other deleterious hepatic events; and iii) extrahepatic clinical manifestations due to accumulation of toxic metabolites in other territories, such as the nervous system, causing neurological disorders. In general, diseases whose primary alteration is a genetic defect in BA synthesis are diagnosed in children or young individuals with a very low incidence. The symptomatology can markedly vary among individuals, ranging from mild to severe conditions. Oral therapy, based on the enrichment of the BA pool with natural C24-BAs, such as CA, CDCA, glyco-CA, or ursodeoxycholic acid (UDCA), depending on the exact deficiency causing the disease, may be beneficial in preventing life-threatening situations. In contrast, in other cases, a liver transplant is the only option for these patients. This review describes the updated information on the genetic and molecular bases of these diseases and the current approaches to achieve a selective diagnosis and specific treatment.
Inborn errors in bile acid (BA) synthesis constitute a group of infrequent genetic disorders with an autosomal recessive transmission. Altogether, they are responsible for up to 2% of persistent cholestasis in children [1]. Although there is no consensus to define rare diseases, for instance, in the European Union these are defined as those that affect no more than 1 in 2,000 individuals while in the United States rare diseases are included in this group if they affect fewer than 200,000 people in the whole country, inborn defects in BA synthesis are globally considered rare diseases.
The most common clinical presentation is cholestasis which develops progressively in childhood, although other clinical settings may also occur, such as neonatal hepatitis and advanced cirrhosis at birth. Cases of cholestasis development in late childhood or even adult-onset of liver disease have been described [2]. The severity of the conditions varies from mild to severe or even life-threatening, depending on the specific enzyme affected and the magnitude of the defect. Failure to synthesize BAs leads to reduced bile flow and decreased intestinal solubilization and absorption of fat and fat-soluble vitamins, resulting in steatorrhea and fat-soluble vitamin deficiency. When the alteration leads to an accumulation of monohydroxylated BAs or unsaturated oxo-BAs, many of which are toxic and potent cholestatic agents, the progression of liver damage is usually rapid [3].
The diagnosis of these disorders is based on the analysis of the profile of BA molecular species and their precursors in body fluids by using mass spectrometric techniques, such as liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) [4, 5] and liquid secondary ionization mass spectrometry [6]. The most common biochemical findings in patients with BA synthetic defects are: i) the marked reduction or complete lack of primary BAs, cholic acid (CA), and chenodeoxycholic acid (CDCA), in serum, bile, and urine, and ii) the presence of elevated concentrations of atypical BAs and intermediate metabolites that retain the structure of the substrates that the deficient enzyme should have biotransformed. Confirmation of the genetic defect is usually achieved by DNA sequencing.
Early diagnosis is critical since many of these patients respond well to oral treatment with BA sequestering resins, such as cholestyramine, but especially with natural BAs, such as CA, glycocholic acid (GCA), CDCA, or ursodeoxycholic acid (UDCA, Figure 1). BA replacement therapy has a dual objective. On the one hand, it is to provide the essential absent primary BAs necessary for the digestion of fats and fat-soluble vitamins. On the other hand, the aim is to suppress the endogenous synthesis of these molecules, thus reducing the formation of abnormal potentially toxic intermediate metabolites. BA therapy is lifelong for responsive conditions. In these cases, the patient’s adherence should be periodically evaluated.
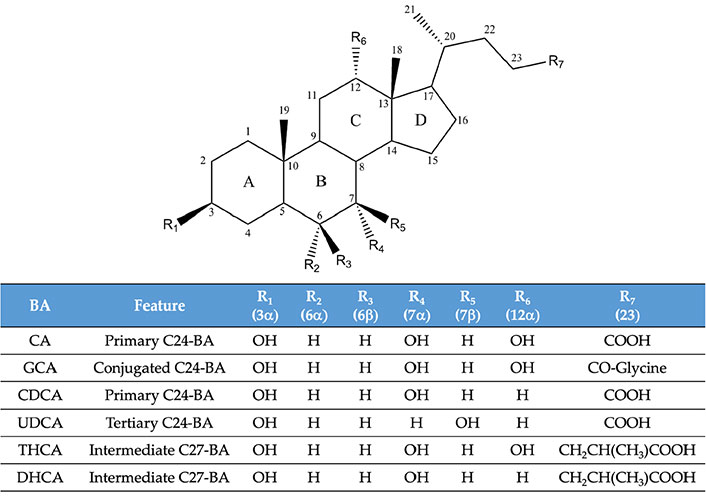
Molecular structure of some of the bile acid (BA) species mentioned in the text. DHCA: 3α,7α-dihydroxy-5β-cholestanoic acid; THCA: 3α,7α,12α-trihydroxy-5β-cholestanoic acid
Regarding the existence of animal models for the study of these diseases, several murine strains have been generated and characterized (Tables 1 & 2). In general, with a few exceptions, these models only partially reproduce the features of the human condition. The differences are mainly accounted for by the dissimilarity in BA pool composition and metabolism between humans and rodents [7, 8].
Inborn defects in BA synthesis: sterol nucleus modifications
| Gene | Protein | Clinical features | Treatment | Ref. | Animal models |
|---|---|---|---|---|---|
| CYP7A1 | CYP7A1 | No signs of liver damage, hypercholesterolemia, hypertriglyceridemia, gallstone disease | N.D. | [24] | [25–28] |
| HSD3B7 | HSD3B7 | Neonatal cholestasis, cirrhosis, malabsorption of fats and fat-soluble vitamins | CA, CDCA | [32, 33, 36] | [35] |
| AKR1D1 (SR5B1) | AKR1D1 | Neonatal hepatitis, cirrhosis, liver failure | CA, CDCA | [37, 40] | [38] |
| CYP7B1 | CYP7B1 | Severe neonatal liver disease, cholestasis, fibrosis, cirrhosis | CDCA, Liver transplantation | [43, 46, 47] | [44] |
AKR1D1: Δ4-3-oxosteroid-5β-reductase; BA: bile acid; CYP7A1: cholesterol 7α-hydroxylase; CYP7B1: oxysterol 7α-hydroxylase; HSD3B7: 3β-hydroxy-Δ5-C27-steroid-oxidoreductase; N.D.: not defined
Inborn defects in BA synthesis: side chain shortening and conjugation
| Gene | Protein | Clinical features | Treatment | Ref. | Animal models |
|---|---|---|---|---|---|
| CYP27A1 | CYP27A1 | Children: neonatal cholestasis, diarrhea, cataracts, growth retardationAdults: xanthomas, neuropsychiatric symptoms (ataxia, dementia) | CDCA | [48, 54] | [50–53] |
| PEX | Several peroxisomal proteins | Liver dysfunction, neurological abnormalities, hearing and vision impairment, adrenocortical dysfunction | Symptomatic or supportive therapies | [56, 57] | [58–60] |
| ABCD3 | PMP70 | Hepatic dysfunction, hepatosplenomegaly | N.D. | [61] | [61, 87] |
| AMACR | AMACR | Children: malabsorption of fat-soluble vitamins, coagulation disorders, cholestasis, hepatitisAdults: sensory and motor degenerative neuropathy | CA | [63–66] | [67] |
| ACOX2 | ACOX2 | Hypertransaminasemia and severe hepatic and neurological alterations | CholestyramineUDCA | [69–72] | [73, 74] |
| HSD17B4 | DBP | Hypotonia, delayed growth and psychomotor development, visual and auditory defects. Hepatomegaly, fibrosis | N.D. | [75] | [76, 77] |
| SCP2 | SCPx | Fertility abnormalities. Brain lesions with motor disorders | Phytanic acid-restricted diet | [78, 79] | [80] |
| SLC27A5 | BACS | No signs of liver disease | N.D. | [81] | [82, 83] |
| BAAT | BAAT | Neonatal cholestasis. Malabsorption of fat-soluble vitamins. Rickets | GCA | [84, 86] | [85] |
ABCD3: ATP-binding cassette subfamily D member 3; ACOX2: acyl-coenzyme A (CoA) oxidase 2; AMACR: α-methyl acyl-CoA racemase; BA: bile acid; BAAT: BA CoA: amino acid N-acyl transferase; BACS: BA-CoA synthetase; CYP27A1: sterol 27-hydroxylase; DBP: D-bifunctional protein; PEX: peroxisomal biogenesis factor or peroxin; PMP70: peroxisomal membrane protein 70; SCP2: sterol carrier protein 2
Biosynthesis of primary BAs from cholesterol involves at least 17 enzymes, one or more intracellular transporters, and different subcellular compartments, including endoplasmic reticulum, mitochondria, peroxisomes, and cytosol. Four main pathways of BA biosynthesis have been described: the classic or neutral pathway, the alternative or acidic pathway, the Yamasaki pathway, and the 25-hydroxylation pathway.
In this metabolic route, the modification of the cholesterol ring precedes the cleavage of its side chain (Figure 2). The pathway starts with the hydroxylation at C7, which is catalyzed by the microsomal enzyme CYP7A1, a member of the cytochrome P450 family of proteins. CYP7A1 is exclusively expressed in hepatocytes and, owing to its critical role in this biosynthetic pathway, is the element on which most regulatory factors converge [9]. The product of the CYP7A1-mediated reaction is 7α-hydroxycholesterol, which is then converted into 7α-hydroxy-4-cholesten-3-one (7αC4) by the action of microsomal HSD3B7. Then the microsomal enzyme sterol 12α-hydroxylase (CYP8B1) can add a hydroxyl group at C12, generating a series of intermediates whose final product is CA. If 12α-hydroxylation does not occur, the final compound generated by this route is CDCA, which highlights the role of CYP8B1 in controlling the CA/CDCA ratio and justifies its tight regulation [10]. In both cases, the double bond at the C4 position is then reduced by the cytosolic enzyme AKR1D1, resulting in the presence of a hydrogen at the 5β position. The last steroid ring modification is catalyzed in the cytosol by 3α-hydroxysteroid-dehydrogenase (AKR1C4), which reduces the 3-oxo group to a 3-hydroxyl with α-stereochemistry.
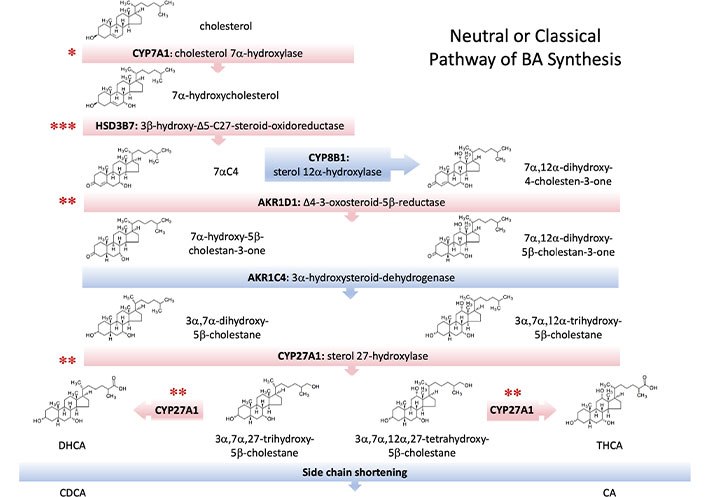
Enzymes involved in the main, neutral, or classical pathway of bile acid (BA) synthesis. Several inborn errors leading to autosomal recessive disorders have been described to affect some of these enzymes (red bars). Although all resulting diseases are infrequent, the level of incidence is indicated by the number of asterisks as low (***), very low (**), and extremely low (*). 7αC4:7α-hydroxy-4-cholesten-3-one; CA: cholic acid; CDCA: chenodeoxycholic acid; DHCA: 3α,7α-dihydroxy-5β-cholestanoic acid; THCA: 3α,7α,12α-trihydroxy-5β-cholestanoic acid
The shortening of the cholesterol side chain begins with the 26-hydroxylation due to the mitochondrial enzyme CYP27A1 [11]. Subsequent oxidation to an aldehyde and then to a carboxylate group allows the formation of the C27-BA intermediates DHCA and THCA. The hydroxylation of the pro-chiral side chain by CYP27A1 results in the exclusive formation of the 25R-stereoisomer of both compounds, which must then be activated by a thioester bond with an acetyl-CoA molecule. Both BACS and very long-chain acyl-CoA synthetase (VLCS) are involved in the generation of C27-BA-CoAs, which are taken up by the peroxisome through the ABCD3 transporter, also called PMP70. The reactions in this organelle start with the racemization of the C25 chiral center from the R- to the S-configuration by AMACR. Next, the stereospecific enzyme ACOX2 oxidizes the 25S-isomers, generating a double bond at position C24, which is then hydroxylated and subsequently oxidized to generate a 24-keto group by the action of HSD17B4 or DBP. SCPx or SCP2 accounts for the thiolytic cleavage of this ketone into propionyl-CoA and CA-CoA or CDCA-CoA. Finally, the conjugation of these BA-CoAs with glycine or taurine is carried out by BAAT, generating the four primary human BAs, i.e., taurocholic acid (TCA), GCA, taurochenodeoxycholic acid (TCDCA) and glycochenodeoxycholic acid (GCDCA).
In this metabolic pathway, which is quantitatively more important during early life and infancy, the first step is the formation of a C27-carboxylic acid, which is then followed by modifications of the steroid nucleus and further peroxisomal chain shortening to finally generate C24-BAs, preferentially CDCA [12]. The acidic pathway is initiated by CYP27A1, which carries out the hydroxylation of the C26 position generating the 25R-stereoisomer. Despite catalyzing the first reaction of the process, CYP27A1 is not its limiting enzyme. Instead, the crucial step appears to be the steroidogenic acute regulatory protein-related lipid transfer domain 1 (STARD1)-mediated transport of cholesterol to the inner mitochondrial membrane [13]. Once (25R)-3β-hydroxy-5-cholestenoic acid is obtained, its 7α-hydroxylation is carried out by CYP7B1. This constitutes a difference from the neutral pathway in which this hydroxylation is mediated by CYP7A1. Next, the same enzymes involved in the classic pathway (HSD3B7, AKR1D1, and AKR1C4) complete the ring modifications. Other oxysterols such as 24- and 25-hydroxycholesterol, highly relevant in regulating lipid metabolism, can also be incorporated into the biosynthesis of BAs at the beginning of this pathway. Both metabolites can be obtained by the action of CYP27A1. Moreover, brain CYP46A1 can also minimally contribute to this metabolic route through the synthesis and release to the blood of 24-hydroxycholesterol [14].
This route shares with the acidic pathway the initial formation of (25R)-3β-hydroxy-5-cholestenoic acid through CYP27A1. Subsequently, the shortening of the side chain is carried out at the peroxisome to obtain 3β-hydroxy-5-cholenoic acid, whose steroidal nucleus is then modified to finally generate C24-BAs, mainly CDCA. Although the contribution of this route to the biosynthesis of BAs is unknown, the presence of 3β-hydroxylated intermediates in amniotic fluid suggests its importance during intrauterine life [15].
This alternative route of BA synthesis, in which the side chain shortening is independent of peroxisomal β-oxidation, begins with the 25-hydroxylation of 5β-cholestane-3α,7α,12α-triol by the action of the microsomal enzyme CYP3A4 [16]. The product of this reaction, 5β-cholestane-3α,7α,12α,25-tetrol is then hydroxylated to 5β-cholestane-3α,7α,12α,24S,25-pentol [17]. This hydroxylation is likely catalyzed by CYP27A1, because patients with cerebrotendinous xanthomatosis (CTX), who lack active CYP27A1, accumulate cholestanetetrol when the direct precursor is administered [18, 19]. Finally, conversion to CA would be carried out rapidly through a 24-keto intermediate by one or more cytosolic enzymes with stereospecific selectivity for the 24S isomer [18].
Besides the amidation with taurine or glycine, under certain circumstances mature C24-BAs can be additionally conjugated with other polar polyatomic moieties, which results in compounds with enhanced water-solubility, reduced cytotoxicity, and facilitated renal elimination. More than 70% of BAs in urine are conjugated at 3α-position with a sulfate group, being this reaction carried out by sulfotransferase family 2A member 1 (SULT2A1) [20]. This biotransformation is particularly relevant in the case of lithocholic acid (LCA), a very hydrophobic and toxic BA, which is sulfated in high proportion, thus increasing its water solubility and facilitating its renal and fecal excretion [21]. Glucuronidation is mediated by uridine-diphospho (UDP) glucuronosyltransferase family 1 member A3 (UGT1A3), as well as other enzymes of the UGT family. This conjugation reaction can be performed at different sites of the molecule, depending on the involved isoenzyme, e.g., UGT1A3 mainly generates glucuronide-BAs at C24 position [22]. These conjugated BA species are a minority in healthy individuals but can become quantitatively relevant in patients with cholestasis [20, 23].
Up to now, four inborn errors affecting the enzymes involved in transforming the cholesterol ring during BA biosynthesis have been identified (Table 1, Figure 2).
Deficient CYP7A1 activity was described in three siblings with statin-resistant hypercholesterolemia. Interestingly, two of these patients were male and had hypertriglyceridemia and a history of biliary lithiasis. One presented an accumulation of cholesterol in the liver biopsy, along with decreased fecal excretion of BAs and an increased proportion of CDCA in feces. All three individuals were homozygous for a mutation in the CYP7A1 gene, leading to a frameshift (L413fsX414) that markedly alters the active site of the enzyme and subsequent results is the lack of function [24]. CYP7A1 deficiency does not lead to acute liver disease, as the reduction in BA synthesis by the neutral pathway can be compensated by an increment of the alternative acidic route. However, pathological repercussions appear in adulthood. As its inheritance seems co-dominant, individuals with this defect in heterozygosis also have high cholesterol levels, early gallstones formation, and even premature coronary and peripheral vascular disease [24]. Mice lacking CYP7A1 display reduced BA synthesis, while cholesterol serum levels and mortality differ between models probably due to their different genetic backgrounds [25–28].
Deficiency in HSD3B7 activity [Online Mendelian Inheritance in Man (OMIM): 607765] is the most frequent genetic defect affecting BA synthesis. In an American study from 1987 to 2016, HSD3B7 deficiency was found in 34% out of 297 patients with inherited errors of BA metabolism included in that cohort [29]. The inheritance of this disorder, characterized by a diverse spectrum of mutations in the HSD3B7 gene, is autosomal recessive [30]. In these patients, there are low levels of mature BAs due to their inability to reduce the double bond in the Δ5 position of the cholesterol ring, preventing the epimerization of the hydroxyl in 3β and causing the accumulation of unsaturated 3β,7α-dihydroxy-5-cholenoic acid and 3β,7α,12α-trihydroxy-5-cholenoic acid, which are usually sulfated in the C3 position. It has been proven that 3β-hydroxy-5-cholenoic acids fail to act as agonists of the main nuclear receptors involved in BA homeostasis farnesoid X receptor (FXR), pregnane X receptor (PXR) and constitutive androstane receptor (CAR), resulting in their impairment to exert hepatoprotective actions and thus explaining the mechanism for progressive cholestatic liver disease [31]. Sulfation of these toxic Δ5-intermediates will favor their elimination, as is the case for LCA [21]. Serum aminotransferases [alanine aminotransferase (ALT), aspartate aminotransferase (AST)] are elevated, while gamma-glutamyl transpeptidase (GGT) levels are normal. Clinically, HSD3B7 deficiency is characterized by neonatal cholestasis with jaundice, impaired absorption of lipids and fat-soluble vitamins, and steatorrhea, which progresses to more severe liver alterations, such as cirrhosis and hepatomegaly, with or without splenomegaly [1, 32]. However, in some cases, the clinical presentation is atypical [33] or even asymptomatic [34]. HSD3B7 deficiency in mice leads to cholesterol malabsorption and a very high mortality rate [35]. Early treatment with CA or CDCA is effective in normalizing liver function in these patients [33, 36]. Accurate determination of 3β-hydroxy-Δ5-BA sulfates is critical for both diagnosis and dose titration and monitoring response to therapy with primary BAs [4, 5].
Deficiency in AKR1D1 activity (OMIM: 235555) is the second most frequent disorder of BA metabolism, accounting for 20% of cases in the American cohort mentioned above [29]. Patients with this disorder show similar symptomatology to those with HSD3B7 deficiency. Still, in this case, there is an accumulation of 4-cholenoic BAs, which may have a 3-oxo group, along with the presence of 5α-BAs (flat “allo” BAs) [37]. Liver function tests show elevated serum aminotransferases and GGT. The typical clinical presentation resembles neonatal hepatitis, with rapid progression of liver disease, leading to cirrhosis and liver failure in infancy. In contrast to patients, mice with AKR1D1 deficiency do not show overt signs of cholestasis, hepatic inflammation, or liver damage [38]. Due to the reduced synthesis of primary BAs, treatment based on BA replacement therapy could be beneficial for these patients [37]. However, conversely to HSD3B7 deficiency, treatment of pronounced AKR1D1 deficiency with CDCA worsen cholestasis and liver function tests. This may be due to the enhanced generation in these patients of 3-oxo-4-cholenoic acids which cannot be sulfated at the C3 position, and which inhibit the secretion of exogenously administered CDCA [3]. Relief of cholestasis and improvement of liver function in these patients is achieved by administration of the less toxic CA in combination with CDCA [39]. Different autosomal recessive mutations in the AKR1D1 gene, both in homozygosis and heterozygosis, have been associated with this disorder. Patients with a longer prothrombin time have a worse prognosis and a poorer response to treatment with BAs [40]. Importantly, secondary AKR1D1 deficiency may occur due to several liver diseases, highlighting the need for genotyping AKR1D1 deficient patients to elucidate the actual etiology of the disease [41].
This enzyme is specific to the alternative acidic pathway. The lack of CYP7B1 enzymatic activity (OMIM: 613812) is one of the rarest defects in BA synthesis, as it has been reported in few children [29]. However, a recent study on Taiwanese infants suggested that the incidence of this disorder may be higher than believed due to its misdiagnosis in children [42]. The accumulation of Δ5-monohydroxylated and saturated 3β-monohydroxylated BAs leads to severe neonatal liver disorders such as progressive cholestasis, fibrosis, and cirrhosis with fatal consequences [43]. The serious clinical consequences of CYP7B1 deficiency highlight the importance of the alternative acidic pathway in early life in humans. Conversely, in Cyp7b1−/− mice, BA metabolism as well as liver function are normal, and the loss of this enzyme is compensated by increases in the synthesis of BAs by other pathways [44]. Along with 3β-hydroxy-Δ5-cholenoic acid, which makes up 90% of total plasma BAs, various oxysterols such as 25-hydroxycholesterol and 27-hydroxycholesterol accumulate and help to differentiate this disorder from AKR1D1 deficiency [45]. Serum aminotransferases are markedly elevated, while GGT levels are normal. Unlike other defects in enzymes of BA synthesis involved in the biotransformation of the steroid ring, CYP7B1 deficiency is a particularly severe condition that does not respond to CA. As therapeutic options, liver transplantation [46] or CDCA treatment [47] has been effective in some patients.
Altered shortening of cholesterol side chain to biotransform C27 steroid molecules to mature C24-BAs can be due to dysfunctional organelles or specific enzymes involved in the cleavage process (Table 2).
Deficient mitochondrial CYP27A1 activity (OMIM: 213700) causes CTX, a rare autosomal recessive lipid storage disease. Its clinical manifestations in affected children include neonatal cholestasis, diarrhea, cataracts, and growth retardation. The situation worsens in adults with the onset of xanthomas and neuropsychiatric symptoms such as ataxia and dementia [48]. This alteration is characterized by a deficit of BAs and a deposit of cholesterol and cholestanol in different tissues. The blockade of the neutral pathway at this level generates an accumulation of 5β-cholestane-3α,7α,12α-triol, which is diverted through the 25-hydroxylation route for its biotransformation into CA [19]. However, this alternative pathway cannot entirely compensate for the lack of CYP27A1-mediated side chain shortening for C24-BA production. Hence, bile alcohols are generated and accumulated, which is a helpful plasma biomarker for the diagnosis of this disorder [49]. On the contrary, mice lacking CYP27A1 show a milder phenotype with lower accumulation of oxysterols and BA intermediates, probably due to a more active 25-hydroxylation pathway in this species [50, 51]. Consistently, these animals do not develop metabolic and neurologic CTX-related pathological abnormalities [52, 53]. The treatment of CTX patients with CDCA restores normal C24-BA levels and decreases those of cholestanol and bile alcohols, thus preventing severe clinical symptoms if the treatment is started early enough [54].
Peroxisomes are involved in many cellular functions, e.g., long-chain fatty acid β-oxidation, pipecolic and phytanic acid oxidation, synthesis of plasmalogens, cholesterol, BAs, and regulation of the oxidation-reduction status. Therefore, disorders accounting for impaired peroxisome biogenesis or specific defects in any particular peroxisomal enzyme involved in these metabolic pathways lead to the accumulation of various metabolites, which may be deposited in several tissues, consequently causing pathophysiological alterations [55]. These defects affecting BA metabolism are described below.
Zellweger spectrum disorders (ZSDs) are a group of diseases characterized by the absence of functional peroxisomes. Several mutations affecting members of the PEX gene family are involved in ZSDs. These mutations impair the transport of certain proteins to the peroxisomal membrane and matrix during the biogenesis of these organelles. This group of disorders includes Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease. Although ZSDs are clinically heterogeneous, they share features such as liver dysfunction, developmental delay, adrenocortical dysfunction, and neurological abnormalities (e.g., hearing and vision impairment). ZSD patients lack mature C24-BAs, whereas their precursors DHCA and THCA accumulate. There are also increased levels of C29-dicarboxylic acid, which is likely generated by elongation of THCA-CoA. The detection of these BA intermediates in serum can be used in the diagnosis of these patients [56]. Their treatment is based on symptomatic and supportive therapies [57]. Several animal models lacking different PEX proteins have been developed. Their phenotypes resemble those typical of patients with ZSDs. Thus, Pex5 and Pex13 knockout mice show intrauterine growth retardation, hypotonia and high neonatal mortality, associated with alterations in neuronal maturation and migration, together with biochemical alterations similar to those of ZSDs [58, 59]. Mice lacking PEX2 develop early intrahepatic cholestasis leading to steatohepatitis, with reduction of C24-BAs and accumulation of C27 intermediates [60].
Other peroxisomal disorders are characterized by the alteration in the transport of substrates into the peroxisomal matrix via the ATP-binding cassette (ABC) pump ABCD3, also known as PMP70, accounting for the peroxisomal uptake of THCA-CoA and DHCA-CoA. This disorder has been described in a patient with hepatosplenomegaly who presented plasma accumulation of THCA-CoA, DHCA-CoA, and C29-dicarboxylic acid [61]. Abcd3 knockout mice also show high levels of these BA intermediates in serum and an increase of liver weight [61]. On the other hand, although X-linked adrenoleukodystrophy (X-ALD) due to mutations in the ABCD1 gene has never been associated with alterations in BA metabolism, a decrease in mature C24-BA levels has been reported in a recent case of X-ALD [62]. However, the absence of inactivating mutations in other peroxisomal enzymes of the BA biosynthetic pathway remains to be demonstrated and the actual link between BA synthesis and X-ALD elucidated.
Deficiency in AMACR activity (OMIM: 614307) is an autosomal recessive disorder characterized by plasma accumulation of pristanic acid and C27-BAs. A feature that facilitates the diagnosis is that, due to the lack of racemase enzymatic activity, only the 25R-isomers of THCA, DHCA, and pristanic acid are accumulated. The clinical presentation of this disease is highly variable. Affected children have malabsorption of fat-soluble vitamins, vitamin K deficiency-associated coagulation disorders, cholestasis, and hepatitis, although they respond favorably to treatment with primary C24-BAs [63, 64]. Adult presentation is characterized by sensory and motor degenerative neuropathy, with high interindividual variability. Thus, peripheral neuropathy, cognitive deficits, retinitis pigmentosa, and cerebellar dysarthria, among other alterations, may appear [65, 66]. Surprisingly, AMACR deficient mice do not develop clinical symptoms, probably due to a metabolic adaptation that reduces intestinal absorption of cholesterol and liver biosynthesis of different sterols, keeping the overall cholesterol levels in normal ranges [67].
Deficiency of ACOX2 activity (OMIM: 617308) results in impaired peroxisomal metabolism of branched-chain fatty acids and BA intermediates. A few cases of ACOX2 deficiency have been described so far. The first patient reported was a 16-year-old male diagnosed with persistent hypertransaminasemia upon exposure to non-steroidal anti-inflammatory drugs [68]. The finding of the homozygous mutation c.673C>T (p.Arg225Trp) in the ACOX2 gene was associated with decreased activity of the enzyme, leading to serum and urine accumulation of THCA and DHCA [69]. Different mutations in the same gene reported in two additional patients have been associated with more severe clinical manifestations. Both children had consanguineous parents and died suffering from severe hepatic and neurological alterations. One of these patients carried a homozygous premature termination mutation in ACOX2 (p.Y69*) [70], whereas the other one was homozygous for a four nucleotide deletion leading to a premature stop codon (p.Thr154Serfs*25) [71]. Both patients presented elevated serum levels of C27-BAs. A more recent study has detected a new mutation and identified several mutations with a potential dysfunctional impact on ACOX2 activity. Moreover, in two patients, the coexistence of two different mutations in heterozygosis but on different alleles also resulted in metabolic disorder leading to C27-BA accumulation [72]. Interestingly, this study revealed that patients with ACOX2 deficiency-associated hypertransaminasemia (ADAH) could benefit from treatment with UDCA, which normalizes serum levels of aminotransferases [72]. Two Acox2 knockout mice models have recently been reported. In one of these studies, the animals spontaneously developed steatosis, liver inflammation with fibrosis, and eventually liver cancer [73]. Unfortunately, the authors did not provide information regarding BA metabolism in this case. In contrast, in the other murine model of ACOX2 deficiency, developed in our laboratory, young animals do not show signs of severe liver disease. These mice partially mimic the phenotype observed in ADAH patients as far as BA metabolism is concerned [74].
Because DBP is a complex enzyme with more than one activity, which justifies its name, inborn errors affecting the HSD17B4 gene (OMIM: 261515) can impair the two sequential reactions catalyzed by this enzyme during the peroxisomal step of BA metabolism. Consequently, DBP disorders can be classified into three groups: type I, characterized by the absence of both hydratase and dehydrogenase activity; type II, in which only the hydratase activity is impaired; and type III, in which only dehydrogenase activity is deficient. The three phenotypes have similar severe clinical symptomatology, with most patients not surviving beyond the age of two years. Although the clinical manifestations show marked interindividual variability, the most frequent alterations are neonatal hypotonia accompanied by seizures and cranial dysmorphia, delayed growth and psychomotor development, visual and auditory system failures, and hepatomegaly. Postmortem liver biopsy carried out in some cases has shown fibrosis, steatosis, hemosiderosis, and signs of cholestasis. The few patients with prolonged survival reported so far had a severe delay in acquiring motor skills, lack of the ability to use language, audiovisual defects, and signs of demyelination. Regarding biochemistry data, elevated plasma levels of very long-chain fatty acids, THCA and DHCA have been found. Although the accumulation of these C27-BA intermediates does not occur in all patients, there seems to be an inverse correlation between their plasma levels and patient survival [75]. Concerning mice models of HSD17B4 deficiency, the absence of this protein and the accumulation of the mentioned metabolites cause growth retardation during the first weeks of life and lead to premature death [76, 77].
Only two cases of patients with a deficiency of SCPx activity (OMIM: 613724), a peroxisomal enzyme required for the last step of β-oxidation of branched-chain fatty acids, have been reported [78]. The first patient was a male with a medical history of spasmodic torticollis, dystonic head tremor, dysphemia, and fertility disturbances such as hypergonadotrophic hypogonadism and azoospermia. Worsening neurological symptoms revealed hypothalamic, pontine, and occipital brain lesions, as well as hyposmia, slight hypoacusis, saccadic eye movements, diminished reflexes of lower extremities, slight cerebellar ataxia, and gait and balance impairment. Nevertheless, liver biochemical markers were normal, and traces of C27-BAs (DHCA and THCA) were barely detected in plasma. In contrast, serum levels of pristanic acid were elevated. In addition, large amounts of bile alcohols were detected in urine [78]. Although the second patient carried in heterozygosis a different mutation affecting SCPx, the clinical features of the disease were similar, with motor disturbances in the hands, balance problems, and deafness. Subsequent medical examinations revealed alterations in proprioception, mild dysmetria, and dysdiadochokinesis. In addition, serological tests showed an increase in liver markers such as aminotransferases, GGT, and creatine kinase, as well as the accumulation of pristanic acid [79]. In both cases, symptoms improved after starting a phytanic acid (a precursor of pristanic acid) restricted diet. Cholesterol and BA metabolism alterations, without signs of neuropathy, have been reported in SCP2 knockout mice [80].
Conjugation of the C24-BAs with the amino acids glycine and taurine is the final step of BA synthesis by hepatocytes. This process requires the sequential action of two enzymes, BACS (encoded by SLC27A5), essential for re-conjugation of BAs deconjugated by gut bacteria, and BAAT (encoded by BAAT). Firstly, BACS catalyzes the activation of BAs via the formation of BA-CoA thioesters, which are needed for the glycine or taurine addition to BAs, a reaction catalyzed by BAAT [9]. Inborn errors affecting these two enzymes have been reported (Table 2).
So far, the only patients described with deficiency concerning BACS activity were two sisters carrying a homozygous mutation in the SCL27A5 gene. Although both girls had a similar abundance (85%) of unconjugated BAs in plasma and urine, only one of them developed liver disease. The reason for this discrepancy likely was that in the patient with more severe manifestations, BACS deficiency coexisted with another homozygous deleterious mutation in the ABCB11 gene, encoding the bile salt export pump (BSEP), which plays an essential role in the canalicular secretion of BAs. In contrast, her sister was heterozygous for this mutation [81]. Mice lacking BACS show a high proportion of unconjugated BAs in bile, without signs of disease. However, these animals do no gain extra weight when fed with a high-fat diet [82, 83].
In ten pediatric patients harboring a lack of enzymatic activity in BAAT (OMIM: 619232), this deficiency caused malabsorption of fat-soluble vitamins and increased prothrombin time. The rest of the clinical manifestations showed marked interindividual variability. Thus, neonatal cholestasis ranged from mild to very severe, with liver transplantation being necessary for one of the patients who developed biliary cholangiopathy, eventually leading to cirrhosis and liver failure. Data obtained from the liver biopsy of several of these patients showed idiopathic neonatal hepatitis with ductular proliferation and fibrosis. In addition, four patients suffered from rickets because of vitamin D deficiency. As could be expected, biochemical signs included the absence of glyco- and tauroconjugated BAs in plasma, urine, and bile, where unconjugated CA was the most abundant BA [84]. BAs in the liver of BAAT-deficient mice are mostly unconjugated. As a response to facilitate their detoxification, BA polyhydroxylation is markedly enhanced. These mice also display defective body weight gain [85]. The similarity in the BA pattern of patients with BAAT or BACS deficiency determines that the only way to discriminate between both disorders is through genotyping or immunolabeling analysis of liver tissue obtained by biopsy. Therapy based on the oral administration of conjugated CA (GCA) has improved growth rate and fat-soluble vitamin absorption in patients with BA amidation defects [86].
The general conclusion of this review is that, in patients with the clinical characteristics described above, after ruling out the presence of more frequent pediatric liver diseases, the existence of an inborn error in BA metabolism must be elucidated. Despite the difficulty in discriminating between different types of cholestatic conditions associated with these rare genetic defects, it is evident that a crucial goal in the diagnosis of these patients is to identify the exact cause of the failure in this aspect of liver function as soon as possible. An accurate and early diagnosis will direct the specific pharmacological treatment, which can prevent the progression to more severe stages of liver damage. Fortunately, the advance in analytical techniques such as LC-MS/MS to determine BA profiles, particularly measuring in a sensitive manner these rare and less abundant species in the serum of normal individuals, and available sequencing methods to identify the underlying genetic defect permit us to carry out a rapid and helpful screening among patients with typical clinical manifestations and biochemical signs of cholestasis and liver damage.
ABCD3: ATP-binding cassette subfamily D member 3
ACOX2: acyl-coenzyme A oxidase 2
AKR1C4: 3α-hydroxysteroid-dehydrogenase
AKR1D1: Δ4-3-oxosteroid-5β-reductase
AMACR: α-methyl acyl-coenzyme A racemase
BAAT: bile acid coenzyme A: amino acid N-acyl transferase
BACS: bile acid-coenzyme A synthetase
BAs: bile acids
CA: cholic acid
CDCA: chenodeoxycholic acid
CoA: coenzyme A
CTX: cerebrotendinous xanthomatosis
CYP27A1: sterol 27-hydroxylase
CYP7A1: cholesterol 7α-hydroxylase
CYP7B1: oxysterol 7α-hydroxylase
CYP8B1: sterol 12α-hydroxylase
DBP: D-bifunctional protein
DHCA: 3α,7α-dihydroxy-5β-cholestanoic acid
GCA: glycocholic acid
GGT: gamma-glutamyl transpeptidase
HSD3B7: 3β-hydroxy-Δ5-C27-steroid-oxidoreductase
OMIM: Online Mendelian Inheritance in Man
PEX: peroxisomal biogenesis factor or peroxin
PMP70: peroxisomal membrane protein 70
SCP2: sterol carrier protein 2
THCA: 3α,7α,12α-trihydroxy-5β-cholestanoic acid
UDCA: ursodeoxycholic acid
UGT1A3: uridine-diphospho glucuronosyltransferase family 1 member A3
X-ALD: X-linked adrenoleukodystrophy
ZSDs: Zellweger spectrum disorders
REE, EH, ASM and PSS played a main role in collecting information; JJGM coordinated the study; MJM wrote the first draft.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This study was funded by the CIBERehd (EHD15PI05/2016) and “Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III”, Spain (PI19/00819 and PI20/00189, co-funded by European Regional Development Fund/European Social Fund, “Investing in your future”); “Junta de Castilla y Leon” (SA074P20); Fundació Marato TV3 (Ref. 201916-31), Spain; AECC Scientific Foundation (2017/2020), Spain; “Centro Internacional sobre el Envejecimiento” (OLD-HEPAMARKER, 0348_CIE_6_E), Spain. REE was supported by a predoctoral contract funded by “Junta de Castilla y Leon” and “Fondo Social Europeo” (EDU/574/2018). ASM and PSS are supported by a predoctoral scholarship (FPU) funded by the Ministry of Science, Innovation and Universities, Spain. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Maitane Asensio ... Jose J. G. Marin
Grégory Merlen ... Thierry Tordjmann
Beatriz Sanchez de Blas ... Marta R. Romero
Carola Dröge ... Verena Keitel
Vasiliy Ivanovich Reshetnyak, Igor Veniaminovich Maev
Jose M. Pinazo-Bandera ... Miren García-Cortés
Elias Kouroumalis ... Argyro Voumvouraki
