Review
Review

Affiliation:
Department of Propaedeutics of Internal Diseases and Gastroenterology, A.I. Yevdokimov Moscow State University of Medicine and Dentistry, 127473 Moscow, Russian Federation
†These authors share the first authorship.
Email: vasiliy.reshetnyak@yandex.ru
ORCID: https://orcid.org/0000-0003-3614-5052
Affiliation:
Department of Propaedeutics of Internal Diseases and Gastroenterology, A.I. Yevdokimov Moscow State University of Medicine and Dentistry, 127473 Moscow, Russian Federation
†These authors share the first authorship.
ORCID: https://orcid.org/0000-0001-6114-564X
Explor Dig Dis. 2023;2:149–171 DOI: https://doi.org/10.37349/edd.2023.00024
Received: March 20, 2023 Accepted: May 02, 2023 Published: August 27, 2023
Academic Editor: Jose J. G. Marin, University of Salamanca, Spain
The article belongs to the special issue CHOLESTASIS
Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, is a rare chronic autoimmune cholestatic liver disease, affecting mostly females. With PBС develops chronic cholangiopathy, this is accompanied by the development of gradually progressive liver fibrosis, which leads to intrahepatic cholestasis. Defects in autoimmune tolerance are critical factors in the emergence of the disease. Biochemical signs in PBС appear already in the asymptomatic stage of the disease and they are associated with a disturbance of the secretion of bile acids. Understanding the pathophysiological mechanisms of these signs is essential to both the early diagnosis and treatment of PBC. Early diagnosis of the disease contributes to its more effective treatment. There are many scientifically based modern data on the pathophysiology of clinical and laboratory signs developing in PBС. The purpose of this review is to summarize the data available in the literature and those obtained by the authors on the mechanisms for the development of biochemical criteria for PBC and their diagnostic significance. The opportunity to present the pathophysiological mechanisms of the development of biochemical signs in patients with PBC is associated with the success in the development of modern research methods in biochemistry, molecular biology, and genetics.
Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, is a chronic progressive cholestatic granulomatous, and destructive inflammatory damage in the biliary epithelial cells of the small interlobular and septal bile ducts associated with a loss of tolerance to mitochondrial antigens and the presence of serum antimitochondrial antibodies (AMAs) as well as a potential tendency to progress to cirrhosis [1, 2]. PBC develops due to a defect in autoimmune tolerance [3, 4]. As a result, damage occurs to the cholangiocytes lining the small and medium branches of the bile ducts. Bile excretion is disrupted, intrahepatic cholestasis develops [5]. There is insufficient intake of bile acids into the intestine. Cholestasis in the small bile ducts leads to the entry of bile acids into the systemic circulation and their accumulation in liver cells. Enterohepatic circulation of bile acids is disrupted. It is precisely these changes in the processes of bile formation and excretion in patients with PBC that should be considered as the underlying cause of biochemical signs of this disease.
There is a change in many biochemical parameters of blood in patients with PBС [3]. Patients’ blood serum shows the enhanced activity of alkaline phosphatase (ALP), γ-glutamyl transferase/γ-glutamyl transpeptidase (γ-GT), 5’-nucleotidase (5’-NT), and leucine aminopeptidase (LAP), the higher levels of bile acids, cholesterol, phospholipids (PhLs), copper, γ-globulins, and bilirubin, and the lower level of total protein mainly at the expense of albumin fractions [3, 6].
A serum biochemical test shows an enhanced activity of the enzymes ALP, 5’-NT, γ-GT, and LAP in patients with PBC just in its early stages [3, 6].
ALP is present in many tissues. ALP isoenzymes are found in the liver, intrahepatic and extrahepatic bile ducts, osteoblasts, intestinal mucosa, kidneys, placenta, and lactating breast. However, more than 80% of serum ALP is released from the liver and bone tissue [6]. A significant (2–3 or more times higher than the upper limit of the norm) increase in ALP activity is observed in patients with PBC already at an early stage of the disease [3]. At the same time, there is no correlation between the level of increased ALP activity and the degree of disease progression or stage [7]. It was noted that the increase in enzyme activity occurs later than itching appears in patients [8]. It should be emphasized that in PBC, there is an increased activity of predominantly the liver isoenzyme fraction that is produced in both hepatocytes and endothelial cells lining the bile ducts and tubules [9]. In cholestasis, serum ALP levels may increase in response to the accumulation of bile acids in the systemic circulation [9]. The level of 5’-NT is relatively specific to liver diseases and strongly correlates with serum ALP of hepatic origin [6]. A moderate increase in the activity of the hepatic fraction of ALP and 5’-NT just in the early stages of PBC indicates changes in phosphorus metabolism in this disease [2].
The mechanism of the increase in ALP and 5’-NT levels in PBC is the subject of debate. There is a theory that states that ALP ensures the formation of orthophosphate where the latter is needed [10]. Based on this theory, the enhanced activity of ALP and 5’-NT in PBC suggests that there is an increased need for and synthesis of orthophosphate in the liver. But at the same time, the quantity of orthophosphate is shown to be reduced in the hepatic bile portion from PBC patients [11]. The secretion of orthophosphate from the hepatocytes and epithelial cells lining the bile ducts into bile is passively affected by the concentration gradient [12]. Consequently, the concentration of orthophosphate is also reduced in the hepatocytes and epithelial cells of bile duct. This indicates the intensive use of the phosphorus group in the metabolic processes occurring in the hepatocytes and bile duct epithelial cells in PBC. The biosynthesis of PhLs is one of the possible ways of using orthophosphate [13, 14].
In patients with PBC, the elevated level of ALP and 5’-NT has been noted to be associated with the higher plasma level of phosphatidylcholines [15, 16]. Orthophosphate, glycerol, palmitic, and oleic fatty acids are the base of lecithin’s (PhLs) in PBC [17]. The formation of orthophosphate is known to depend on the activity of the enzymes ALP and 5’-NT involved in the hydrolysis of phosphomonoesters. ALP activates the hydrolysis of glycerophosphate, glucose-1-phosphate, and glucose-6-phosphate to form the corresponding carbohydrates and orthophosphate. Under the effect of 5’-NT, the ribonucleotides [adenosine monophosphate (AMP), guanosine monophosphate (GMP), cytidine monophosphate (CMP), and uridine monophosphate (UMP)] are hydrolyzed to ribonucleosides (adenosine, guanosine, cimetidine, uridine) and orthophosphate.
Studies convincingly demonstrate that the enhanced activity of ALP and 5’-NT in PBC is associated with an increase in the synthesis of these enzymes [6]. Hatoff et al. [18] have shown that on day 5 after rat bile duct ligation, ALP activity comes to be determined in the endoplasmic reticulum and Golgi apparatus membranes. Kryszewski et al. [19] reported that in rats with a ligated bile duct, the serum level of ALP did not increase when a translation and transcription inhibitor of protein synthesis was used. Both of these studies point to the fact that there is a process for inducing ALP synthesis in chronic cholestasis. At the same time, there is an increase in the translation of messenger RNA (mRNA) responsible for the synthesis of ALP, followed by a rise in the serum secretion of the enzyme [6]. The increased biosynthesis of ALP and 5’-NT in PBC requires the higher delivery of amino acids into the cell, which is provided by γ-GT. The latter is a transmembrane protein that provides transport and delivery of amino acids into the cell, followed by their active use in the biosynthesis of proteins, including enzymes [20]. Similarly, to the activity of ALP, that of γ-GT increases by 5 or more times the upper limit of normal in PBC [3].
Moreover, the enhanced activity of γ-GT precedes that of ALP and 5’-NT in PBC patients, which may indirectly indicate the involvement of γ-GT in the synthesis of the enzymes ALP and 5’-NT.
The elevated serum level of γ-GT in combination with the enhanced activity of ALP and 5’-NT in PBC reflects damage to the biliary tract to a greater extent than damage to hepatocytes [6]. The higher level of γ-GT is a very sensitive biochemical sign that can be used to increase the predictive value of measuring ALP and 5’-NT in PBC [21].
It should be taken into account that γ-GT activity stimulation can occur under the influence of alcohol, medicines, and other microsomal oxidation-inducing preparations. In addition, γ-GT is a microsomal protein that is present not only in the liver, but also in the kidneys, pancreas, and intestines [20]. The induction of this enzyme in these organs is generally due to chronic alcohol consumption [9]. Therefore, it is important to exclude the effect of alcohol and/or the drugs when detecting the enhanced activity of γ-GT.
γ-GT is a sensitive but non-specific indicator of damage to the bile canaliculi and ducts. An increase in the activity of this enzyme has been proven in the asymptomatic stage of the disease. This indicates that PBC begins with damage to the bile canaliculi and ducts [22]. The available data indicate that for unknown reasons, damage to cholangiocytes lining the bile ducts occurs [23]. Cholangiocytes are normally protected from the effects of bile acids by a bicarbonate “umbrella” [24]. The secretion of bicarbonate ions protects cholangiocytes from uncontrolled transmembrane current of glycochenodeoxycholic acid, which can induce apoptosis of biliary epithelial cells [25]. With PBC, the bicarbonate “umbrella” becomes “leaky” and passes bile acids toxic to cholangiocytes, triggering the process of fibrogenesis. The latter leads to the development of ductulopenia already in the early stages of PBC. A significant increase in the activity of γ-GT is a sign of fibrosis and can be used as an indirect marker of bile duct fibrosis in PBC [26–28]. γ-GT is one of the first biochemical signs of PBC, along with an increase in the activity of ALP and the appearance of AMAs, which is used to diagnose this disease [29].
LAP is found in the highest concentrations in the liver, kidneys, and small intestine. The activity of LAP increases due to bile duct epithelial damage in all forms of intrahepatic and extrahepatic cholestasis. LAP has approximately the same clinical significance as ALP. However, the activity of LAP does not practically change in bone tissue diseases. Therefore, the determination LAP is used for the differential diagnosis of diseases of the hepatobiliary system and bone tissue, when the activity of ALP is enhanced. Since the determination of γ-GT activity has been introduced into clinical practice, there is a noticeably decreasing interest in LAP studies. In clinical practice, the determination of ALP and γ-GT activities is most common in patients with PBC. The European Association for the Study of the Liver (EASL) and the American Association for the Study of Liver Diseases (AASLD) recommend using ALP and γ-GT activities in combination with AMAs for the diagnosis of early-stages PBC [30, 31].
In the hepatic bile portion of patients with PBC, there is a decrease in the levels of bile acids, cholesterol, and phosphatidylcholine, while increasing their content in hepatocytes and blood plasma [11]. These data indicate impaired enterohepatic circulation of bile acids in PBC.
As a result of the autoimmune process, ductulopenia gradually develops and enterohepatic circulation of bile acids is disrupted. The intake of bile acids into the intestine decreases. In liver cells, the synthesis of bile acids and cholesterol increases compensatory, through a feedback mechanism. Primary bile acids are the end product of cholesterol catabolism. Insufficient intake of bile acids into the duodenum stimulates increased cholesterol biosynthesis by hepatocytes. Progressive ductulopenia and increased synthesis of cholesterol and bile acids lead to their accumulation in hepatocytes. The reabsorption of bile acids from the portal venous blood is decreased. Bile acids begin to enter the systemic circulation. Stasis of bile in the bile capillaries also leads to the reflux of bile acids into the general bloodstream through the interhepatocyte space. An increase in the concentration of bile acids in the blood plasma of patients with PBC is noted already at an early (asymptomatic) stage of the disease. This is accompanied by the deposition of bile acids in the epidermis and the development of skin itching, the earliest clinical sign of the disease [13]. All fractions of conjugated bile acids are detected in the blood plasma of patients with PBC. On the contrary, unconjugated bile acids are rarely detected.
Conjugated and especially unconjugated bile acids are strong detergents. Entering into the systemic circulation, they are able to damage the cell membranes of blood cells and vascular endotheliocytes. Getting into the epidermis, they cause an irritating effect on the nerve endings. Accumulation of bile acids in hepatocytes causes their damage and induces apoptosis. In vitro experiments have shown that the mechanism of development of the latter is associated with the formation of reactive oxygen species generation by mitochondria [29, 32–34].
An increase in the ratio of tri-/dihydroxy bile acids was noted in PBC patients’ blood plasma [13]. On the contrary, the glycine/taurine ratio is decreased [13]. By Greim et al. [35, 36], this is due to the fact that trihydroxycholanic (cholic) acid is more hydrophilic and has fewer detergent properties than the more hydrophobic deoxycholic and chenodeoxycholic bile acids.
Conjugation of bile acids with glycine and taurine, their sulfation, and glucuronidation leads to an increase in their water solubility and a decrease in their toxic and detergent properties. The resulting increase in the hydrophilic properties of bile acids contributes to their removal from the systemic circulation through the skin, kidneys, and intestines. Along with this, taurine conjugates of bile acids form micellar-lamellar structures with cholesterol and lecithin more actively than glycine conjugates [37]. The penetration of primary bile acids conjugated with glycine into the epithelial cells lining the bile ducts depends on the presence of a “carbonate umbrella” [29]. Insufficient synthesis and intake of bicarbonate into the lumen of the bile duct can lead to increased entry of glycine-conjugated bile acids into cholangiocytes. This contributes to increased oxidative stress, accelerated senescence of cholangiocytes, and their apoptosis [29]. This is probably the reason for the decrease in the glycine/taurine ratio in PBС.
Thus, the higher ratio of trihydroxy-/dihydroxycholanic bile acids, lower glycine/taurine coefficient of bile acid conjugates, and the appearance of sulfated, glucuronidated bile acids in the systemic circulation in PBC may be considered as the body’s compensatory, detoxifying response to cholestasis and the entry of cholanic acids in circulating blood.
Atypical, non-physiological bile acids appear in the blood and urine of patients with PBC [38]. The latter has more potent detergent effects on cell membranes and a stronger irritant effect on nerve receptors than primary and secondary bile acids. The atypical, non-physiological bile acids can be released from the systemic circulation through the skin, taking part in the mechanisms of skin itching [13]. The intensity of the latter may depend on the amount of atypical bile acids in the skin of PBC patients.
In the hepatic bile portion of patients with PBC is noted not only decreases concentration of bile acids, but also the content of lecithin and cholesterol [11]. At the same time, cholesterol decreases to a lesser extent, which changes the ratio between bile acids, lecithin, and cholesterol in favor of the latter [11]. As a result, there is an increase in the lithogenicity of bile, which in 35–40% of cases are accompanied by the formation of gallstones in PBC.
The excretion of lecithin from the hepatocyte into the bile capillary depends on the secretion of bile acids. A decrease in the concentration of lecithin in the hepatic bile portion of patients with PBC is associated with a change in the relative amount of bile acids [39, 40]. The relative content of the hydrophobic and hydrophilic pool of bile acids is also of great importance for the secretion of phosphatidylcholine into bile [41].
At the same time, a 1.5-fold increased PhL content (P = 0.044) was observed in the membranes of hepatocytes from hepatic biopsy specimens of patients with PBC [42]. This is accompanied by an almost 2-fold decrease in the ratio of cholesterol/PhLs. At the same time, the content of lysophosphatidylcholine, sphingomyelin, phosphatidylserine (PS), phosphatidylinositol, and phosphatidylethanolamine (PE) in the same membranes is reduced [42, 43].
The insufficient intestinal release of primary bile acids in PBC leads to a change in the composition of the microbiome, causing gut dysbiosis [44–48]. According to DiBaise and Paustian [49], dysbiosis in patients with PBC determines the development of steatorrhea. Therefore, these patients should be necessarily evaluated for bacterial overgrowth [45, 49].
Insufficient entry of bile acids into the duodenum in PBC patients is accompanied by a decrease in the absorption of dietary cholesterol. The latter undergoes solubilization by bile acids and PhLs with the formation of micelles and subsequent absorption. As a result, hepatic cholesterol synthesis is induced via a feedback system [50]. The intake of low-density lipoprotein cholesterol (LDL-C) from blood plasma into hepatocytes is reduced [50]. This leads to an increase of total cholesterol (TC) levels in the blood.
Gradually progressive cholestasis in PBC leads to the accumulation of bile acids in hepatocytes and their entry into the blood plasma. The latter requires the neutralization of the detergent effect of bile acids on the membranes of the blood corpuscles and endotheliocytes. As a result, there is an impairment of lipid metabolism. Dyslipidemia occurs, which is manifested by changes in the synthesis and transport of cholesterol, as well as PhLs. The appearance of bile acids in the blood in the patients with PBC leads to an increase in the expression of fibroblast growth factor 19 (FGF19) through a feedback system. Fibroblast growth factor receptor 4 (FGFR4) is activated on hepatocyte membranes, followed by inhibition of the bile acids synthesis from cholesterol. This contributes to the development of hypercholesterolemia [51–54].
Dyslipidemia is detected in patients with PBС already in an early stage of the disease. It is manifested by an increased content of PhLs, TC, LDL-C, and high-density lipoprotein cholesterol (HDL-C) in the blood plasma [55]. An unusual lipoprotein-X (LP-X) appears in the blood plasma [53]. At the same time, the level of neutral lipids (triglycerides) remains almost unchanged [56]. A slight increase in the quantities of triglycerides is possible, which is associated with a decrease in the activity of LP lipase [56]. The latter cleaves neutral lipids in chylomicrons, in very low-density lipoproteins (VLDL) and low-density lipoprotein (LDL).
Knowledge of the physicochemical properties of lipids and their transport processes in the human body allows us to understand the mechanisms of dyslipidemia development in PBC.
Blood and bile are an aqueous medium in which both food lipids and lipids (triglycerides, PhLs, cholesterol, and fatty acids) synthesized in the human body cannot exist in monomeric form. The transport of lipids in these media is carried out due to the formation of special micellar and lamellar structures, LPs.
Apolar triglycerides, esterified cholesterol, and a weakly polar non-esterized cholesterol molecule are hydrophobic structures. In this regard, they cannot form either micellar or lamellar structures in the aquatic environment and need special transport structures for transportation through the body. Such structures are blood LPs and micellar-lamellar structures of bile. They are formed by polar PhLs, which together with fatty acids are amphiphilic (consist of a hydrophilic and hydrophobic part in their molecule) compounds. Nonpolar triglycerides, esterified cholesterol, and a weakly polar non-esterized cholesterol molecule are hydrophobic structures. In this regard, they cannot form either micellar or lamellar structures in the aquatic environment and need special transport structures for transportation through the body. Such structures are blood LPs and micellar-lamellar structures of bile. They are formed by polar PhLs, which together with fatty acids are amphiphilic (consist of a hydrophilic and hydrophobic part in their molecule) compounds. Blood LPs are unilamellar hollow PhL structures [57–59]. Solubilization (capture) of non-esterified cholesterol by PhLs occurs and integrates it into the PhL monolayer of LP particle. PhLs are a kind of “solvent” for cholesterol (Figure 1). Fully hydrophobic triglycerides and esterified (fatty acid-bound) cholesterol are located in the internal hydrophobic cavity of the LP particle, and thus are transported in blood.
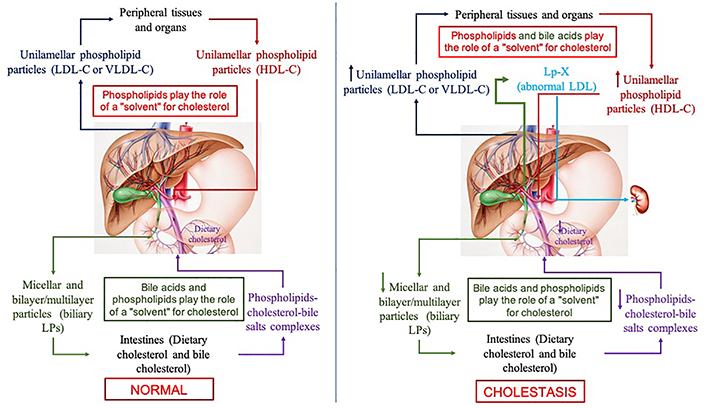
Schematic diagram of cholesterol transport in the human body in health (the left part of the figure) and in cholestasis (the right part of the figure) [60]. VLDL-С: very low-density lipoprotein cholesterol
Note. Adapted from “Features of lipid metabolism disorders in primary biliary cholangitis”, by Reshetnyak VI, Maev IV. Biomedicines. 2022;10:3046 (https://www.mdpi.com/2227-9059/10/12/3046). CC BY.
Cholesterol homeostasis largely depends on its conversion into bile acids and its excretion with bile. The main components of bile, in addition to lecithin and cholesterol, are primary bile acids. The latter are the end product of cholesterol catabolism in the human body. Bile acids are amphiphilic molecules. This allows them to form micellar-lamellar structures in an aqueous medium, both independently and with PhLs. During the formation of these structures, bile acids can solubilize cholesterol and integrate it into these structures (Figure 1). In this regard, bile acids can also be considered as a “solvent” for cholesterol. The ratio of cholesterol:PhLs:bile acids is important for the solubility of cholesterol in micellar-lamellar structures. The presence of bile acids in these structures leads to the formation of bilayer or multilayer vesicular particles [61, 62]. These are the so-called LP particles of bile [62]. Thus, blood LPs differ from bile LPs both in composition and structure. Differences in structural organization are determined by the presence or absence of bile acids in LP particles, as well as the concentration of albumin in bile and blood plasma [61, 62]. Both blood LPs and bile ones are transport systems for lipids and, first of all, for cholesterol. The liver is the central organ where these two transport systems intersect (Figure 1). Herewith, PhLs serve as a “solvent” for cholesterol in plasma LPs, while bile acids and PhLs do this in bile, which determines the unilamellar and bilayer/multilayer vesicular structure of blood LPs and bile LPs, respectively.
Changes in the enterohepatic circulation of bile acids and their entry into the systemic circulation in PBC lead to disruption of cholesterol transport and metabolism (Figure 1). Bile acids that have entered the plasma due to cholestasis begin to form micellar-lamellar structures with PhLs and cholesterol in the blood. The detergent and physicochemical properties of bile acids contribute to the formation of bilayer/multilayer LP particles in the blood similar to those formed in bile. In the blood of patients with PBC, already at an early stage of the disease, an unusual LP-X appears, resembling both LDL and bile LPs [63]. There is an increase in the level of PhLs and the amount of cholesterol in LDL, VLDL, and high-density lipoproteins (HDLs), as well as an increased content of TC [40].
LP-X is found in the blood plasma of patients with both intrahepatic and extrahepatic cholestasis [64–67]. Its appearance in the blood plasma is always associated with the entry of bile acids into the systemic circulation. The composition of LP-X always contains bile acids, there is a high content of PhLs and free cholesterol [61]. The presence of lithocholic acid in the composition of LP-X has been shown [67, 68]. An obligatory component of LP-X is albumin [61].
Elevated formation of micellar-lamellar structures in the blood, when bile acids enter the systemic circulation in PBC, occurs due to additional synthesis of cholesterol and PhLs. The synthesis of fatty acids in the liver increases, as well as the formation of orthophosphate necessary for the biosynthesis of additional PhLs in cholestasis [69]. In the blood of patients with PBC, an increase in the level of palmitic and oleic fatty acids, the main components of bile lecithins, was noted [70].
The physico-chemical properties of LP-X and LDL are similar in many ways. Therefore, LP-X is called “abnormal” LDL. It is assumed that LDL is involved in the mechanism of LP-X formation. It was noted that the PhL composition of LDL and HDL in the blood plasma of patients with cholestasis is largely similar to the PhL composition of LP-X [69]. But the content of esterified cholesterol in LP-X is sharply reduced. It has been shown in vitro and in vivo that LP-X does not inhibit de novo cholesterol synthesis in the liver [71–73].
Bile acids, entry in the systemic circulation, initiate the solubilization of cholesterol and PhLs from the surface of LPs with the involvement of plasma albumin in this process and the subsequent formation of LP-X. This mechanism makes it possible to explain the increase in all LP fractions in the blood of patients with PBC. The latter is the result of increased synthesis of cholesterol, PhLs, palmitic and oleic fatty acids in the liver. Their increase in the blood plasma of patients with PBC already in the early stages of the disease was noted [2, 74].
The increase in the level of TC and LDL-C in patients with PBC is alarming for many authors, since these indicators are considered as risk factors for the development of cardiovascular events [56, 75, 76].
However, hypercholesterolemia developing with cholestasis is aimed at neutralizing the detergent properties of bile acids entering the systemic circulation. And therefore, the developing dyslipidemia should be considered as a compensatory reaction of the body in response to cholestasis and the entry of bile acids into the blood. At the same time, hypercholesterolemia is accompanied by an increased content of HDL. And in this regard, dyslipidemia in PBC is “anomal” and is not accompanied by an increase in the frequency of cardiovascular events [51, 55, 64, 75, 76]. The absence of cardiovascular events in cholestasis on the background of hypercholesterolemia can serve as a model system for the development of new methods of treatment of dyslipidemia. These patients also have a low degree of hepatic steatosis [51, 55]. This is promoted by the malabsorption of fats due to insufficient intake of bile acids into the intestine and developing steatorrhea [2, 17]. Along with this, inhibition of lipogenesis and activation of oxidation of free fatty acids is observed, which is associated with high expression of FGF19 [77]. All this reduces the accumulation of triglycerides in the liver and in blood plasma [77].
In liver diseases, one of the sensitive and specific biochemical markers of cholestasis is the determination of LP-X [65]. There was more than 95% coincidence between the detection of LP-X and the determination of cholestasis syndrome by histological methods [78]. However, the laboratory determination of LP-X for the diagnosis of cholestatic conditions is rarely used. This is due to the complex method of determining LP-X and the high informative value of ALP and γ-GT as biochemical markers of cholestasis.
During PBC development, the serum activity of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), in contrast to that of γ-GT, ALP, and 5’-NT, does not exceed the upper limit of normal for a long time or increases slightly. ALT is present only in the hepatocyte cytoplasm, whereas AST is in both the hepatocyte cytoplasm and mitochondria [79]. Both aminotransferases belong to the class of transaminase enzymes and play an important role in amino acid metabolism. They are involved in enzymatic transamination by the following overall reaction: amino acid1 + keto acid2 ⇄ aminotransferase ⇄ amino acid2 + keto acid1.
In a healthy being, these enzymes are normally released from the cells at a constant rate due to programmed liver cell death (apoptosis), followed by release of the enzymes into plasma and their clearance from the body [79]. There is equilibrium between the normal turnover of hepatocytes and the clearance of these enzymes from the plasma [79]. The increased release of ALT and AST from the liver cells into the bloodstream is usually associated with either cytoplasmic membrane damage or enhanced hepatocyte death. In early-stage PBC, the preserved AST and ALT levels within the normal range indicate the integrity of the hepatocyte cytoplasmic membrane and the normal rate of hepatocyte apoptosis [16, 79]. It was noted that the level of ALT activity in PBC is higher in men than in women [80, 81]. It is assumed that this is due to biological and physiological characteristics, with the level of male and female sex hormones [80, 82, 83].
In clinical practice, the AST:ALT ratio (De Ritis ratio) is often used for differential diagnosis. When concurrent with a cholestatic pattern, an AST:ALT ratio < 1.5 indicates extrahepatic obstruction [8]. In cholestatic liver diseases, the De Ritis ratio is important at late stages of the disease (the cirrhosis stage) or in monitoring the efficiency of ursodeoxycholic acid treatment in PBC patients. The AST:ALT ratio can serve as a predictor of liver cirrhosis in patients with PBC. At the same time, this ratio is not a prognostic factor. Alempijevic et al. [84] have shown that the increased AST:ALT ratio correlates with disease stage and outperforms other non-histological findings for assessing the presence of cirrhosis in PBC, but has a low sensitivity and specificity of 65–79%. The development of fibrosis in the early stages of the disease is caused by immunopathological processes in the small and medium bile ducts. At the same time, liver cells remain intact for quite a long time. Therefore, in the early stages of the disease and before the development of pronounced clinical signs of cholestasis, ALT and AST cannot be used as indirect markers of early stages of liver fibrosis. At the stage of developed pronounced cholestasis, bile acids accumulating in hepatocytes trigger the processes of damage to the cytoplasmic membranes of hepatocytes, apoptosis, and fibrosis. During this period, ALT and AST can be used to assess all these damages. Additionally, the AST to platelet ratio index (APRI) can be used to predict the development of fibrosis [85]. It has been shown that the APRI parameter has a certain value for the diagnosis of fibrosis in patients with PBC [86].
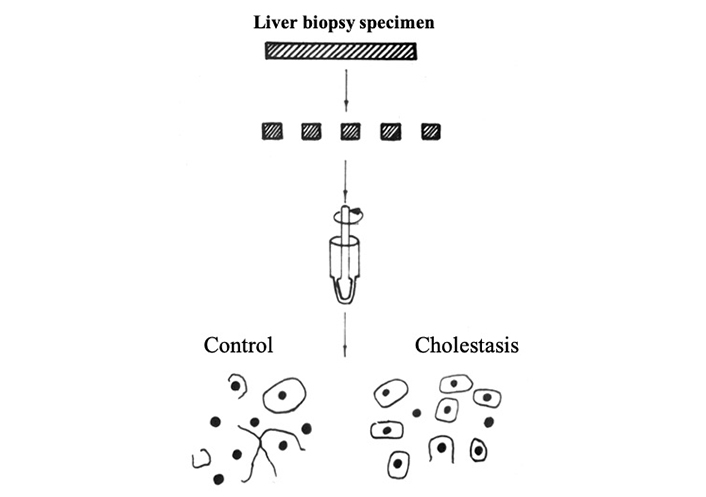
Despite the fact that the cytoplasmic membranes of hepatocytes in PBC retain their integrity for quite a long time, the electron microscopic studies show impaired cell-cell junctions [87, 88]. Tight junctions are an important structure that provides mechanical cell adhesion and maintains tissue barrier properties. The tight junction area between hepatocytes limits the bile capillary lumen, preventing bile from penetrating the interhepatocyte space and sinusoidal region. The disrupted integrity of the tight junction between the liver cells results in the entry of bile components into the systemic circulation. Experimental animal models of cholestasis were found to have pronounced changes in the tight junction area, which led to a twofold decrease in the area of contacting hepatocyte surfaces [89]. A decrease in the interaction between liver cells in PBC may determine higher values of AST:ALT proportion [90]. In PBC is revealed a decrease in inter-hepatocyte interactions at the site of a tight junction, which is associated with an increase in pressure in the bile capillaries due to cholestasis [90]. PBC patients were noted to have an impaired interhepatocytic interaction that was revealed by electron microscopic examination and adhesiometric method [90]. The reduction of cell-cell interactions leads to the fact that during mechanical dispersion of a liver biopsy specimen, most hepatocytes move away from each other, without breaking the cytoplasmic membrane (Figure 2). As a result, a large number of isolated single cells and significantly fewer single nuclei are detected under the microscope. The nuclei are formed as a result of rupture of the membranes of two hepatocytes connected by tight junction complexes, due to mechanical dispersion of the liver biopsy specimen (Figure 2) [90].
The use of the adhesiometric method makes it possible to quantify the disconnection coefficient (Dc) for hepatocytes in cholestasis. Dc is the ratio of the number of single cells to the sum of the latter and cell nuclei isolated after liver biopsy specimen dispersion: Dc = Ncells/( Ncells + Nnuclei).
The higher the Dc value, the more weakened the interhepatocyte contacts. A study of liver biopsy specimens showed a statistically significant (4–6-fold) increase in the Dc value in patients with PBC compared with other groups of those with chronic liver diseases without cholestasis [90].
The increased Dc value in PBC is associated with a weakening of cell-cell interactions due to an increase bile capillary pressure related to cholestasis. In patients with PBC, cell compression occurs with cholestasis, which leads to a 1.5–2-fold decrease in the size of hepatocytes.
Hyperbilirubinemia is not a characteristic sign of early-stage PBC [8]. Total bilirubin is usually within the normal range in the asymptomatic stage of the disease [3]. In PBC patients, the blood level of total bilirubin gradually increases at the stage of clinically obvious cholestasis and rarely reaches high values [47]. In these patients, an elevation of serum bilirubin primarily occurs due to its conjugated fraction. These findings suggest that hepatocyte glucuronidation of bilirubin remains intact and the process of its secretion with bile through the bile ducts is disturbed [2]. Intrahepatic cholestasis in PBC leads to the development of hyperbilirubinemia at the expense of the conjugated fraction, which composes a large proportion of total bilirubin [91]. The cause of the development of conjugated hyperbilirubinemia in PBC may be a difficulty in the outflow of bile through the bile ducts or a change in the activity of bilirubin transporter proteins on the apical membrane of hepatocytes [91]. Since there is a disorder of the integrity of the inter-hepatocyte interaction and the development of ductulopenia in PBC, conjugated hyperbilirubinemia is associated with difficulty in bile outflow [91]. The latter is associated with damage to the small and medium bile ducts. The resulting stasis of bile leads to an increase in pressure in the bile capillaries. There is a weakening of intercellular interactions and reflux of bile components, including conjugated bilirubin, through tight junction into the sinusoidal space, and then into the systemic circulation.
An increase in the content of conjugated bilirubin in the bile capillary leads to a decrease in the activity of bilirubin transporter proteins on the apical membrane of hepatocytes. As a result, conjugated bilirubin accumulates in liver cells, which was noted in liver biopsy specimens of patients with PBC [92].
Hyperbilirubinemia in the late stages of PBC may be additionally caused by increased hemolysis of red blood cells. The entry of a significant amount of bile acids into the blood causes hemolysis of erythrocytes due to the detergent effect on cytoplasmic membranes. As a result of increased hemolysis in the blood of patients with PBC, the unconjugated fraction of bilirubin also increases in the end stage of the disease. The detergent action of bile acids is also exposed to other blood cells: leukocytes, platelets.
An increase in the level of direct bilirubin in the blood plasma of patients with PBC leads to its excretion in the urine. The amount of bilirubin entering the duodenum with bile determines the amount of urobilinogen excreted in the urine. PBC patients are observed to have pronounced individual fluctuations in the values of bilirubin. However, its content generally corresponds to the stage of the disease and the degree of activity of the pathological process [55]. Therefore, one of the most important biochemical indicators used to assess the severity of PBC is bilirubin [55]. In this connection, the AASLD considers that the serum bilirubin level in patients with PBC is the most important factor in predicting the development of the disease [93]. Several non-invasive prognostic scores, such as Child-Pugh score, model of end-stage liver disease (MELD), Mayo risk score, and Newcastle model, are used in patients with PBC to identify those at high risk of complications [94–97]. All these scores include serum bilirubin levels [98]. Studies were conducted at the Mayo Clinic to predict the course of the disease; an evaluation scale was developed that excluded biopsy as an invasive method [99]. It is proposed to use age, blood bilirubin and albumin content, prothrombin time, and the presence of peripheral edema [99]. An increase in the level of bilirubin on the background of a decrease in the level of albumin and platelets may be a harbinger of the development of cirrhosis and portal hypertension [55]. In 2015, to evaluate liver function, a new simple score was developed and proposed, which was calculated using only serum albumin and bilirubin levels: the albumin-bilirubin (ALBI) score [100, 101]. The advantage of the latter is that it can be easily calculated using objective blood tests without subjective factors or invasive procedures. The ALBI score and the Mayo risk score are of the highest prognostic value in predicting the disease outcomes, determining the treatment strategy, and timing of liver transplantation in patients with PBC [98]. A progressive rise in bilirubin levels should cause concern, as it is a poor prognostic sign and is used as one of the important signs in determining the timing of liver transplantation in a patient with PBC [3, 102]. It is believed that any increase in the level of bilirubin in patients with PBC is an important prognostic factor requiring optimization of the management of these patients [103, 104].
Copper entering from food is metabolized in the liver to form a complex with ceruloplasmin [105]. As a result of the formation of the copper-ceruloplasmin complex, the toxic properties of copper decrease, its solubility in the aqueous medium (blood, bile) increases, its transportation and excretion from the body is facilitated. Most (about 80%) of copper is removed from the body by bile.
The copper content in liver cells in PBC patients significantly increases and can reach 25 mg/100 g of dry liver tissue (normally up to 6 mg/100 g) [106, 107]. This is due to the development of cholestasis and a decrease in the rate of copper excretion through the apical membrane of the hepatocyte into the bile capillary. Along with this, there is a reflux of bile components into the systemic circulation, which is accompanied by an increase in the level of copper in blood plasma [108]. The accumulation of copper in hepatocytes reduces its absorption from the portal vein venous blood, which also increases the level of copper in blood plasma. Copper accumulation in PBC patients leads to the activation of the copper-containing enzyme tyrosinase. This results in increased melanin production. When patients with PBC are examined, attention is drawn to the loss of elasticity in the skin and its dryness, as well as hyperpigmentation in many cases. Along with this, the body attempts to remove excess copper not only through the kidney, but also through the skin. This brings about the deposition of copper in the epidermis, which imparts a bronzed hue to the hyperpigmented skin in PBC patients.
It is believed that copper accumulation in hepatocytes in patients with PBC does not have a clinically significant toxic effect on liver cells [108]. Typical histological signs of the toxic effects of copper on liver cells in PBC patients are not observed [108]. There is no Kayser-Fleischer ring in patients with PBC. The level of ceruloplasmin in blood plasma is preserved [105]. All this indicates the preservation of copper metabolism in the liver. Nevertheless, an increase in the concentration of copper in liver cells correlates with the stage of the disease, but does not correlate with changes in biochemical parameters [108, 109].
The serum level of blood proteins in patients with early stage of PBC remains within physiological limits for a long time [2, 110]. At the same time, the sera from patients already at the asymptomatic stage show AMAs at a diagnostic titer of 1:40 or higher. As the disease progresses, there is an increase in the level of γ-globulins, primarily of the immunoglobulin M (IgM) class [2, 111]. The development of hepatocellular failure in the advanced stage of PBC causes a decline in different liver functions, and primarily a protein synthesizing function. In this case, the biochemical signs are hypoalbuminemia, lower serum prothrombin and fibrinogen levels, and reduced coagulation factor V and VII concentrations. Albumin is a major plasma protein synthesized by the liver cells. Albumin plays a key role in the maintenance of plasma colloidal osmotic (oncotic) pressure and circulating blood volume, as well as in the transport and deposition of various substances. The occurrence of hypoalbuminemia in PBC indicates protein synthesizing dysfunction in hepatocytes [48]. Studying the level of the alkaline fraction of albumin belongs to high-sensitivity tests to identify hepatocellular failure [112]. The synthesis of alkaline (only 3%) and other fractions of albumin occurs in the liver. Disorder of the protein-synthetic function of hepatocytes at a late stage of cirrhosis leads to a decrease in serum total albumin levels. However, the proportion of the alkaline fraction can increase from 3% to 50% due to a longer half-life of this albumin fraction. Whereas there are albumin losses in urine and feces without damaging the liver, the proportion of the alkaline fraction does not change [112].
The development of hypoalbuminemia in PBC is favored by an increase in the processes of catabolism due to a rise in the resting metabolic rate and total thermogenesis [48, 70]. There is a metabolic situation of resource redistributions, which intensifies as cholestasis increases and hepatocellular failure develops. In parallel with decreased serum protein synthesis, the rate of conversion of ammonia to urea reduces in the liver of PBC patients. All leads to a sharp decrease in the level of circulating plasma albumins and an increase in urinary nitrogen excretion [78, 113].
A decrease in the protein synthesizing function of liver cells and an increase in the processes of protein catabolism lead to a deficiency of the visceral protein pool, followed by edema and ascites. Portal hypertension plays a certain role in the development of ascites. In portal hypertension, there is an elevated capillary filtration pressure; hence, fluid transudes into the abdominal cavity. The occurrence of ascites in patients with PBC at the stage of cirrhosis is accompanied by an obvious retention of sodium and its accumulation in the intercellular space. There is a gradual abdominal enlargement and may be dyspnea. The cause of abdominal enlargement is not only ascitic fluid accumulation, but also gas distended bowel loops. The development of ascites in PBC indicates hepatocellular failure and portal hypertension.
In early-stage PBC, procoagulation, anticoagulation, and fibrinolytic laboratory parameters remain within normal limits or may be minimal and practically undetectable [114–116]. During the development of the disease, a balance between the coagulation and anticoagulation systems is generally maintained, but often with a reduced balance [117, 118]. The hepatocyte protein synthesizing function that is preserved at this time makes it possible to maintain the balance of the procoagulant and anticoagulant blood systems [119]. In terminal stage hepatocellular failure, the balance is easily disturbed in one direction or another, which can lead to both bleeding and thrombosis [120, 121]. In some patients with PBC, reduced hepatocyte protein-synthesizing function has been shown to cause a decrease in the synthesis of coagulation factors (II, VII, IX, X, and XIII) to a lesser extent than natural anticoagulants (protein S, protein C, and/or antithrombin III) [115, 122]. There is an imbalance between the decreased level of the natural anticoagulants and that of the blood coagulation factors. The lower concentration of the vitamin K-dependent glycoproteins C and S may contribute to increased thrombus formation.
Hypercoagulability is detected in some PBC patients, by using thromboelastography (TEG) long before the stage of cirrhosis [123–125]. One of the signs of cholestasis detected in some patients with PBC is the activation of the blood coagulation system [123, 124, 126, 127]. The opportunity to identify this condition appeared after the introduction of TEG and rotational thromboelastometry (ROTEM®) into clinical practice [128, 129]. The methods allow to assess the state of the blood coagulation system taking into account the simultaneous contribution of its cellular (platelets, vascular endotheliocytes) and plasma (plasma coagulation proteins) components [128, 129]. As a result, the TEG allows to determine the state of hypo- or hypercoagulation in patients with PBC. Based on thromboelastograms, Ben-Ari et al. [123] have shown that hypercoagulability is observed more frequently in patients with PBC than in healthy volunteers (P = 0.005). To date, the mechanism for hypercoagulability remains unclear in patients with PBC.
The hemostatic system is in a delicate balance between the prothrombotic and antithrombotic processes aimed at preventing spontaneous thrombosis [120]. The initiation of blood coagulation requires highly specific interactions between the coagulation factors and the cell membrane components of platelets and vascular endothelial cells [130–135].
Hemostasis considerably depends on the cell membrane PhLs interacting with blood coagulation factors [136–138]. In health, the cytoplasmic membrane of platelets and endothelial cells lining the vessels has transverse asymmetry: the outer leaflet of the cytoplasmic membrane contains predominantly neutral PhLs (phosphatidylcholine and sphingomyelin), while the inner leaflet has negatively charged PE and PS [136–138]. That is to say, the outer surface of the cell plasma membranes is uncharged in health [139]. The highest coagulation activity is seen in negatively charged PhLs (PS and PE) that are present in high concentrations on the inner leaflet of the cytoplasmic membrane [138]. And this makes it difficult for them to come into contact with clotting factors. Membrane enzymes, such as floppase, scramblase, and aminophospholipid translocase (flippase), regulate the movement of PhLs and maintain this dynamic asymmetry in a stable state. If the cytoplasmic membrane is damaged, PS and PE become available for the interaction with the factors of coagulation, by initiating its process.
The triggers for hypercoagulability already in the early stages of PBC can be bile acids that appear in the systemic circulation. While entering the systemic circulation, bile acids in patients with intrahepatic cholestasis can affect the PhL composition of cytoplasmic membranes in the blood corpuscles, in particular platelets and vascular endothelium, by disrupting transverse membrane asymmetry. Bile acids as strong detergents can solubilize the neutral PhLs from the outer leaflet of membrane of the platelets and vascular endotheliocytes (Figure 3). Here, the negatively charged PhLs from the inner leaflet of the membrane “jump” to the outer one, occupying the freeing place on it.
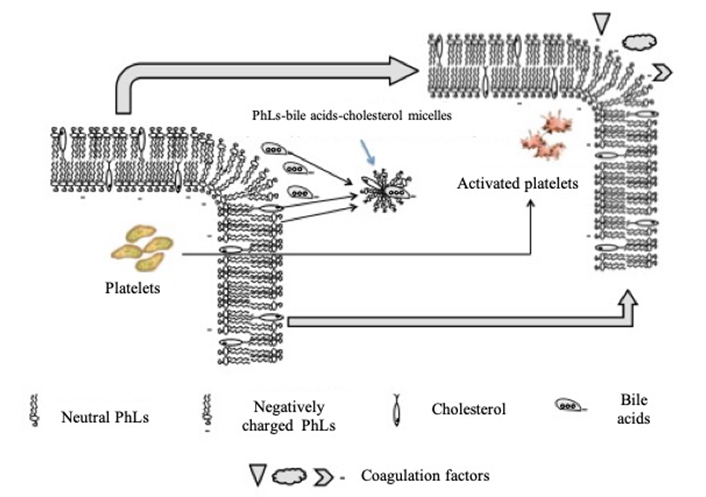
Model for activation of the membranes of platelets and endothelial cells by bile acids in cholestatic liver diseases (explanation in the text)
A change in the transverse asymmetry of platelet and endotheliocyte membranes leads to the appearance on their outer surface of the negatively charged (thrombogenic) areas, which are exposed to tissue factor (TF) apoprotein III. The appearance of a negative charge on the outer leaflet of the platelet membrane leads to its activation. Changing the neutral charge of the outer leaflet of the vascular endotheliocytes membrane to negative creates favorable conditions for interaction with clotting factors. The interaction of TF with the coagulation factor VII/VIIa protease is the initiator of the coagulation system in vivo [140]. A procoagulant complex (TF-VII-Ca2+) can is formed on the membranes surface. It promotes the adhesion of activated platelets to the vascular wall. The process of thrombus formation is triggered.
In addition, the plasma of patients with a pre-thrombotic state displays an increased level of microparticles (MPs) that contain activated TF+ and PS+ on their surface [141, 142]. MPs are extracellular vesicles and represent submicron and intact membranous vesicles that are released from the cells after their activation or during apoptosis [143]. MPs are vesicles of cellular origin, 0.1–1 µm in size, which have neither a nucleus nor a synthetic ability, but may contain cytoskeletal proteins, whereas some amount of PS may be present on the outer leaflet of the membrane [144]. MPs are found in the blood of both in healthy people and in patients with various diseases [145]. In the human body there is a tightly controlled balance between living, proliferating, and dead cells. A reflection of this balance is the physiological level of MPs in the blood [145]. MPs play an important role in many biological processes of the body, including hemostasis [146]. MPs are considered to be a blood circulating repository of bioactive molecules, which, if properly interpreted, can provide valuable information about the body’s state and can be used as biomarkers for various pathological conditions [139, 145]. MPs arise from blebbing on the membranes of blood cells and endothelial cells, followed by bleb detachment, MPs formation and release [141]. Platelets can be the source of MPs [147]. MPs have formed from platelets can contain TF and PS on their surface and serve as signaling molecules for the hemostasis system [148]. Cytokines and/or endotoxins, including bile acids, can stimulate the formation of MPs from platelets [149]. In PBC patients, bile salts that enter the blood due to cholestasis may be presumed to serve as inducers of the formation of TF- and PS-enriched MPs. In this case, loss of PhL asymmetry with PS exposure to the outer leaflet of the cell membrane seems to be important to form these MPs in the presence of simultaneously activated TF+ and PS+ on their surface [141, 150]. Blebbing as an integral indicator for the influence of inducing and damaging factors on cell membranes, as well as loss of PhL asymmetry in the cytoplasmic membranes of blood corpuscles, in particular platelets, can lead to the formation and release of MPs [141]. Platelet-derived MPs have been shown to have a procoagulant activity that is 50–100 times higher than that of the activated platelets themselves [151]. Both PS exposure and the presence of TF in the membranes of MPs can explain their procoagulant properties: PS acts as a catalytic surface for the assembly of coagulation enzyme complexes [141], while TF is deemed to be the main initiator of the clotting cascade in the extrinsic pathway [152]. Cellular MPs and their putative functions in relation to coagulation, angiogenesis, inflammation, and vascular function are reviewed by El-Gamal et al. [145]. However, there is currently no data on the involvement of MPs in the hemostatic imbalance during the development of hypercoagulability in patients with cholestatic liver diseases. The study of MPs in cholestatic liver diseases can provide new insights into the mechanism of the development of a thrombotic tendency in these patients.
A developing disturbance in the processes of bile secretion and enterohepatic circulation of bile acids in patients with PBC already in its early stages leads to the entry of bile acids into the systemic circulation, which, as cholestasis increases, is accompanied by a change in biochemical parameters: there is an elevation in the quantities of bile acids, cholesterol, PhLs, fatty acids, LDLs and HDLs, bilirubin, copper, γ-globulins; LP-X appears; the total protein level falls mainly due to the albumin fraction, and there is also an enhanced activity of the enzymes ALP, γ-GT, LAP, and 5’-NT.
The entrance of bile acids into the blood plasma of patients with PBC is one of the causes of the development of pruritus already in the asymptomatic stage of the disease and lipid metabolism disorders. In the blood of patients with PBC, the level of TC, PhLs, fatty acids, LDL-C, HDL-C increases, and abnormal LP-X appears. The latter is an LP particle, in which, in addition to PhLs and cholesterol, there are bile acids that have entered the systemic circulation as a result of cholestasis. LP-X performs a transport function in blood plasma, like other blood LPs. Synthesis of LP-X is an important mechanism for inactivation of bile acids in blood plasma. The content of LP-X correlates well with the level of bile acids in the blood plasma, but not with the TC level. The formation of LP-X can be considered as a protective mechanism in response to developing cholestasis. Hypercholesterolemia in PBC is not accompanied by an increase in the frequency of cardiovascular events. Therefore, hypercholesterolemia is called “anomal”.
The orthophosphate required for increased synthesis of PhLs is formed as a result of raising activity of the enzymes ALP and 5’-NT. The enhanced activity occurs, by increasing the synthesis of these enzymes in the liver and bile ducts, which requires a higher delivery of amino acids to hepatocytes and bile capillary endothelial cells. In this connection, there is an increase in γ-GT activity just in the asymptomatic stage of PBC. The main clinical value of measuring the serum ALP, 5’-NT, and γ-GT levels lies in the early diagnosis of PBC. The activity of LAP in the presence of enhanced ALP activity is determined for the differential diagnosis of hepatobiliary and bone tissue diseases.
A developing disturbance in the bile secretion processes already in the early disease stages causes a higher bile capillary pressure that rises as cholestasis increases, which is accompanied by disturbed intercellular contacts in the presence of the remaining integrity of hepatocyte cytoplasmic membranes. In the late stages of the disease, the accumulation of bile acids in the hepatocytes is followed by impaired permeability of their cytoplasmic membranes, which is manifested by the enhanced activity of ALT and AST, as well as disrupted copper and bilirubin metabolism. The latter is attended by skin hyperpigmentation and conjugated hyperbilirubinemia with jaundice, respectively. In PBC, development of the stage of decompensated cirrhosis may also occur with an elevation in unconjugated bilirubin. The development of liver cirrhosis at a late stage of PBC is accompanied by a disturbance of the protein-synthesizing function of liver cells. A deficiency of the visceral protein pool is developed. This leads to a change in oncotic pressure and the development of edema and ascites. Protein-synthesizing dysfunction of liver cells results in diminished protein synthesis in the coagulation and anticoagulation systems with a shift in the balance between these systems, which can be accompanied by both hypo- and hypercoagulation. The imbalance associated with the lower concentration of the vitamin K-dependent glycoproteins C and S than that of the decreased level of blood clotting factors may contribute to increased thrombosis. The severity of hyperbilirubinemia and hypoalbuminemia permits assessment of hepatocellular failure, which is used in prognostic scales.
5’-NT: 5’-nucleotidase
ALP: alkaline phosphatase
ALT: alanine aminotransferase
AMAs: antimitochondrial antibodies
AST: aspartate aminotransferase
Dc: disconnection coefficient
HDLs: high-density lipoproteins
HDL-C: high-density lipoprotein cholesterol
LAP: leucine aminopeptidase
LDL: low-density lipoprotein
LDL-C: low-density lipoprotein cholesterol
LP-X: lipoprotein-X
MPs: microparticles
PBC: primary biliary cholangitis
PE: phosphatidylethanolamine
PhLs: phospholipids
PS: phosphatidylserine
TC: total cholesterol
TEG: thromboelastography
TF: tissue factor
VLDL: very low-density lipoproteins
γ-GT: γ-glutamyl transferase/γ-glutamyl transpeptidase
VIR and IVM equally contributed to: Conceptualization, Formal analysis, Investigation, Writing—original draft, Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Maitane Asensio ... Jose J. G. Marin
Ricardo Espinosa-Escudero ... Maria J. Monte
Grégory Merlen ... Thierry Tordjmann
Beatriz Sanchez de Blas ... Marta R. Romero
Carola Dröge ... Verena Keitel
Jose M. Pinazo-Bandera ... Miren García-Cortés
Elias Kouroumalis ... Argyro Voumvouraki