 Perspective
Perspective

Affiliation:
1Department of Cell Death and Proliferation, Institute of Biomedical Research of Barcelona (IIBB), CSIC, 08036 Barcelona, Spain
2Liver Unit, Hospital Clinic I Provincial de Barcelona, Instituto de Investigaciones Biomédicas August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
3Center for the Study of Liver and Gastrointestinal Diseases (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
†These authors share the last authorship.
Email: sandra88tn@gmail.com
ORCID: https://orcid.org/0000-0002-2894-3188
Affiliation:
1Department of Cell Death and Proliferation, Institute of Biomedical Research of Barcelona (IIBB), CSIC, 08036 Barcelona, Spain
2Liver Unit, Hospital Clinic I Provincial de Barcelona, Instituto de Investigaciones Biomédicas August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
3Center for the Study of Liver and Gastrointestinal Diseases (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
4Center for ALPD, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, US
†These authors share the last authorship.
ORCID: https://orcid.org/0000-0003-3422-2990
Affiliation:
1Department of Cell Death and Proliferation, Institute of Biomedical Research of Barcelona (IIBB), CSIC, 08036 Barcelona, Spain
2Liver Unit, Hospital Clinic I Provincial de Barcelona, Instituto de Investigaciones Biomédicas August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
3Center for the Study of Liver and Gastrointestinal Diseases (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
4Center for ALPD, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, US
†These authors share the last authorship.
Email: cgrbam@iibb.csic.es
ORCID: https://orcid.org/0000-0002-2652-6102
Explor Dig Dis. 2022;1:170–186 DOI: https://doi.org/10.37349/edd.2022.00012
Received: June 14, 2022 Accepted: September 26, 2022 Published: December 30, 2022
Academic Editor: Kenichi Ikejima, Juntendo University Graduate School of Medicine, Japan
Metabolic zonation in the liver carries out the maintenance of organ and body homeostasis. Hypoxia is an inherent physiological feature of the liver and contributes to the zonal properties of the hepatic parenchyma. As a master regulator of hypoxia, the transcription factor hypoxia-inducing factor (HIF) is stabilized primarily by oxygen availability, and it is thought to contribute to steatohepatitis due to alcohol-related (ASH) and non-alcohol-related liver disease (NASH). Cholesterol has emerged as an important player in both diseases, and hypoxia increases hepatic cholesterol levels. Steroidogenic acute regulatory protein 1 (STARD1) is a mitochondrial outer membrane protein that transfers cholesterol to mitochondrial inner membrane for metabolic processing and acts as the rate-limiting step in the alternative pathway of bile acid synthesis in hepatocytes. STARD1 expression increases in ASH and NASH and determines the accumulation of cholesterol in mitochondria, which impacts the physico-chemical mitochondrial membranes properties and as a consequence impairs the activity of specific mitochondrial solute carriers, such as the 2-oxoglutarate carrier (2-OGC), limiting the exchange between cytosolic glutathione and mitochondrial 2-oxoglutarate (2-OG). Although HIF-1 is stabilized in hypoxia largely due to the requirement of prolylhydroxylases (PHDs) for oxygen to signal HIF degradation, PHDs are also dependent on 2-OG, and therefore it is conceivable that impairment of 2-OGC by STARD1-mediated cholesterol accumulation may contribute to HIF-1 stabilization due in part to decreased availability of cytosolic 2-OG. In this perspective, this review explores the interplay between HIF-1 stabilization and STARD1 induction and the potential contribution of this functional relationship to ASH and NASH.
Hypoxia is a stressful condition determined by the limited availability of oxygen. Although primarily recognized as a risk factor for cancer progression with poor prognostic outcome, hypoxia is an inherent feature of the liver acinus and is thought to contribute to the specific functions and differential gene expression of periportal (PP) and perivenous (PV) hepatocytes, which are exposed to a differential oxygen environment determined by liver physiology, with PV hepatocytes being relatively hypoxic with respect to PP hepatocytes [1]. The liver has zonal properties characterized by several factors, including oxygen homeostasis coming from the blood. The hepatic artery supplies highly oxygenated blood compared to hepatic portal vein, leading to a physiological oxygen gradient of 4–8% between the PP and pericentral areas, respectively [1]. Although the hepatic artery and portal vein are mixed towards the central vein of the hepatic lobule, this oxygen gradient leads to a liver zonation where PP and PV hepatocytes have different functional and gene expression signatures. Hypoxia signaling in hepatocytes is initiated by the stabilization of the master transcription factor, hypoxia-inducing factor-1 (HIF-1), which comprises a stable β subunit (HIF-1β) and a labile α subunit (HIF-1α) [2]. HIF-1 is stabilized primarily when oxygen availability is low, as prolylhydroxylases (PHDs) require oxygen to initiate a signaling pathway leading to HIF degradation. HIF-1α activation induces a specific gene program, which helps cope with the stress imposed by oxygen limitation [3].
Steroidogenic acute regulatory protein 1 (STARD1) is a cholesterol transporter protein that is located in the mitochondrial outer membrane (MOM). STARD1 translocates cholesterol from MOM to the mitochondrial inner membrane (MIM) for metabolic processing [4]. For instance, in the liver, the cholesterol transport into MIM acts as the rate-limiting step in the alternative pathway of bile acid (BA) synthesis [5], while in steroidogenic tissue of certain neurons, mitochondrial cholesterol is metabolized to pregnenolone [6, 7]. Compared to steroidogenic tissues, the expression of STARD1 in hepatocytes is low [8–10]. However, STARD1 becomes upregulated in acute liver injury and chronic liver diseases, causing the accumulation of cholesterol in mitochondria, which in turn leads to an increase in generation of reactive oxygen species (ROS) and mitochondrial dysfunction [11–15]. Glutathione (GSH) is a low-molecular-weight thiol with antioxidant properties and it is synthesized in the cytoplasm and transported to other organelles such as the mitochondria by specific solute carriers [16]. Cumulative evidence in kidney, brain, and liver pointed to the 2-oxoglutarate carrier (2-OGC, Slc25A11) and dicarboxylate carrier (DIC, Slc25A10), as two important carriers with mitochondrial GSH (mGSH) transport activity [17]. However, recent candidates, such as Slc25A39, have been proposed to contribute to mGSH transport [18, 19]. An important feature of the transport of mGSH is its sensitivity to the loss of mitochondrial membrane fluidity due to increased cholesterol accumulation. In this regard, mitochondrial cholesterol impairs Slc25A11 function and results in mGSH depletion [20, 21]. Whether the newly identified Slc25A39 carrier is sensitive or not to changes in mitochondrial membrane fluidity remains to be established. Besides their primary function as oxygen sensors, PHDs also require iron (II) and 2-oxoglutarate (2-OG) for activity, and hence can regulate HIF-1 stability independently of oxygen [22]. Therefore, since STARD1-mediated cholesterol accumulation decreases membrane fluidity and impairs Slc25A11 [23–25], it is conceivable that the increased ROS generation and limitation of 2-OG may contribute to HIF-1 stabilization. In this perspective, we present evidence supporting a potential interplay between HIF-1 stabilization and the cholesterol/STARD1 axis, contributing to mitochondrial damage and the development of metabolic liver diseases, in particular steatohepatitis due to alcohol-related liver disease (ASH) and steatohepatitis due to non-alcohol-related liver disease (NASH), two advanced stages of fatty liver disease. NASH is of special concern due to its association with obesity and insulin resistance and has become one of the most prevalent forms of chronic liver disease.
Hypoxia is a stressful condition that reflects a limitation in the availability of oxygen to cells. Hypoxia is a characteristic feature of tumor microenvironment, which is known to contribute to cancer cell survival and proliferation. Hypoxia is of particular relevance in the development of solid tumors, which ensues due to the disorganized architecture of tumor vasculature, resulting in poor oxygen delivery particularly to inner core of cancer cells [26]. The development of hypoxia in tumors represents a poor prognostic value for cancer patients [27–29]. One of the cardinal features of hypoxia development as solid tumor grows is the stabilization of HIF-1, which activates a genetic program related to angiogenesis, glucose metabolism, apoptosis, tumor growth, invasion, and metastasis, leading to the survival and expansion of cancer cells [30–32]. The active HIF-1 complex comprises a heterodimer constituted by a stable β subunit (HIF-1β) and a labile α subunit (HIF-1α), which encompasses three members (HIF-1α, HIF-2α, and HIF-3α) [33]. As inferred, the biological action of the HIF-1α/β heterodimer is dependent on the stability of the labile α subunit, which is determined largely by the availability of oxygen. In normoxia, HIF-1α is hydroxylated in proline residues by the PHDs in the cytosol, followed by HIF-1α ubiquitination by Von Hippel Lindau (VHL) tumor suppressor and the recruitment of E3 ubiquitin ligase, which signals the degradation of hydroxylated HIF-1α by the proteasome. PHDs, which encompass several isoforms (PHD1–3), are dioxygenases whose activity is dependent largely but not exclusively on oxygen availability [22], and therefore under hypoxia, HIF-1α cannot be hydroxylated by PHDs and becomes stabilized to dimerize with HIF-1β/Arnt, which then translocates to the nucleus where recruits the co-activators p300/CBP to bind to hypoxia response elements (HREs) within the promoters of target genes to complete transcriptional activity [2, 34–36]. Despite oxygen, PHDs are also dependent on iron and 2-OG [22], and changes in these cofactors also regulate PHDs activity, and hence the fate of HIF-1α (stabilization/degradation). Moreover, it has been shown that the activity of PHDs can be regulated during normoxia by oxidative stress and ROS generation determined from mitochondrial function [37–39]. Although its role in the activation of specific gene programs [e.g., epidermal growth factor (EGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF)] aimed to protect cancer cells during tumor development is well-recognized [40–48], HIF-1α has also been shown to regulate mitochondrial function [49, 50]. HIF-1α stabilization impairs mitochondrial function resulting in a paradoxical increase in ROS generation, leading to an amplification of further HIF-1α activation. Considering the multiple roles of HIF-1α in tumor progression and metastasis, various strategies have been developed to antagonize HIF-1α at different levels, including inhibition of messenger RNA (mRNA) expression, protein translation and stabilization, dimerization, DNA binding, and transcriptional activity [51]. Despite its emergence as a target in cancer cell biology, HIF-1α can affect early stages of cancer development, including hepatocellular carcinoma (HCC), which represents the end stage of chronic liver diseases, and thus a better understanding of the pathways of HIF-1α stabilization and role in liver diseases may offer an opportunity for intervention in pathophysiology of chronic metabolic diseases, including ASH and NASH.
Consistent with the crucial role of HIF-1α in cancer progression and in the determination of tumor diversity, there is clinical and experimental evidence involving hypoxia as an important player not only in hepatocarcinogenesis but also in the early phases of chronic liver diseases, such as ASH and NASH [52]. For example, 25–35% of obese individuals manifest recurrent apnea episodes during sleep, leading to nocturnal intermittent hypoxia, which as discussed below, can contribute to the increase in hepatic cholesterol levels [53–55], indicating that hypoxia signaling is involved in the regulation of hepatic lipid metabolism. For instance, it has been shown that the inactivation of the PHD1 led to hepatic steatosis and insulin resistance in mice fed with a low-fat diet, indicating that the stabilization of HIF-1α promoted liver steatosis [56, 57]. However, intriguingly PHD2 inhibition protected mice against diet-induced obesity and metabolic dysfunction [58], suggesting the specificity of PHD2 as a potential target for pharmacologic PHD inhibition. Additional experimental studies showed that HIF-1α and HIF-2α were involved in hypoxia-induced lipid accumulation in hepatocytes, being HIF-2α the major regulator [59]. In this regard, it has been shown that HIF-2α induced CD36 and adipose differentiation-related protein (ADRP), both involved in free fatty acids uptake [60, 61]. Moreover, reduction of hepatic HIF-2α by oxygen therapy in mice fed with high-fat diet decreased lipogenic gene expression [62], and the HIF-2α stabilization led to β-oxidation suppression via peroxisome proliferator-activated receptor α (PPARα) in steatotic hepatocytes [63]. Unlike HIF-2α, the role of HIF-1α in fatty liver disease, particularly in ASH, is less established. For instance, using an identical approach to cause ASH, characterized by steatosis and mild inflammation, recent data provided opposite findings regarding the role of HIF-1α in ASH progression. While Nath et al. [64] indicated that HIF-1α is a determinant of alcohol-induced lipid accumulation and liver injury, Nishiyama et al. [52] showed a protective effect of HIF-1α induction in alcohol-induced liver damage and steatosis. The underlying causes of this conundrum remain to be fully understood. Moreover, a similar protective role for HIF-1α was found in a different context of hepatic steatosis caused by feeding a choline-deficient diet [65]. Despite the unestablished role of HIF-1α in liver steatosis, the specific deletion of HIF-1α can also protect against liver fibrosis [66], in different contexts of liver diseases, including NASH [67, 68]. Moreover, additional evidence demonstrated that VHL overexpression that affects HIF-1α/HIF-2α stability reduces liver inflammation and fibrosis [69]. Recent research showed that hypoxia signaling in hepatic stellate cells (HSCs) induces VEGF-dependent angiogenesis, leading to the acceleration on liver regeneration [70]. Furthermore, a protective role of hypoxia and HIF-1α stabilization has been demonstrated recently in murine models of acute hepatic inflammation, where the specific deletion of HIF-1α in T-cells impairs their function to resolve inflammation [71]. Finally, HIF-1α is activated though adenosine receptors, particularly ADORA2B, and its activation results in an increased signal transduction, leading to excessive inflammation that facilitates injury resolution and ischemia tolerance [72]. Therefore, the complex functions of the different HIFs proteins in each context and their potential as therapy targets with beneficial effects on liver diseases remain to be fully elucidated.
Cholesterol is an essential component of membrane bilayers and a key determinant of their physical-chemical properties. As many different signaling platforms reside in the plasma membranes of cells, cholesterol fulfills essential functions in cell metabolism [73, 74]. All cells synthesize cholesterol de novo from acetyl-coenzyme A (CoA) in the endoplasmic reticulum (ER) in the mevalonate pathway in which farnesyl-PP serves as a branching point for the sterol arm starting with squalene, which is committed to synthesizing cholesterol in a series of steps that require oxygen consumption (Figure 1). The pathway of cholesterol synthesis is regulated by the master transcription factor sterol regulatory element protein 2 (SREBP-2), which induces key enzymes in the mevalonate pathway such as hydroxymethylglutaryl-CoA (HMG-CoA) reductase, the target of statins. The final product, cholesterol, turns off the mevalonate pathway by signaling the degradation of SREBP-2 to prevent unnecessary cholesterol synthesis. Besides this synthetic pathway, cholesterol can also be taken up from the diet as a component of lipoproteins followed by its distribution to different organs, including the liver. Once cholesterol arrives to the cell is delivered to endolysosomes, which in turn distributes it to other organelles by vesicle-mediated inter-organelle cholesterol transport or non-vesicular protein-mediated transfer, and incorporates it in the membrane where cholesterol plays its crucial structural and functional roles [73, 75]. Compared to plasma membrane, which represents about 25–30% of the total cholesterol pool, cholesterol is also transported to mitochondria, which represents a minor membrane destination accounting for about 2–4% of the total cholesterol levels in hepatocytes [75, 76].
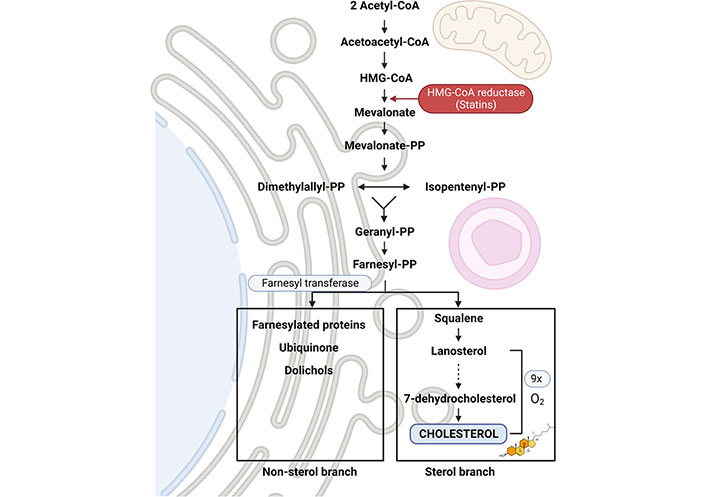
Diagram of the mevalonate pathway for the synthesis of cholesterol. The synthesis of cholesterol encompasses a large number of enzymes that convert acetoacetyl-CoA and acetyl-CoA to squalene. Among them, 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR) is the only limiting step for cholesterol synthesis, and the target of statins, which block the active site of the enzyme. Then, squalene is subsequently converted to lanosterol, which in turn is biotransformed to cholesterol after an oxygen-intensive process of 9 successive reactions (represented in a dot-lined arrow) on ER membrane in the sterol branch, which indicates that the availability of oxygen exerts another level of regulation of cholesterol synthesis. In parallel, farnesyl-PP is transformed into other compounds in the non-cholesterol branch
Despite its limited levels, the content of cholesterol in mitochondria plays important physiological roles, including the synthesis of steroid hormones and neurosteroids in steroidogenic tissues and specific neurons, respectively, and in the generation of BAs in hepatocytes in an alternative pathway [7, 12, 77, 78]. Importantly, the transport of cholesterol to the MIM is the rate-limiting step in the synthesis of steroids and BAs. Although the mechanism of this transport process is not completely elucidated, cholesterol transport to MIM occurs via a non-vesicular protein-mediated transfer that involves proteins marked with the steroidogenic acute regulatory protein-related lipid transfer (START) domain [74, 75, 77–79]. START proteins are a family of proteins that transport cholesterol and other lipids, such as oxysterols, phospholipids, sphingolipids, and fatty acids. The START members include STARD1, STARD3, also known as metastatic lymph node protein 64 (MLN64), and STARD4 subfamily (STARD4, STARD5, and STARD6). STARD1 (also known as StAR), the first member discovered, plays an essential role in the transport of cholesterol from the MOM to the MIM, where it is converted by cytochrome P450scc into pregnenolone, a common precursor of steroid hormones. The relevance of STARD1 in cholesterol transport to MIM has been best illustrated by the consequences of mutations in the human StAR gene, which cause the development of congenital adrenal hyperplasia (CAH) [9, 80]. In addition, mice with global StAR deletion die by lethal CAH within 7–10 days after birth despite hormone replacement therapy [81–83]. These findings not only evidence the key role of STARD1 in cholesterol transport to MIM for metabolism and generation of steroids, but also indicate that other members of the STAR family cannot compensate for the lack of STARD1, suggesting a key role for this founding member for cholesterol metabolism. STARD1 is a highly conserved nuclear-encoded phosphoprotein that contains a mitochondrial localization sequence, an amino-terminal amphipathic helix, and a sterol-binding domain. STARD1 is synthesized in the cytosol as a precursor with a calculated molecular mass of ~32 kDa, which is finally processed into a mature 30 kDa form that is localized within the mitochondrial matrix [84, 85]. Despite significant advances, the exact molecular mechanism whereby STARD1, which is located in MOM, transports cholesterol to MIM is incompletely unknown. In this respect, it has been shown that specific MOM proteins such as voltage-dependent anion channel 1 (VDAC1) and the phosphate carrier protein (PCP) may have a partnership to maximize STARD1 activity so that once it is phosphorylated by protein kinase A, requires PCP followed by the contact of phospho-STARD1 with VDAC1 [86]. Moreover, it was shown that overexpression of MLN64 in mice liver increased mitochondrial cholesterol levels, leading to an increase of superoxide anion, which in turn contributes to the decrease of the membrane potential (ΔΨm) and ATPase activity, with mild effect in metabolism and intracellular distribution of cholesterol [77, 87, 88]. Furthermore, MLN64 promotes lipidation of apoA-I and de novo lipid synthesis, and regulates genes influencing hepatic insulin resistance [89]. Besides MLN64, STARD4 is a soluble sterol transporter, transcriptionally regulated by SREBP-2 that appears to move cholesterol between liposomes as well as organelles, such as the mitochondria and the ER for cholesterol esterification [79]. Moreover, translocator protein (TSPO), a protein abundant in steroidogenic tissues and highly expressed in many types of cancer, is localized in the MOM. Interestingly, TSPO was proposed that together with STARD1 and VDAC1 form a transduceosome complex that transfers cholesterol into mitochondria [90]. In support for this role of TSPO in the transfer of cholesterol into mitochondria, recent studies showed that GL261 cells deficient in TSPO exhibit mitochondrial damage, accompanied by decreased ATP production and reduced mitochondrial oxidative phosphorylation [91]. However, other studies showed unimpaired steroidogenesis in MA-10 and primary Leydig cells deficient in TSPO, suggesting that the transport of cholesterol to mitochondria for steroid hormones synthesis was independent of TSPO [92–95]. Conversely, TSPO seems to play a crucial role in the murine development associated with hormone-induced steroidogenesis [95]. Therefore, further investigations are needed to clarify the interplay between STARD1 and TSPO and perhaps other mitochondrial partners in the regulated transport of cholesterol in mitochondria.
As mentioned above, most of the functional studies with STARD1 have been performed in steroidogenic cells and tissues where availability of cholesterol in mitochondria regulates the generation of steroid hormones [4]. In the liver, the basal expression of STARD1 is lower with respect to steroidogenic tissues, and thus its function in mitochondrial cholesterol metabolism and impact in the development of liver diseases have been much limited [96, 97]. Nevertheless, there has been evidence correlating the expression of STARD1 in the progression of NASH. For instance, using a cohort of an obese subject with simple steatosis or full-blown NASH, Caballero et al. [98] reported an increase in the expression of STARD1 in patients with NASH compared to those with simple steatosis. Interestingly, this increase in the expression of STARD1 paralleled the higher levels of free cholesterol content in liver samples from NASH subjects compared to individuals with simple steatosis [98]. As NASH can progress to HCC, a leading cause of cancer-related death whose impact is expected to raise due to its association with the obesity pandemic, recent findings have explored the expression of STARD1 in patients with NASH-driven HCC. In this regard, Conde de la Rosa et al. [12] recently reported an increase in the expression of STARD1 in liver samples from patients as well as in experimental models of NASH-HCC. Like in the cohort of obese patients with NASH, the increased expression of STARD1 was paralleled with higher levels of free cholesterol, indicating the coincidence of both events in NASH and that this tandem precedes the onset of HCC. In further analyzing the potential role of STARD1 in the development of NASH-driven HCC, there was a correlation between the levels of STARD1 and the increase in the profile of hepatic BAs content being higher in patients with NASH-driven HCC. Thus, while the alternative mitochondrial pathway of BAs synthesis is considered to contribute to a minor degree (< 10%) to the total BAs synthesized in hepatocytes mostly via the classic pathway regulated by CYP7A1 [99], the findings of Conde de la Rosa et al. [12] suggest that the higher rate of total BAs in patients with NASH-HCC could be derived from the mitochondrial alternative pathway regulated by STARD1. The recent generation of STARD1 floxed (STARD1f/f) mice allowed the production of the hepatocyte-specific STARD1 knockout (STARD1ΔHep) mice to assess the contribution of STARD1 to the BA generation and impact in NASH progression towards HCC. In this regard, STARD1ΔHep mice subjected to a nutritional approach to cause NASH-driven HCC exhibited less tumor burden and lower levels of BAs compared to STARD1f/f mice fed a high-fat high-cholesterol diet. Thus, these findings revealed a critical and early role of STARD1 in the progression of NASH to HCC. To further explore the role of STARD1 in other liver diseases, recent findings have shown a key role for STARD1 in acetaminophen (APAP) hepatotoxicity. While STARD1f/f mice exhibited APAP-mediated liver damage, STARD1ΔHep mice were protected against APAP-induced liver injury. In examining the underlying mechanisms, Torres et al. [11] showed that APAP-induced ER stress increased STARD1 expression as an early step that caused mitochondrial cholesterol accumulation and subsequent mGSH depletion. An interesting feature of these studies was the dissociation between STARD1 and MLN64 in the increase of mitochondrial cholesterol in APAP hepatotoxicity, where hepatocyte-specific STARD1 deletion has been shown to protect against APAP-mediated liver failure despite unchanged MLN64 expression, indicating the specific role of STARD1 in APAP liver toxicity. Prevention of STARD1 expression by chaperones (e.g., tauroursodeoxycholic acid) that prevent ER stress protected against APAP hepatotoxicity. Thus, taken together STARD1 induction emerges as an early event in NASH and HCC as well as in drug-induced liver injury, suggesting it as a potential target for liver disease. The role of STARD1 in other liver diseases, including ASH, is currently being investigated.
As mentioned above, the committed pathway of cholesterol synthesis from squalene is an oxygen intense process. There are several enzymes that require oxygen as cofactor for the step-wise progression towards the generation of the final product, cholesterol [100]. Indeed, the final steps from lanosterol to cholesterol conversion require nine molecules of dioxygen (Figure 1) [101]. Thus, as it can be inferred, cholesterol synthesis is a self-fulfilling pathway in normoxia, where the adequate oxygen tension ensures the efficient generation of cholesterol. However, cancer cells, including HCC cells, which are dependent on the synthesis of lipids to match tumor growth, exhibit an increased content of cholesterol levels, and statins use has been associated with a reduction in cancer-related mortality for different types of cancers [102, 103]. Thus, the oxygen-dependent generation of cholesterol posits a conundrum in growing solid HCC tumors, raising the question as to how tumor cells maintain a high-cholesterol tone in a hypoxic environment. The paradox of maintaining high-cholesterol levels despite low oxygen availability is reflected in subjects living at high altitude, who are at a risk of coronary heart disease due to elevated low-density lipoprotein (LDL) and total cholesterol levels caused by high-altitude hypoxia, disclosing the activation of an adaptive response that controls cholesterol homeostasis [104]. While the mechanisms underlying the increase in systemic cholesterol levels are not completely understood, there have been important advances accounting for the increase of hepatic cholesterol content associated with intermittent hypoxia, which is of relevance in obese patients [105, 106]. In this regard, the stabilization of HIF-1α and HIF-2α signal the increase of hepatic cholesterol content by suppressing its major route of catabolism into BAs. For instance, Ramakrishnan et al. [55] reported that Vhl loss of function in hepatocytes activates HIF-1α/HIF-2α and results in the increase in systemic and hepatic cholesterol levels. Intriguingly, Vhl disruption does not affect genes involved in the de novo cholesterol synthesis (e.g., HMGCoAR) nor intestinal cholesterol absorption (e.g., NPC1L1). Cholesterol conversion into BAs in the classical pathway is regulated primarily by the expression of Cyp7A1, which is feedback repressed by BAs through the enterohepatic circulation and FXR signaling. Importantly, Vhl disruption markedly repressed the circadian expression of Cyp7A1 and this effect was accompanied by a paralleled activation of the stress kinase c-Jun N-terminal kinase (JNK) [55]. The effect of hepatocyte-specific Vhl deletion on cholesterol homeostasis and Cyp7A1 repression was mediated by HIF-2α stabilization as shown in mice with deletion of Vhl and Hif-2α in hepatocytes. Using a model of chronic systemic hypoxia in which mice were exposed to 10% O2 for 3 weeks, Ramakrishnan et al. [55] revealed a 2-fold increase in liver cholesterol levels that was accompanied by a mild (about 30%) but significant decrease in the total pool of liver BAs. Interestingly, this outcome was accompanied by a robust repression of Cyp7A1 (> 90%) but not of Cyp27A1. As STARD1 expression is regulated by HIF-1α, consistent with the well-recognized activating effect of hypoxia in steroidogenesis and the presence of HRE in Star [107–109], these findings suggest that the maintenance of hepatic BAs during systemic hypoxia could be derived from the alternative mitochondrial pathway of BA synthesis governed by STARD1. Therefore, further investigation on the regulation of STARD1 expression by HIF-1α in the liver is needed.
Although hypoxia and HIF-1 signaling are closely linked as described above, hypoxia can alter the pattern of gene expression in both HIF-1-dependent and independent manners. Moreover, HIF-1 can be activated independently of hypoxia, involving different mechanisms including the induction of oxidative stress and generation of ROS through the inactivation of PHDs. In this regard, HIF-1α activation has been linked to mitochondrial ROS generation from diet-induced cholesterol increase [110, 111]. In this process, food-derived cholesterol and cholesterol esters contained within LDL are delivered to lysosomes through endocytosis via the LDL receptor (LDLR) [112]. Lysosomal hydrolysis of LDL-derived cholesterol esters releases cholesterol, which in excess can result in mitochondrial cholesterol deposition, leading to mitochondrial impairment, and ROS generation. An important event in the overproduction of ROS from mitochondria due to cholesterol accumulation is the depletion of mGSH, caused by the sensitivity of the Slc25A11 carrier to the disruption of mitochondrial membrane fluidity imposed by cholesterol [113]. This process has been characterized as an early event in NASH development as mGSH depletion sensitizes steatotic livers to inflammatory cytokines-mediated liver injury and further aggravation of liver steatosis due to mitochondrial dysfunction [110, 113]. As steatosis has been suggested to impair oxygen distribution in the liver acinus [114], the impairment in mitochondrial function and ROS generation and subsequent HIF-1α stabilization could engage in a self-amplifying cycle in steatotic livers. To further support the regulation of HIF-1α by cholesterol in normoxia, Anavi et al. [115] incubated cultured hepatocytes with cholesterol, which resulted in an early accumulation of cholesterol in mitochondria that preceded the subsequent HIF-1α stabilization, involving mitochondrial ROS production, and an increase in nitric oxide (NO) levels. Moreover, as the induction of HIF-1α activation was abolished upon mitochondrial DNA depletion, the requirement for mitochondrial ROS and NO in the HIF-1α stabilization suggests the involvement of peroxynitrite generation [115]. Besides ROS generation through mGSH depletion, the impairment of Slc25A11 by cholesterol accumulation can also result in decreased cytosolic 2-OG levels [116]. This event can have a functional impact on the activity of PHDs and subsequent HIF-1α stabilization, as PHDs are also dependent on 2-OG for optimal function (Figure 2). In line with these findings, disruption of 2-OG dehydrogenase (OGDH) complex and lipoic acid synthase (LIAS) stabilizes HIF-1α in a non-hydroxylated form in aerobic conditions [117]. Besides the impact of impairment of mitochondrial function and ROS generation in HIF-1α stabilization, there are also data suggesting that HIF-1α regulates mitochondrial function, establishing a vicious cycle [42, 43]. For instance, it has been shown that HIF-1α represses iron-sulfur cluster assembly proteins ISCU1/2 impairing the mitochondrial respiration and electron transfer chain activity by the activation of the microRNA-201 transcription [118]. In addition, HIF-1α can induce mitochondrial autophagy by the activation of the apoptotic protein BCL-2/adenovirus E1B 19KD interacting protein 3 (BNIP3) and the constitutive expression of Beclin-1 and Atg5, inducing mitochondrial autophagy in response to hypoxia [119]. Moreover, several studies have demonstrated that hypoxia induces the localization of HIF-1α with mitochondria [111, 120], where it may exert a beneficial mechanism for the regulation of mitochondrial function and metabolism. In this regard, it has been demonstrated that the mitochondrial protein VDAC1 works as part of the HIF-1α-mortalin-VDAC1-HK-II complex, which mediates the inhibition of hypoxia-induced apoptosis and promotes cancer cell resistance to chemotherapy [121]. These findings suggest a mutual upregulation between HIF-1α and STARD1, where STARD1-mediated cholesterol trafficking to mitochondria results in HIF-1α stabilization by the increment of mitochondrial ROS generation and NO, affecting the levels of 2-OG and consequently the PHD activity. The HIF-1α stabilization, in turn, leads to a self-amplifying cycle favoring the STARD1 overexpression and followed mitochondrial cholesterol accumulation. These findings identify important mechanistic connections between hypoxia and mitochondrial cholesterol leading to changes in cell metabolism and function.
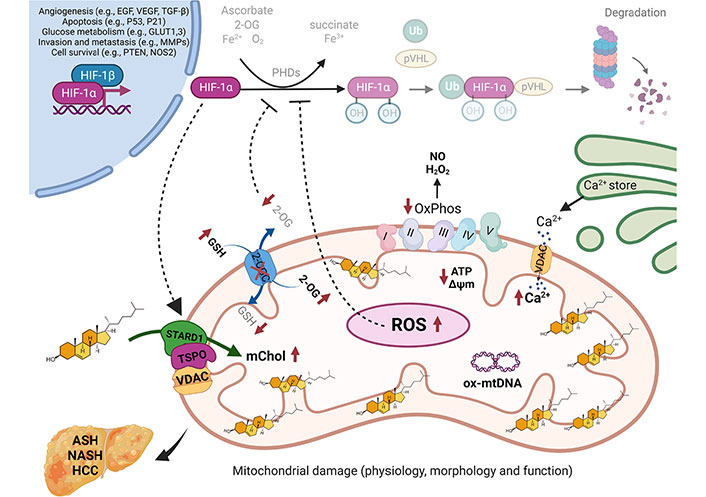
Regulation of HIF-1α by mitochondrial cholesterol loading. Under physiological conditions, the continuous presence of ROS production is coped by mGSH antioxidant mechanism, maintaining the mitochondrial functions. HIF-1α is hydroxylated by the PHDs in the presence of O2, Fe2+, 2-OG, and ascorbate, in the cytosol, and consecutively, HIF-1α is ubiquitinated by VHL and degraded by the proteasome. In cholesterol-enriched mitochondria, mediated by STARD1 and the transduceosome, mitochondrial membrane fluidity decreases, besides regulates the assembly of respiratory supercomplexes, results in an impaired activity of the OGC, which in turn leads to a depletion of mGSH and consequently to a depletion of cytosolic 2-OG. The subsequent increase in ROS and the cytosolic 2-OG depletion (dot-lined arrows) impact PHD, resulting in their inhibition and VHL is no longer able to bind and target HIF-1α for proteasomal degradation, which leads to HIF-1α stabilization, which in turn, leads to a self-amplifying cycle favoring the STARD1 overexpression (dot-lined arrow). There, HIF-1α dimerizes with HIF-1β in the nucleus, inducing the transcription of target genes related to angiogenesis, apoptosis, glucose metabolism, invasion, metastasis, and cell survival, genes related to the progression of liver diseases, such as ASH, NASH, and HCC. GLUT1,3: glucose transporter1,3; mChol: mitochondrial cholesterol; MMPs: matrix metalloproteinases; NOS2: nitric oxide synthase 2; ox-mtDNA: oxidized mitochondrial DNA; OxPhos: oxidative phosphorylation; PTEN: phosphatase and tensin homolog; TGF-β: transforming growth factor-β; Ub: ubiquitin
Hypoxia is an important event in several pathologies, and of particular relevance in liver diseases in which the functional segregation of PP and PV hepatocytes is determined in part by the relative hypoxia along the acinus imposed by the anatomical characteristics of the liver. Although hypoxia can induce gene expression by several independently of HIF-1, this transcription factor is a master regulator of the cellular responses to hypoxia, and is of particular relevance in cancer cell biology, including the progression towards HCC developed by different etiologies, including ASH and NASH. While HIF-1 is induced primarily by the availability of oxygen, HIF-1 activation can occur in normoxia involving mechanisms independent of oxygen, consistent with the dependence of PHDs on other cofactors, including 2-OG and iron. In addition, ROS generation and the subsequent inactivation of PHDs by oxidative stress can also lead to HIF-1 stabilization in normoxia, involving the generation of ROS predominantly from mitochondria. Since STARD1 expression transfers cholesterol to the MIM, STARD1 induction over physiological levels in ASH and NASH can result in mitochondrial cholesterol accumulation, which impacts the impairment of specific mitochondrial solute carriers, including Slc25A11 causing the depletion of mGSH levels. The subsequent ROS generation amplified in part by the limitation in the GSH content in mitochondria can cause the stabilization of HIF-1. As STARD1 transcription is regulated by hypoxia through HRE, both HIF-1 stabilization and STARD1 induction can engage in a self-amplifying vicious cycle, which may contribute to the perturbation of mitochondrial function and impairment in antioxidant defense of relevance to ASH and NASH [122], particularly in the early phases of development. Thus, targeting HIF-1 and STARD1 may be of relevance in the treatment of metabolic liver diseases, of which NASH is of particular concern due to its association with the obesity and insulin resistance pandemic.
2-OG: 2-oxoglutarate
2-OGC: 2-oxoglutarate carrier
APAP: acetaminophen
ASH: steatohepatitis due to alcohol-related liver disease
BA: bile acid
CoA: coenzyme A
ER: endoplasmic reticulum
GSH: glutathione
HCC: hepatocellular carcinoma
HIF: hypoxia-inducing factor
HMG-CoA: hydroxymethylglutaryl-coenzyme A
HREs: hypoxia response elements
LDL: low-density lipoprotein
mGSH: mitochondrial glutathione
MIM: mitochondrial inner membrane
MLN64: metastatic lymph node protein 64
MOM: mitochondrial outer membrane
NASH: steatohepatitis due to non-alcohol-related liver disease
NO: nitric oxide
PHDs: prolylhydroxylases
PP: periportal
PV: perivenous
ROS: reactive oxygen species
SREBP-2: sterol regulatory element protein 2
STARD1: steroidogenic acute regulatory protein 1
STARD1f/f: steroidogenic acute regulatory protein 1 floxed
STARD1ΔHep: hepatocyte-specific steroidogenic acute regulatory protein 1 knockout
START: steroidogenic acute regulatory protein-related lipid transfer
TSPO: translocator protein
VDAC1: voltage-dependent anion channel 1
VEGF: vascular endothelial growth factor
VHL: Von Hippel Lindau
ST, JCFC, and CGR: writing – review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
We acknowledge the support from the grants PID2019-111669RB-I00 and PID2020-115055RB-I00 from Plan Nacional de I+D funded by the Agencia Estatal de Investigación (AEI) and the Fondo Europeo de Desarrollo Regional (FEDER) and from the CIBEREHD; the center grant P50AA011999 Southern California Research Center for ALPD and Cirrhosis funded by NIAAA/NIH; as well as support from AGAUR of the Generalitat de Catalunya SGR-2017-1112, European Cooperation in Science & Technology (COST) ACTION CA17112 Prospective European Drug-Induced Liver Injury Network, and the Red Nacional 2018-102799-T de Enfermedades Metabólicas y Cáncer and the Project 201916/31 ‘Contribution of mitochondrial oxysterol and bile acid metabolism to liver carcinogenesis’ 2019 by Fundació Marató TV3.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

