 Review
Review

Affiliation:
1Department of Gastroenterology, PAGNI University Hospital, University of Crete School of Medicine, 71500 Heraklion, Crete, Greece
2Laboratory of Gastroenterology and Hepatology, University of Crete School of Medicine, 71500 Heraklion, Crete, Greece
Email: kouroumi@uoc.gr
ORCID: https://orcid.org/0000-0002-6875-906X
Affiliation:
2Laboratory of Gastroenterology and Hepatology, University of Crete School of Medicine, 71500 Heraklion, Crete, Greece
31st Department of Internal Medicine, AHEPA University Hospital, 54621 Thessaloniki, Central Macedonia, Greece
ORCID: https://orcid.org/0000-0002-8595-2750
Affiliation:
31st Department of Internal Medicine, AHEPA University Hospital, 54621 Thessaloniki, Central Macedonia, Greece
ORCID: https://orcid.org/0000-0002-2725-6028
Explor Dig Dis. 2023;2:223–245 DOI: https://doi.org/10.37349/edd.2023.00028
Received: May 07, 2023 Accepted: July 03, 2023 Published: October 16, 2023
Academic Editor: Jose J. G. Marin, University of Salamanca, Spain
The article belongs to the special issue CHOLESTASIS
The pathogenesis of primary biliary cholangitis (PBC) is particularly complicated as both intrinsic and extrinsic factors are implicated. Several forms of cellular death, both programmable and non-programmable, operate leading biliary epithelial cells (BECs) to elimination. The precise role of critical pathways like autophagy, apoptosis, senescence, and their interplay has not been fully clarified. Therefore, in this review, data on these important mechanisms are presented and their implication in PBC is discussed. The interplay of the three mechanisms is examined and the factors that drive them are analyzed. Moreover, the upstream drivers of autophagy, apoptosis, and senescence are presented. They include the loss of the protective bicarbonate umbrella in BECs due to the reduction of activity of the anion exchanger 2 (AE2) with the resultant activation of the intracellular soluble adenylyl cyclase (sAC). The role of toxic bile acids is also presented. A sequence of events is proposed including involvement of the gut-liver axis and the possible role of ferroptosis. Finally, a brief account of the initial trigger of the disease is given.
Primary biliary cholangitis (PBC) is a chronic non-suppurative destructive cholangitis of small bile ductules, leading to bile duct loss and biliary cirrhosis. Several antimitochondrial antibodies (AMAs) are detected in patients’ sera [1, 2]. Emphasis has been given to autoimmunity and the critical factor responsible for disease pathogenesis is the presence of autoreactive CD4+ and CD8+ lymphocytes accumulating in the liver of patients. Moreover, a T helper (Th) 1 immune response mediated by interleukin (IL)-12 is found in the early stages of PBC, while in the fibrotic stages, a Th17 response driven by IL-23 is evident [3]. AMAs are not pathogenic, but macrophages from PBC patients secrete pro-inflammatory cytokines such as IL-6, and tumor necrosis factor alpha (TNFα) on incubation with apoptotic bodies from biliary epithelial cells (BECs) possibly indicating an M1 polarization of the phagocytes [4].
However, unlike other autoimmune diseases, PBC does not respond to immunosuppression, but rather to ursodeoxycholic acid (UDCA), a bile salt that induces a bicarbonate-rich choleresis or to obeticholic acid (OCA) which is a farnesoid X receptor (FXR) agonist. Fibrates can also be used as an alternative to UDCA-resistant patients, but they are discouraged when cirrhosis is decompensated liver disease [5].
Earlier studies have indicated that genetically predisposed patients develop PBC when epigenetic mechanisms reduce the expression of the Cl–/HCO3– exchanger anion exchanger 2 (AE2) BECs leading to disruption of the alkaline umbrella that inhibits access of toxic bile salts into BECs [6–9]. In addition, the accumulated bicarbonate increases the intracellular pH of BECs resulting in the activation of soluble adenylyl cyclase (sAC), and sensitization of cholangiocytes to apoptosis induced by toxic bile acids. On the other hand, AE2 deficiency and the increased pH impair mitophagy (a specific form of autophagy) in BECs, leading to the accumulation of damaged mitochondria, oxidative stress, endoplasmic reticulum (ER) stress, and aberrant presentation of immune-reactive mitochondrial antigens to the immune cells [2]. In support of this idea, a study reported that AE2 knockout (KO) mice spontaneously develop a PBC-like disease, with the appearance of specific AMAs against pyruvate dehydrogenase complex E2 (PDC-E2) [10]. An extensive review of PBC pathogenesis has recently been published [11].
The reason for BECs death is not clear as multiple cellular death mechanisms are operational in PBC. Moreover, the precise role of critical pathways like autophagy, apoptosis, and senescence, and their interplay have not been clarified. Therefore, the effort of this review is to present the data of these important mechanisms that participate in the crucial problem of BECs death in PBC. Additionally, the interplay of these pathways will delineate a possible sequence of events that lead to disease progression.
Apoptosis is a pathway of programmed cell death without the liberation of cellular elements into the cell microenvironment. Apoptosis is regulated by a series of activated caspases. After an initial signal, pro-caspases activate the initiator caspases-8 and -9 leading to the activation of the executioner caspases-3, -6, and -7.
There are two apoptotic pathways. An internal cellular signal initiates the intrinsic pathway. Initially, the activity of the pro-apoptotic Bcl-2 homology 3 (BH3)-only proteins is increased and suppresses the pro-survival members of the Bcl-2 family, while the pro-apoptotic members such as the Bcl-2 homologous antagonist/killer (BAK) and Bcl-2-associated X protein (BAX) assemble and cause the mitochondrial outer membrane permeabilization (MOMP). MOMP permanently damages mitochondria and is a critical factor in apoptotic death. A complex of cytochrome c, apoptosis protease activator factor 1 (Apaf-1), deoxyadenosine triphosphate (dATP), and procaspase-9, called apoptosome is formed as a consequence of MOMP. The initiator caspase-9 is activated by the apoptosome and cleaves the pro-caspases-3 and -7 leading to the execution of apoptosis. Furthermore, activated caspases-3 and -7 cleave and activate several additional pro-caspases such as caspases-2, -6, -8, and -10 leading to an apoptosis-amplifying cascade [12].
The extrinsic pathway is activated when a ligand binds to membrane death receptors such as tumor necrosis factor receptor 1 (TNFR1) or tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). After initiation, the pathway is mediated by the formation of the complex I comprised of the protein TNFR1-associated death domain protein (TRADD), the three ubiquitin ligases TNF receptor associated factor (TRAF) 2/5, cellular inhibitor of apoptosis proteins (cIAPs), linear ubiquitin chain assembly complex (LUBAC), and the kinase receptor-interacting serine/threonine-protein kinase 1 (RIPK1). Ubiquitination of RIPK1 is the scaffold that recruits transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) and its binding partners TAK1-binding proteins (TAB1, TAB2) and the complex of inhibitors of NF-B kinase a/b with the NF-B essential modulator (IKKα/β-NEMO) leading to nuclear factor-kappa B (NF-κB) activation and suppression of apoptosis through the transcription of pro-survival genes. On the other hand, complex II, comprised of Fas-associated death domain protein (FADD), TRADD, RIPK1, and caspase-8, is formed when RIPK1 is deubiquitinated leading to activation of the executioner caspases-3 and -7. Alternatively, caspase-8 may indirectly cleave the BCL-2 family member BH3-interacting domain death agonist (BID) into truncated BID (tBID) translocate into the mitochondria, and bind BAX/BAK resulting in MOMP. Activation of caspase-8 is inhibited by the caspase-like protein FLICE-inhibitory protein (FLIP) [12, 13]. A variety of cellular proteins are cleaved by the effector caspases causing morphological changes in the cell including DNA fragmentation and actin reorganization resulting in the creation of membrane blebs among others. Phosphatidylserine (PS) molecules expression in the plasma membranes function as “eat me” signals for macrophages [14–16]. The integrity of complex I and RIPK1 ubiquitination prior to the initiation of apoptosis is important. Apoptosis was blocked when TRADD was inhibited. Furthermore, autophagy was activated through activation of beclin-1 [17]. The tumor suppressor p53, a responder to DNA damage is also a pro-apoptotic regulator regulating the expression of several pro-apoptotic BCL-2 proteins [14, 18, 19].
Autophagy is a pathway necessary for the survival of cells. It is a recycling mechanism that depends on lysosomal degradation regulating the handling of intracellular damaged organelles or pathogens. Degradation products are re-used by the cell to synthesize new proteins, lipids, and enzymes.
There are several stages in autophagy. The first stage is initiation, followed by phagophore formation, autophagosome formation, fusion with lysosomes, and degradation. Autophagy starts with the activation of the unc-51-like kinase 1 complex (ULK1), leading to the activation of beclin 1 and autophagosome formation and the implication of light chain protein 3 (LC3) which initially forms LC3-I, and then LC3-II. Blocking of the late stages of autophagy will increase autophagosomes leading to autophagy-dependent cell death. The complex molecular steps of autophagy were recently described in detail [20, 21]. The mammalian target of rapamycin complex 1 (mTORC1) and the 5’ AMP-activated protein kinase (AMPK), are the metabolic sensors that initiate autophagy. mTORC1 is a negative controller of autophagy through phosphorylation and inhibition of ULK1. AMPK, on the other hand, increases autophagy by suppression of the mTORC1 activity [22]. The transcription factor EB (TFEB), is the long-term controller of lysosomal biogenesis [23].
Mitophagy is a form of autophagy that removes damaged mitochondria. Mitophagy is important in many liver diseases including cholestatic syndromes. The first step of mitophagy is similar to canonic autophagy and is controlled by mTORC1 and AMPK. Mitochondrial reactive oxygen species (ROS) suppress mTORC1 and increase mitophagy. Adenosine triphosphate (ATP) depletion activates AMPK and increases mitophagy. The second step is associated with a molecular modification of the mitochondria which are recognized by the autophagosomes [24]. Mitophagy of damaged mitochondria reduces pro-apoptotic proteins and the production of ROS [25]. A recent study demonstrated that accumulation of ROS and p62 were observed due to the loss of forkhead box proteins O1 and O3 (FOXO1/3) when autophagy is impaired. The p62-FOXO1/3 axis is probably the underlying mechanism for the repression of antioxidant defense in deficient autophagy [26].
PTEN-induced putative kinase 1 (PINK1)/465-amino acid residue E3 ubiquitin ligase (Parkin)-dependent and PINK1/Parkin-independent are the two recognized mechanisms of mitophagy. PINK1/Parkin-dependent mitophagy is activated when mitochondrial depolarization inhibits PINK1 degradation leading to PINK1 sequestration on the outer membrane of damaged mitochondria. Then, PINK1 phosphorylates Parkin and promotes the mitochondrial translocation of Parkin. PINK1/Parkin-independent mitophagy is induced when ubiquitin E3 ligases mediate the ubiquitination of mitochondrial proteins [27, 28].
The association between autophagy and inflammasome activation is variable. Autophagy could suppress the assembly stages of the nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome [29] or may eliminate active inflammasomes, particularly in macrophages [30]. This negative interplay between autophagy and inflammasome activation is not always constant as the association can become positive. Autophagy may promote NLRP3 activation by initiating NF-κB nuclear translocation leading to cell pyroptosis [31].
Αpoptosis and autophagy may cooperate to kill, or their correlation may be negative. Similar signals can initiate either apoptosis or autophagy. Usually, autophagy precedes apoptosis as an effort of the cell to survive [32] but apoptosis will finally eliminate the cell when autophagy is impaired. The apoptotic program will be inhibited by the initiation of autophagy and vice versa [33]. There are several gatekeepers between autophagy and apoptosis.
Bcl-2 is a significant controller of the interplay. Bcl-2 suppresses the pro-apoptotic proteins such as BAX and interacts with the phosphatidylinositol-3 kinase (PI3K) complex of the autophagy process, favoring cell proliferation. Phosphorylation of Bcl-2, on the other hand, represses the binding to BAX favoring apoptosis [34].
The Beclin-1/Bcl-2 interaction was the first association between autophagy and apoptosis to be identified. Beclin-1 participates in autophagosome formation [35]. Several BH3-only proteins have the ability to activate either autophagy or apoptosis. The BH3-only proteins inhibit the anti-apoptotic proteins of the Bcl-2 family and activate those with a pro-apoptotic effect to initiate apoptosis [36–38]. To initiate autophagy, BH3-only proteins dissociate the Beclin-1/Bcl-2 complex and allow Beclin-1 to increase autophagic activity. However, Bcl-2 interacting mediator of cell death (BIM), a particular BH3-only protein, decreases autophagy acting the opposite way with the other BH3-only proteins. Jun N-terminal kinase (JNK) induces either autophagy or apoptosis through phosphorylation of Bcl-2 by JNK inactivates Bcl-2 favoring apoptosis but phosphorylation of BIM is leading to autophagy [33].
Mammalian target of rapamycin (mTOR) can regulate autophagy as mentioned before. There are two mTOR signaling pathways: the PI3K/Ak strain transforming-protein kinase B (Akt)/mTOR and the liver kinase B1 (LKB1)/AMPK/mTOR signaling pathway [39]. mTOR also has variable effects on apoptosis. The final effect depends on the cells involved and the downstream targets such as p53 and Bcl-2 family members. mTOR either inhibits apoptosis by activation of the anti-apoptotic Bcl-2 or induces apoptosis by activating the nuclear p53 [39, 40].
p27Kip1 is a significant mediator of autophagy, and apoptosis [41] depending on its cellular location. The cytoplasmic location of p27Kip1 favors cellular survival through autophagy while the nuclear location of p27Kip1 makes the cell susceptible to apoptosis or senescence [42, 43].
The cellular FLICE protein (c-FLIP) and the viral FLIP (v-FLIP) are significant anti-apoptotic proteins in the extrinsic apoptotic pathway as mentioned before [44]. Moreover, c-FLIP inhibits autophagy interfering with the autophagy-related protein (Atg) 3 association with the LC3 protein, which is a critical element for autophagosome formation [45]. FLIPs, therefore, are both anti-apoptotic factors and also inhibitors of autophagy.
Death-associated protein kinases (DAPK) comprise a family of five kinases that are implicated in both apoptosis and autophagy [46]. DAPK2 increase leads to extensive apoptosis of cells not attached to the extracellular matrix [46, 47]. DAPK2 may also induce autophagy by mTORC1 activity inhibition [48]. A second mechanism of autophagy induction is the phosphorylation of Beclin-1 by DAPK, leading to the separation of Beclin-1 from the inhibitory effect of Bcl-2 [49]. On the other hand, DAPK1 attenuates oxidative stress and reduces autophagy and inflammation [50].
A diagram of the interplay between autophagy and apoptosis is presented in Figure 1.
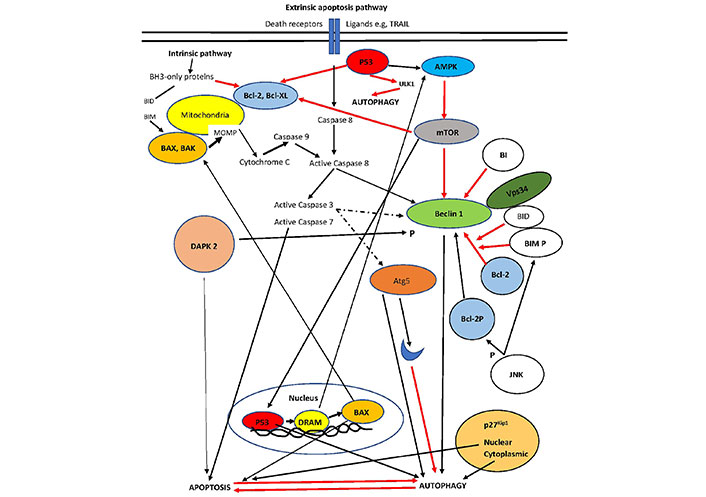
The interplay of autophagy and apoptosis. p53 localization in the nucleus is the master controller that increases autophagy via damage-regulated autophagy modulator (DRAM) activation of AMPK. In contrast, p53 in the cytoplasm represses autophagy. p53 may also lead to apoptosis through either suppression of Bcl-2 (cytoplasmic) or overexpression of BAX (nuclear). Bcl-2 association with Beclin1 represses the complex increasing autophagy (only Vps34 is shown). Phosphorylation of Beclin-1 by DAPK or Bcl-2 by JNK activates the pro-autophagy complex. DAPK directly favors apoptosis. Cleavage of Beclin-1 and/or Atg5 (an autophagy inducer initiated by ER stress) by apoptosis-associated caspases leads to inhibition of autophagy. BH3-only proteins such as BID dissociate Bcl-2 from the pro-autophagy complex and increase autophagy. BIM inhibits Beclin 1, but when phosphorylated dissociates Bcl-2 from the complex. Intermediate proteins are not shown for clarity. Intermittent line: cleavage; black line: activation; red line: inhibition [51]. The blue moon represents a cleaved part of Atg5. Bcl-XL: B-cell lymphoma-extra large; Vps34: class III phosphoinositide 3-kinase; BIM P: phosphorylated BIM; Bcl-2P: phosphorylated Bcl-2
Note. Adapted from “Pathogenesis of hepatocellular carcinoma: the interplay of apoptosis and autophagy,” by Kouroumalis E, Tsomidis I, Voumvouraki A. Biomedicines. 2023;11:1166 (https://doi.org/10.3390/biomedicines11041166). CC BY.
Apart from autophagy and apoptosis, cells may respond to stress with cellular senescence. Senescent cells are permanently arrested at the G1 or G2 phase of the cycle being unable to grow, but they are still metabolically active [52]. The senescence-associated secretory phenotype (SASP) is characterized by hypersecretion of pro-inflammatory cytokines, chemokines, and metalloproteases [53, 54]. It has been suggested that a change in terminology may be more appropriate to describe what a senescent cell is. Apart from aging, senescence is in fact a process of cellular adaptation and a mechanism of tissue remodeling. The replacement of the term cellular senescence with remodeling activation and SASP with remodeling-associated secretory phenotype (RASP) has been recently proposed [55].
Different cells can activate both apoptosis and senescence. However, in most cells, these processes seem to exclude each other. Interference with Bcl-2 or caspase levels is the key to switching from apoptosis to senescence [56]. The critical gatekeeper for the crosstalk between these pathways seems to be the level of p53 [57]. In addition, senescence blockade by telomerase overexpression protects cells from apoptosis [58]. Moreover, initiation of senescence seems to be protective against p53-dependent apoptosis [57] and increased oxidative stress [59, 60]. To achieve this, senescent cells overexpress anti-apoptotic proteins such as Bcl-2 that inhibit apoptosis [61]. On the other hand, inhibition of the same anti-apoptotic proteins induces apoptosis of senescent cells [61, 62]. Other mechanisms of apoptosis resistance in senescence, are the inhibition of the p53-mediated apoptosis through upregulation of the cell cycle inhibitor p21 [63, 64] or inhibition of the effector caspase-3 and the activation of the JNK pathway, both mandatory for apoptosis [65]. IL-6 maintains senescence by inhibiting the intrinsic pathway of apoptosis and by stimulating the pro-survival nuclear translocation of NF-κB [66]. Mitochondria are key modulators in the progression of the pro-inflammatory SASP. SASP hypersecretion causes ROS overproduction by mitochondria thus completing a circle. Molecular details of senescence and mitochondria were recently reviewed [67].
Cells under stress initiate autophagy to maintain metabolism through lysosomal degradation of cellular waste. Autophagy can either favor senescence or delay apoptosis [68]. Autophagy has a dual effect on cellular senescence. When damage is accumulated in cells, senescence and SASP production are triggered. At the same time, a different pathway might operate leading to the induction of autophagy and cell survival. However, autophagic degradation of cellular components provides building blocks such as amino acids and nucleotides that are used by the already senescent cells in SASP production and reduced survival [69, 70]. Senescent cells have a significant increase in lysosomal mass and elevated autophagic activity [71].
Mitophagy and cell senescence also crosslink. ROS, hypoxia, and unfolded proteins activate the mitophagic pathways PINK/E3 ubiquitin ligase Parkin and Bcl-2 and adenovirus E1B 19kD interacting protein 3 (BNIP3)/BNIP3-like protein (NIX)/LC3. On the other hand, mitochondrial dysregulation, induced by ROS and increased AMPK activity, leads to senescence by activating tumor suppressor pathways such as p53/p21. Inhibition of the PINK/PARK pathway by the cytoplasmic p53 stabilizes senescence. Therefore, defective mitophagy favors cell senescence, and restoration of mitophagy delays cellular senescence. The regulatory role of mitophagy in senescence seems to be partly independent of alterations in general autophagy [72, 73]. Detailed reviews on cellular senescence have been published [67, 70, 74, 75].
The reasons and mechanisms of hepatocyte death in cholestasis are still under investigation [12]. Autophagy and lysosomal degradation were reported to be impaired in the cholestasis model of bile duct ligation [76]. Moreover, lysosomal inhibition caused by chloroquine or deletion of Atg7 and Atg5 increased liver damage in experimental cholestasis [77].
Autophagy is involved in the pathogenesis of PBC [78, 79] more so as autophagy is also implicated in the presentation of several antigens to the immunocytes. Cellular senescence in BECs was also associated with deregulated autophagy [80, 81]. A pathogenetic hypothesis has been proposed, implicating deregulated BECs autophagy associated with cholangiocyte senescence and increased apoptosis [82, 83].
Light chain 3β (LC3B) and p62 protein accumulation are in close relation with senescence markers in damaged BECs, suggesting that autophagy could induce BECs senescence [84–86]. LC3 and p62 were demonstrated in vesicular elements of the cytoplasm in bile duct lesions in PBC [87]. At first, it was interpreted as a reflection of increased autophagy [88], but it turned out to be an abnormal sequestration of autophagosomes as a result of impaired autophagy.
Autophagy also interferes with PBC therapy. UDCA is the first-line treatment of PBC [89]. Experimental evidence has shown that hydrophobic bile acids impair autophagy and initiate senescence and aberrant presentation of mitochondrial antigens in BECs, through induction of ER stress. Pretreatment with UDCA suppressed ER stress and partially corrected impaired autophagy and senescence [90]. UDCA is a FXR antagonist. OCA, an alternative treatment of PBC, is an FXR agonist. It reduces the synthesis of endogenous bile acids and regulates the bile acid transporters of hepatocytes [89, 91]. OCA impairs autophagy a fact that is not consistent with current data showing aggravation of cholestasis when autophagy is blocked [77]. It seems, therefore, that other properties of OCA compensate for its negative effect of suppressed autophagy. An interesting explanation was recently proposed. Autophagy is impaired in human cholestasis where hydrophobic bile acids induce Rubicon in an FXR-dependent fashion that suppresses the fusion of autophagosomes with the lysosomes and inhibits lysosomal degradation. Rubicon was also overexpressed after treatment with OCA while inhibition of Rubicon restored the impairment of autophagy [92].
In addition to the involvement of canonic autophagy in PBC, mitophagy is implicated in PBC as shown by the co-localization of the mitochondrial protein PDC-E2 with LC3. Mitophagy may reduce inflammatory cytokine secretion and regulate mitochondrial antigen presentation and immune cell infiltration of the liver. Mitophagy has been shown to prevent the autoimmunity implicated in PBC, while mitochondria-derived vesicles (MDV) facilitate mitochondrial antigen presentation as autoantigens [93].
Autophagy may also be involved in the fibrotic late stages of PBC, as autophagy is indeed associated with liver fibrosis and the final result varies according to the liver cells involved. Autophagy restricts fibrosis acting in hepatocytes, macrophages, and liver sinusoidal endothelial cells (LSECs). Autophagy reduces the production of inflammatory cytokines by macrophages and LSECs, thus limiting hepatocyte damage. However, in hepatic stellate cells (HSCs), autophagy favors fibrogenesis first by increasing lipophagy and release of the energy required to activate HSC, and second by p62 loss leading to impairment of the vitamin D receptor/retinoid X receptor (VDR/RXR) complex which is critical for the maintenance of HSC in a quiescent state [94, 95].
Terminal deoxynucleotidyl transferase deoxyuracil triphosphate (dUTP) nick end labeling (TUNEL) staining of liver sections has demonstrated activation of apoptosis in BECs of PBC patients compared to the normal and patients with chronic cholestasis with similar degrees of inflammation [96–99]. Significantly higher expressions of Fas, Fas ligand (FasL), perforin, granzyme B, and TRAIL were also observed indicating the implication of the extrinsic pathway of apoptosis [96, 100]. TNFα is one of the central inflammatory mediators of apoptosis initiation in BECs [101, 102].
Immunologically active PDC-E2 resistant to cleavage by caspases, was recognized in cholangiocytes after apoptosis [103]. Moreover, PDC-E2, one of the autoantigens of PBC, is localized in the apoptotic bodies where it is recognized by immune cells and produces AMAs [4, 104]. Glutathione modifies a PDC-E2 sulfhydryl group being the reason for the normal loss of recognition of this epitope in BECs. Loss of recognition was abolished by either overexpression of Bcl-2 or depletion of glutathione pointing to glutathiolation of PDC-E2, as the reason for it [105]. Furthermore, macrophages from PBC patients co-cultured with apoptotic blebs in the presence of AMAs produce large amounts of inflammatory cytokines [4, 100].
The implication of apoptosis in cholangiopathy was additionally shown by studies on ductular reactive cells. Double KO mice for both TRAIL and multidrug resistance 2 (MDR2) displayed overexpressed ductular reaction and advanced fibrosis. This overexpression was associated with impaired apoptosis due to the involvement of the anti-apoptotic protein myeloid cell leukemia 1 (MCL1). Treatment with an MCL1 inhibitor induced apoptosis in these ductal cells and reduced fibrosis [106]. Additional evidence of activation of apoptosis was also reported. Overexpression of activated caspases-3 and -8 in liver tissue of PBC patients has been demonstrated indicating increased apoptosis [107]. KO of caspase-8, but not a pan-caspase inhibitor, attenuated apoptosis without switching to other pathways of cell death [107]. By contrast, the use of pan-caspase inhibitors in bile duct ligated mice, led to decreased apoptosis with reductions of caspases-3 and -7, inflammation, HSC activation, and increased survival [108]. These findings indicate that apoptosis is an important mode of cell death in experimental cholangiopathy.
However, it is uncertain whether increased apoptosis is present in the early stages. In a well-characterized murine autoimmune cholangitis (AIC) model, apoptosis of BECs was significant in the early stages of AIC. Interestingly, this was associated with altered gut microbiota. BECs in AIC mice overexpress the adhesion molecule intercellular adhesion molecule-1 (ICAM-1), the chemokine (C-C motif) ligand 2 (CCL2), chemotactic cytokine ligand (CXCL) 9, CXCL10, and the toll-like receptor (TLR) 2. Activation of the TLR2 increased apoptosis and CXCL10 production. Dysbiosis in the gut microbiota, with increased Firmicutes, induced apoptosis of BECs through TLR2 signaling [109].
Apoptosis may be one additional mechanism of action of UDCA in PBC. UDCA protects against hydrophobic bile acid-induced apoptosis [110]. In vitro, taurine-conjugated UDCA, inhibits apoptosis independently of caspase-8 activation by inducing pro-survival signals through several cellular pathways [110]. Interestingly, increased levels of CXCL9 and CXCL10 were described in the liver tissue and serum of patients with PBC. Treatment of these patients with UDCA led to a significant reduction of these chemokines in serum [111].
The important work of Sasaki and Nakamura [82–84] on BECs senescence in PBC was mentioned before. Interestingly, senescent BECs exhibit overexpression of many chemokines such as CCL2 and fractalkine (CX3CL1) in PBC. These findings indicate that the senescence may modify the microenvironment around bile ducts by SASP, thus contributing to immune cell attraction [112, 113].
As mentioned before, senescence is related to resistance to apoptosis due to overexpression of anti-apoptotic or reduced pro-apoptotic mediators. It has been demonstrated that the anti-apoptotic Bcl-2 family members are upregulated in senescent BECs, while their pharmacological inhibition kills senescent BECs and ameliorates liver fibrosis [114]. A previous report showed that the transcription factor ETS proto-oncogene 1 (ETS1), promotes cholangiocyte senescence [115]. ETS1 and the histone acetyltransferase E1A-binding protein p300 (EP300/p300) increase Bcl-XL transcription making therefore senescent cells resistant to apoptosis [116]. The importance of senescence in cholangiopathies was recently reviewed [117].
Several proteins have been identified as drivers of senescence in PBC. Α recent complementary DNA (cDNA) microarray analysis on senescent cholangiocytes showed overexpression of several chemokines and cytokines. Highly overexpressed genes included the interferon-induced tetrapeptide repeat 3 (IFIT3), encoding for a protein implicated in the anti-viral innate immune response. An upregulation of IFIT3 was demonstrated in ductular reactive cells in PBC patients. Marked reduction of senescence markers p16 and p21 accompanied by increased BECs apoptosis was also demonstrated after inhibition of IFIT3. IFIT3 is therefore a driver senescence [118]. Increased secretion of TGF-β in association with initiation of senescence in neighboring BECs and hepatocytes was observed, indicating a paracrine induction of senescence. Repression of TGF-β disrupted the senescent process. Therefore, TGF-β signaling is also a driver of senescence and disease promotion [119].
Dysfunctional mitochondria and deregulated mitochondrial unfolded protein response (UPRmt) were reported in senescent hepatocytes both in vitro and cirrhosis. Interestingly, compensated cirrhotic liver had a strong UPRmt, associated with high levels of the mitochondrial protease caseinolytic protease P (ClpP). Overexpression of ClpP in vitro inhibited senescence by reducing mitochondrial ROS. Reduction of ClpP, on the other hand, shifted cirrhosis toward decompensation by increasing hepatocyte senescence. If this also applies in cirrhotic PBC should be investigated [120].
Oxidative stress plays a central role in the pathogenesis of PBC as it affects autophagy senescence and apoptosis.
ROS can induce autophagy by inhibiting the splicing of the LC3/Atg8 complex which produces LC3-I. Alternatively, activation of the AMPK pathway under hypoxia by hypoxia-inducible factor (HIF) can also initiate autophagy. ROS-mediated inhibition of PI3K in the PI3K/Akt pathway can suppress autophagy through the control of mTORC1 [70].
Increased expression of the oxidative stress marker 8-hydroxy-2’-deoxyguanosin (8-OHdG) has been found in early PBC, suggesting a connection between oxidative stress with the initiation of senescence. Moreover, expression of the BMI-1 (polycomb complex protein/polycomb group RING finger protein 4) protein is decreased by oxidative stress, and this protein is reduced in the bile ducts of PBC patients, providing additional evidence for the implication of oxidative stress in PBC [79]. Autophagy does not induce oxidative stress, but defective removal of damaged mitochondria due to reduced mitophagy can lead to the accumulation of ROS. As mentioned before, the p62 protein signal is significantly overexpressed in PBC. It is possible that the increased p62 protein is consistent with inefficient mitophagy, and oxidative stress [78, 79, 87].
In line with increased oxidative stress, the corrected total antioxidant capacity (CTAC) was significantly increased in PBC patients from the early stages I and II. Levels of CTAC were restored back to normal after treatment with UDCA. The increased antioxidant capacity might represent an initial adaptation to increased oxidative stress [121].
An important defensive mechanism that allows cells to resist oxidative stress is the nuclear factor-E2-related factor 2 (Nrf2) pathway. Nrf2 promotes the activation of several cytoprotective genes including heme oxygenase-1 (HO-1), catalase, and enzymes participating in glutathione metabolism, such as glutathione S-transferase [122, 123]. Activation of Nrf2 is regulated by the Kelch-like ECH-associated protein 1 (Keap1). Keap1 either accumulates Nrf2 in the cytoplasm or leads to Nrf2 degradation by autophagy. In the first instance, Nrf2 activates the antioxidant genes after translocation to the nucleus. The normal function of the Nrf2/Keap1 system requires the presence of p62. p62 binds to Keap1 and facilitates its degradation [124]. The Nrf2/Keap1 signaling axis is also controlled by microRNAs. Several microRNAs function as modulators of Nrf2 [125, 126]. The Nrf2 protein was elevated in PBC, but Nrf2 gene expression was significantly decreased in PBC-related cirrhosis, a situation in line with the CTAC findings previously mentioned. Nrf2 gene products, such as HO-1, were reduced accompanied by increased levels of microRNA-132 and microRNA-34a. Both Keap1 and p62 protein levels were significantly upregulated in PBC. The impaired Nrf2/Keap1 system affects the defense against oxidative stress in PBC and contributes to the progression of disease [127].
ER stress is the cellular stress that occurs because of the excess sequestration of unfolded and misfolded proteins in the ER. The unfolded protein response (UPR) can compensate, up to a certain point, for this excessive cargo. When the ER stress is severe enough, it overcomes compensation and initiates several pathways of cell death such as autophagy, apoptosis, ferroptosis, and pyroptosis. The UPR is initiated by three pathways, namely, inositol-requiring enzyme 1α (IRE1α), pancreatic ER kinase (PERK), and activating transcription factor 6 (ATF6), that regulate downstream UPR genes to restore ER protein homeostasis [128].
The UPR can activate both autophagy and apoptosis. To initiate autophagy, IRE1α and ATF6 activate autophagy through Beclin1. All three UPR pathways can initiate autophagy by activating the pro-apoptotic factor CCAAT/enhancer-binding protein homologous protein (CHOP) which is a central regulator of autophagy induced by UPR [129, 130]. The UPR may also induce apoptosis. Apoptosis signaling induced by ER stress is mediated by the same UPR pathways, as all three of them PERK, IRE1α, and ATF6 can induce apoptosis [131, 132]. CHOP is again the central regulator of apoptosis. ER stress activates the intrinsic apoptotic pathway through the pro-apoptotic BAX and BAK proteins at the mitochondria and the activation of executor caspase-3. BAX-BAK-double-KO cells or mice are resistant to apoptosis caused by ER stress [133, 134].
In experimental models of intrahepatic cholestasis, an increase in ER stress and upregulation of UPR genes and proteins including CHOP has been reported [135–138]. UPR genes [binding immunoglobulin protein (BIP) and CHOP] are overexpressed in the liver when animals are fed with bile acids [139]. CHOP reduction ameliorates cholestatic liver injury and fibrosis and CHOP-depleted hepatocytes are more resistant to death caused by the bile acid glycochenodeoxycholic acid (GCDCA) [140]. The protective role of CHOP deficiency may also be due to the fact that CHOP impairs intestinal barrier tight junctions leading to increased intestinal bacteria translocation and Kupffer cell-induced inflammation. The gut-liver axis may thus participate in the pathogenesis of cholestatic injury [135]. In the bile duct ligation model of cholestasis, an increased expression of CHOP at all time points, in association with overexpression of caspases-3 and -12 was observed. Treatment with taurine-conjugated UDCA led to restricted expression of CHOP and pro-apoptotic markers. This effect might be due to diminished hepatic UPR and apoptosis [141]. Data indicates that ER stress is involved in human cholestatic syndromes as well. The ER stress markers BIP and protein disulfide isomerase (PDI) are overexpressed in the bile ductular cells of PBC patients [87, 142]. UPR pathways are upregulated in cholestatic human liver biopsy tissue after liver transplantation, data supporting significant participation of the UPR in human cholestatic injury [143].
Nothing is known about iron in PBC, particularly ferroptosis. Ferroptosis is a death mode different from apoptosis and autophagy. Free cytoplasmic iron acts on lipid peroxidation of the unsaturated fatty acids expressed on the cell membrane, accompanied by a significant reduction of glutathione peroxidase 4 (GPX4). Lipid peroxidation is mediated by the acyl-coenzyme A (acyl-CoA) synthetase long-chain family member 4 (ACSL4)-lysophosphatidylcholine acyltransferase 3 (LPCAT3)-arachidonate lipoxygenase 15 (ALOX15) pathway. Antioxidant systems, such as system xc- with its components solute carrier family 7 member 11 (SLC7A11), GPX4, and the Nrf2 system, inhibit ferroptosis. Nrf2 has an important role in ferroptosis [51, 144, 145].
Nrf2 transactivates the metallothionein 1G (a protein with a high affinity for divalent metal ions), SLC7A11, and HO-1, thus limiting ferroptosis [146].
However, excessive activation of Nrf2 leads to HO-1 hyperactivation and induction of ferroptosis increasing the free iron pool through heme metabolism [147, 148]. Therefore, the protective function of HO-1 depends on its antioxidant activity, while the toxic effect is attributed to the generation of free iron that increases the toxic Fenton reaction [149].
Data on the implication of iron metabolism in PBC is scarce. Mass spectroscopic analysis of serum from PBC patients demonstrated alterations characterized by elevation of markers of lipid peroxidation [150] including antibodies against the adduct of malondialdehyde [151]. Alterations in glutathione metabolism and accumulation of protein oxidation products have also been reported in PBC [152]. Nrf2 is involved in PBC as mentioned before, and iron is also increased in PBC. A significant association between evidence of oxidative stress and iron grade after Perl’s stain and PBC stage was observed [151].
Hepcidin, the master regulator of iron metabolism is decreased in PBC patients compared to patients with other liver diseases. Low hepcidin was present even after prolonged treatment [153]. The suppression of signal transducer and activator of transcription 3 (STAT3) phosphorylation by accumulated bile acids may be the reason for low hepcidin [154]. Interestingly, UDCA treatment of PBC seems to increase glutathione levels but not lipid peroxidation [155, 156]. These findings indicate a significant role of ferroptosis induction in PBC but this requires further investigation [157].
Bile acids are the best candidates to interfere with autophagy, apoptosis, and senescence in PBC. Accumulated hydrophobic bile acids are associated with ER stress, mitochondrial dysfunction, inflammasome activation, and apoptosis [158–160].
CHOP-mediated apoptosis has been demonstrated in vitro and in vivo experiments in cholestasis induced by toxic bile acids [161]. Autophagic flux is also repressed by bile acids during cholestasis. FXR KO mice have deregulated autophagic flux by impairment of the autophagosomal-lysosomal fusion [162]. Inclusion bodies or Mallory-Denk bodies (MDBs) are a common finding in patients with PBC [163, 164]. Increased toxic bile acids drive the formation of MDBs in cholestasis [163]. It should be noted that liver MDBs in patients with PBC are located in the acinar zone 1, where bile acids concentrations are highest [165].
Nonetheless, there is an additional factor that acts before the implication of bile acids. Cholangiocyte membrane permeability by bile acids is pH-dependent. The AE2 maintains normal intracellular bicarbonate levels and creates a bicarbonate umbrella to keep toxic bile salts out. After an initial insult, the X-linked miR-506 is upregulated in BECs, binds to the AE2 messenger RNA (mRNA), and represses translation of AE2. Reduced AE2, destabilizes the bicarbonate umbrella and protonated bile salts enter BECs leading to increased cytoplasmic free Ca2+ and activation of sAC promoting thus the sensitization to apoptotic stimuli. Inhibition of sAC prevents bile salt-induced apoptosis [9, 166]. It was recently reported that reduction of AE2 paralleled the abnormal expression of PDC-E2 and the autophagy markers LC3 and p62 [167]. In addition, AE2 knockdown induces cellular senescence [167, 168]. Interestingly, deregulated autophagy can also induce cellular senescence as mentioned before [88]. It is possible then, that in PBC patients, senescence can be initiated through deregulated autophagy due to AE2 downregulation.
Interestingly, abnormalities of the miR-506/AE2-sAC axis may be an explanation for the increased female prevalence of PBC. In females, one of X chromosomes is inactivated and chromosome X-related gene expression is equal in males and females. The unknown initial trigger of PBC may cause reactivation of the silenced chromosome. Females would overexpress miR-506 leading to decreased bicarbonate umbrella and increased sAC activity associated with BECs damage. Pro-inflammatory cytokines increase miR-506 expression and continuous activation of the miR-506-AE2-sAC axis. The created vicious cycle leads to senescence ductopenia and biliary fibrosis [169].
It is interesting to clarify the actual sequence of events as PBC progression may take many years. Sasaki et al. [78] have shown that autophagy precedes senescence in PBC after immunohistochemical evaluation of both processes in PBC livers. Data indicates that an impaired bicarbonate umbrella may precede abnormalities of autophagy, and cellular senescence in BECs. Aberrant expression of mitochondrial antigens may follow [167]. Data also indicate that immune activation and the infiltration of the liver by immunocytes are a direct consequence of the preceding abnormalities. TLRs are expressed in BECs and other liver sinusoidal cells. As mentioned before, microbial components such as lipopolysaccharide (LPS), bind to TLRs, causing injury and release of cytokines and chemokines. Chemokines attract immune cells into the portal tracts of PBC patients [170]. Histopathological assessment of liver tissue from PBC patients demonstrated that CD8+ T cells could infiltrate between BECs, and this entry correlated with TUNEL-positive apoptotic BECs [171]. Chemokines are also implicated in the attraction of regulatory T cells (Tregs) and the pro-inflammatory Th17 and IL-17 secreting CD8 Th cells (Tc17) lymphocytes [172, 173]. In that respect, the role of a subset of innate-like T cells named mucosa-associated invariant T (MAIT) cells, is crucial as they protect the biliary cells from microbial insults [174]. However, MAIT cells are significantly reduced in PBC and the protective role of MAIT cells is restricted limiting the protection of biliary integrity [175]. MAIT cell numbers are not restored to normal even after successful treatment with UDCA, suggesting that the disease may progress despite treatment response [176].
Although the investigation of mechanisms leading to PBC initiation and progress has been advanced, the critical problem of the initial trigger still remains. Interestingly, a human beta-retrovirus (HBRV) has been characterized in patients with PBC. Linking the viral infection with the disease is not an easy task because PBC is a multifactorial disease and genetic, autoimmune, and environmental factors are participating. The evidence to support an autoimmune-only disease is weak. The beta-retrovirus hypothesis, on the other hand, has a lot of evidence to support this idea. Using Hill’s criteria allowed to support that it is the beta-retrovirus infection the initial insult that may trigger autoimmunity and propagate biliary disease [177].
A few years ago, a hypothesis was proposed that initiation and progression of PBC may be due to an increase of circulating endothelins, particularly endothelin 2 (ET2) both in the periphery and in the hepatic vein of patients already present from the early stages of the disease. Such an abnormality may cause vasoconstriction of the peribiliary plexus and hypoxia may induce apoptosis [178, 179]. But even in this theory, the etiology of endothelin overproduction remains unanswered. A graphical demonstration of the sequence of events in the development of PBC is presented in Figure 2.
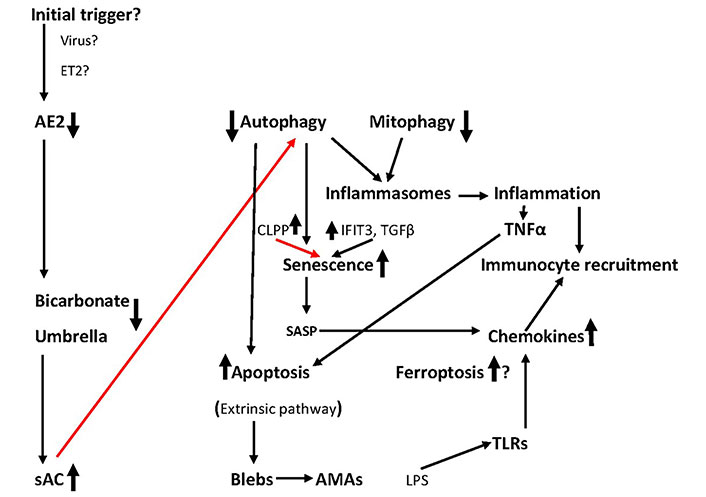
The sequence of events leading to BECs elimination. The role of ferroptosis is under investigation. Initial triggers remain conjectural. Red lines: inhibition; black lines: activation. Question marks indicate a hypothesis. See text for details
The pathogenesis of PBC is multifactorial. It is generally considered an autoimmune disease but the evidence to support this idea is very weak. In fact, it seems that autoimmune involvement is not the primary event that triggers disease initiation. The reason why BECs die leading to ductopenia is not clear. Autophagy, which is a pro-survival pathway, is defective in PBC. Mitophagy, a special form of autophagy is also deregulated, and damaged mitochondria may lead to ER stress and damaged BECs through oxidative stress. In addition, apoptosis of BECs is upregulated from the early stages of PBC, a finding that is in line with the fact that in most instances autophagy and apoptosis are mutually excluded. The implication of ferroptosis has not been investigated in PBC but there is indirect evidence that it may be important. Moreover, a third mechanism of cellular death is also involved. Senescence is extensive in PBC and senescent BECs produce many cytokines and chemokines that attract various immunocytes in the microenvironment of bile ductules. These cells aggravate a local inflammation leading to the progression of the disease. Senescent BECs are resistant to apoptosis. Autophagy, apoptosis, and senescence are interconnected and influence each other. Recently the gut-liver axis was implicated in PBC. To date, the actual participation of these forms of death in every stage of PBC has not been clarified. However, there are upstream initiators of these mechanisms such as the deregulated and defective bicarbonate umbrella of BECs that allows for toxic bile acids to enter the cell and initiate the death mechanisms. It should be noted that the initial trigger of the bicarbonate deregulation is not known.
AE2: anion exchanger 2
AIC: autoimmune cholangitis
AMAs: antimitochondrial antibodies
AMPK: 5’ AMP-activated protein kinase
ATF6: activating transcription factor 6
Atg: autophagy-related protein
BAK: Bcl-2 homologous antagonist/killer
BAX: Bcl-2-associated X protein
BECs: biliary epithelial cells
BH3: Bcl-2 homology 3
BID: Bcl-2 homology 3-interacting domain death agonist
BIM: Bcl-2 interacting mediator of cell death
CHOP: CCAAT/enhancer-binding protein homologous protein
ClpP: caseinolytic protease P
CTAC: corrected total antioxidant capacity
CXCL: chemotactic cytokine ligand
DAPK: death-associated protein kinases
ER: endoplasmic reticulum
FLIP: FLICE-inhibitory protein
FXR: farnesoid X receptor
HO-1: heme oxygenase-1
HSCs: hepatic stellate cells
IFIT3: interferon-induced tetrapeptide repeat 3
IL: interleukin
IRE1α: inositol-requiring enzyme 1α
JNK: Jun N-terminal kinase
Keap1: Kelch-like ECH-associated protein 1
KO: knockout
LC3: light chain protein 3
MAIT: mucosa-associated invariant T
MDBs: Mallory-Denk bodies
MOMP: mitochondrial outer membrane permeabilization
mTOR: mammalian target of rapamycin
mTORC1: mammalian target of rapamycin complex 1
NF-κB: nuclear factor-kappa B
Nrf2: nuclear factor-E2-related factor 2
OCA: obeticholic acid
Parkin: 465-amino acid residue E3 ubiquitin ligase
PBC: primary biliary cholangitis
PDC-E2: pyruvate dehydrogenase complex E2
PI3K: phosphatidylinositol-3 kinase
PINK1: PTEN-induced putative kinase 1
RIPK1: receptor-interacting serine/threonine-protein kinase 1
ROS: reactive oxygen species
sAC: soluble adenylyl cyclase
SASP: senescence-associated secretory phenotype
TGF-β: transforming growth factor-β
Th: T helper
TLR: toll-like receptor
TNF: tumor necrosis factor
TRADD: tumor necrosis factor receptor 1-associated death domain protein
TRAIL: tumor necrosis factor-related apoptosis-inducing ligand
UDCA: ursodeoxycholic acid
ULK1: unc-51-like kinase 1 complex
UPR: unfolded protein response
EK: Conceptualization, Validation, Writing—original draft, Writing—review & editing, Supervision. IT: Investigation, Writing—review & editing. AV: Investigation, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Maitane Asensio ... Jose J. G. Marin
Ricardo Espinosa-Escudero ... Maria J. Monte
Grégory Merlen ... Thierry Tordjmann
Beatriz Sanchez de Blas ... Marta R. Romero
Carola Dröge ... Verena Keitel
Vasiliy Ivanovich Reshetnyak, Igor Veniaminovich Maev
Jose M. Pinazo-Bandera ... Miren García-Cortés
