 Case Report
Case Report

Affiliation:
1Department of Gastroenterology, Hepatology and Infectious Diseases, Medical Faculty, Otto von Guericke University Magdeburg, University Hospital Magdeburg A.ö.R., 39120 Magdeburg, Germany
2Department of Gastroenterology, Hepatology and Infectious Diseases, Medical Faculty, Heinrich Heine University Düsseldorf, University Hospital Düsseldorf, 40225 Düsseldorf, Germany
Email: carola.droege@med.ovgu.de
ORCID: https://orcid.org/0000-0001-7634-5134
Affiliation:
1Department of Gastroenterology, Hepatology and Infectious Diseases, Medical Faculty, Otto von Guericke University Magdeburg, University Hospital Magdeburg A.ö.R., 39120 Magdeburg, Germany
Affiliation:
3Institute for Pharmaceutical and Medicinal Chemistry, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany
Affiliation:
3Institute for Pharmaceutical and Medicinal Chemistry, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany
4Institute of Bio- and Geosciences (IBG-4: Bioinformatics), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
ORCID: https://orcid.org/0000-0001-8613-1447
Affiliation:
1Department of Gastroenterology, Hepatology and Infectious Diseases, Medical Faculty, Otto von Guericke University Magdeburg, University Hospital Magdeburg A.ö.R., 39120 Magdeburg, Germany
2Department of Gastroenterology, Hepatology and Infectious Diseases, Medical Faculty, Heinrich Heine University Düsseldorf, University Hospital Düsseldorf, 40225 Düsseldorf, Germany
Email: carola.droege@med.ovgu.de
ORCID: https://orcid.org/0000-0003-1383-7662
Explor Dig Dis. 2023;2:34–43 DOI: https://doi.org/10.37349/edd.2023.00016
Received: November 29, 2022 Accepted: February 14, 2023 Published: April 21, 2023
Academic Editor: Amedeo Lonardo, Azienda Ospedaliero-Universitaria di Modena, Italy
The article belongs to the special issue CHOLESTASIS
Hereditary cholestasis comprises a broad spectrum of clinical phenotypes of varying severity. Severe forms such as progressive familial intrahepatic cholestasis (PFIC) mostly affect children with disease onset within their first years. Nevertheless, late-onset PFIC forms are increasingly diagnosed. Most adults present with less severe forms of hereditary cholestasis, often suffering from pruritus, gallstone disease, jaundice, or elevated liver enzymes. To identify the underlying genetic background and to rule out potential differential diagnoses, a broad genetic analysis like whole exome sequencing (WES) is recommended. Knowledge of the affected gene may have an impact not only on patient surveillance due to risk for disease progression or tumor development but also on potential therapeutic strategies. This case of the adult patient illustrates the importance of broad genetic analysis, which brought up the potentially relevant rare multidrug resistance protein 3 (MDR3) missense variant p.(Asn489Tyr) underlying the patient’s clinical phenotype of low phospholipid-associated cholelithiasis (LPAC). Patients with MDR3 disease may have an increased risk for cholangiocarcinoma (CCA) development and therefore need an individualized surveillance strategy. Most MDR3-affected patients benefit from life-long therapy with ursodeoxycholic acid (UDCA), which is well tolerated. Bezafibrate treatment can reduce pruritus, one of the main symptoms affecting the quality of life. Whether the administration of ileal bile acid transporter (IBAT) inhibitors is beneficial in adult patients with MDR3 disease is so far unknown.
Intrahepatic cholestasis describes defective hepatic bile formation and comprises a heterogeneous spectrum of liver diseases of varying severity. The underlying molecular mechanisms include defects in bile acid (BA) synthesis, nuclear signaling, vesicular trafficking, canalicular transport, tight junction integrity, and maintenance of cell polarity resulting in impaired bile secretion from hepatocytes into the canalicular lumen [1]. Progressive familial intrahepatic cholestasis (PFIC) represents the severe form of the spectrum of hereditary cholestasis syndromes affecting 1:50,000 to 1:10,000 births, whereby 10–15% of neonatal cholestasis cases in tertiary referral centers are attributed to PFIC [1–3]. PFIC types are subdivided according to the affected gene. Potentially pathogenic variants in the genes encoding the aminophospholipid flippase familial intrahepatic cholestasis 1 [FIC1, adenosine triphosphatase (ATPase) phospholipid transporting, type 8B, member 1 (ATP8B1)], the bile salt export pump [BSEP, ATP binding cassette transporter superfamily, subfamily B, member 11 (ABCB11)], the phospholipid floppase multidrug resistance protein 3 (MDR3, ABCB4), the tight junction protein 2 [TJP2, zona occludens-2 (ZO-2)], the farnesoid X receptor [FXR, nuclear receptor, subfamily 1, group H, member 4 (NR1H4)], the motor protein myosin 5B (MYO5B), as well as SLC51A, USP53, KIF12, ZFYVE19, SEMA7A, VPS33B, were all linked to PFIC (-like) phenotypes as represented on www.omim.org [4–9]. Although most PFIC cases manifest within the first months of life, late-onset severe phenotypes, also requiring liver transplantation, have been described [10, 11]. Especially ABCB4/MDR3-related disease can present in adulthood as late-onset PFIC3 with biliary fibrosis and cirrhosis [12–15]. Genetic variants in these known PFIC-related genes may also underlie less severe and intermittent phenotypes such as intrahepatic cholestasis of pregnancy (ICP), drug-induced cholestasis, benign recurrent intrahepatic cholestasis (BRIC), or ABCB4/MDR3-related low phospholipid-associated cholelithiasis (LPAC) [12, 16–20]. Although patients with genetic variants in ABCB4/MDR3 may only show episodic disease over the years, patients can suffer from chronic liver disease, impaired quality of life, and, moreover, the risk for the development of cholangiocarcinoma (CCA) is increased [21–25]. Since 2021, ileal bile acid transporter (IBAT) inhibitor odevixibat is approved for treatment of PFIC patients older than six months in Europe [26]. Nevertheless, data so far focus on ATP8B1/FIC1- and ABCB11/BSEP-related PFIC.
At the age of 20 years, the female patient showed the first symptoms of cholestatic liver disease, mainly suffering from abdominal pain, and gallstones were identified as the underlying condition. Due to recurrent symptoms, cholecystectomy was performed in the patient’s mid-thirties. At this time point, no hereditary disease was suspected since no family history of gallstone disease, ICP, or liver disease was present and thus, no regular follow up implemented. Forty years after symptom onset, hereditary cholestasis was suspected due to elevated alanine aminotransferase [ALT; 1.9-times the upper limit of normal (ULN)], aspartate aminotransferase (AST; 1.5-times ULN), gamma-glutamyl transferase (GGT; 7.4-times ULN) and alkaline phosphatase (AP; 1.7-times ULN) after viral hepatitis (VH), autoimmune liver disease, primary sclerosing cholangitis (PSC), and primary biliary cholangitis (PBC) were excluded. Bilirubin and liver function tests were normal. Therefore, basic genetics was done for specific cholestasis-related genetic variants that revealed the heterozygous NR1H4/FXR variant c.-1G>T [overall allele frequency in the Genome Aggregation Database (gnomAD) 4.4%, https://gnomad.broadinstitute.org/] [27]. Nevertheless, the patient showed continuously elevated GGT levels, and the clinical phenotype was consistent with LPAC with recurrent choledocholithiasis requiring endoscopic retrograde cholangiography (ERC) and concrement extraction. Due to the clinical presentation with LPAC, the patient was included in the HiChol registry, and whole exome sequencing (WES) was performed. WES analysis [Twist Human Core Exome plus RefSeq Panel (TWIST Bioscience) using a NextSeq550 (Illumina) instrument, data analysis via varvis® 1.20.0 (Limbus Medical Technologies GmbH) data upload] confirmed the heterozygous NR1H4/FXR variant c.-1G>T, the two common polymorphisms ATP8B1/FIC1 c.2855G>A, p.(Arg952Gln) heterozygous; ABCB11/BSEP c.1331C>T, p.(Val444Ala) homozygous and additionally revealed a heterozygous rare missense variant in ABCB4/MDR3 [c.1465A>T, p.(Asn489Tyr)] (Figure 1). The MDR3 variant was also detected in another patient with clinical presentation of LPAC in our cohort but has not been characterized in detail in databases or literature.
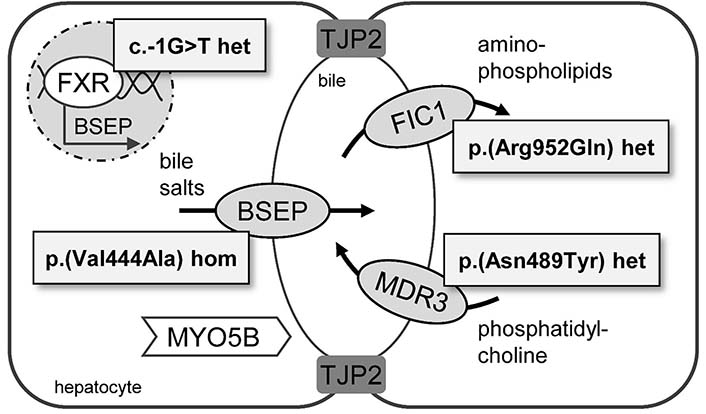
Hereditary cholestasis-related proteins and patient-specific genetic variants. Variants in genes encoding for FIC1 (ATP8B1), BSEP (ABCB11), MDR3 (ABCB4), FXR (NR1H4), TJP2, and MYO5B are related to hereditary intrahepatic cholestasis. Whole exome analysis of the described patient’s DNA revealed the potentially relevant variants: ATP8B1/FIC1 c.2855G>A, p.(Arg952Gln) heterozygous; ABCB11/BSEP c.1331C>T, p.(Val444Ala) homozygous; ABCB4/MDR3 c.1465A>T, p.(Asn489Tyr) heterozygous; NR1H4/FXR c.-1G>T heterozygous
Serum BA and cholesterol levels were in the same range six months ago and recently. Serum BA levels were elevated to 2.3× ULN, whereas cholesterol levels were almost normal (1.2× ULN). Under ursodeoxycholic acid (UDCA) therapy with a daily dose of 1,000 mg, GGT levels decreased to 1.2× ULN most recently while ALT and AST values completely normalized, and neither laboratory values nor ultrasound gives a hint to relevant signs of liver fibrosis so far. Nevertheless, close monitoring of the patient in the outpatient clinic and lifetime UDCA therapy is advisable as heterozygous carriers of ABCB4 variants, and clinical manifestation of ABCB4 disease have an increased risk for the development of CCA [17, 23, 24]. Symptoms such as pruritus can affect the patients’ quality of life and, therefore, need to be minded and treated. Patients with ABCB4 disease reported reduced quality of life in the area of energy/fatigue and general health, similar to patients with PSC [24]. As shown in fibrosing cholangiopathies, the application of bezafibrate can improve pruritus and could be considered for patients with hereditary cholestasis suffering from pruritus [28]. Whether bezafibrate also improves liver biochemistry in patients with ABCB4/MDR3 disease similarly to patients with PBC remains elusive [29].
To evaluate the patient’s clinical course and quality of life related to the specific genetic background, the patient gave written informed consent for participation in the HiChol registry for patients with hereditary intrahepatic cholestasis. The national, multicenter, prospective HiChol registry is part of the Federal Ministry of Education and Research-funded HiChol translational network. The registry is intended for evaluating clinical characteristics in relation to the underlying genotype. This will provide insights into the mechanisms of clinical variability, common and rare symptoms, and their impact on the patient’s quality of life as well as the individual risk of disease progression and tumor development. The achieved results can help to guide more individualized surveillance strategies, estimate therapy response, and guide future therapeutic interventions to improve the long-term outcome in these patients with hereditary cholestasis. Overall, the HiChol registry aims to provide a better understanding of this rare disease group to generate evidence for future guidelines and novel approaches for diagnosis, surveillance, and treatment, thus improving patient care.
The data included in the HiChol registry contain three main parts: clinical, genetic, and quality of life data. Clinical data were collected during baseline and follow-up visit and consist of aspects of medical history regarding first symptoms, first diagnosis, exclusion of other hepatopathies [VH, PBC, PSC, autoimmune hepatitis (AIH), Wilson’s disease (WD)], comorbidities, pregnancy, family history, current symptoms, alcohol-intake, and ultrasound results. For our patient, clinical information on cholecystectomy, and ERC interventions were included. Moreover, information on current medication and lab values were collected. The patient had no known family history of liver disease. For genetic data, variants in cholestasis-related genes ATP8B1, ABCB11, ABCB4, TJP2, NR1H4, MYO5B and potential disease modifiers in SERPINA1, HFE, PNPLA3, TM6SF2, MBOAT7, HSD17B13, and GCKR were documented. First individual evaluation of the patient’s quality of life data using the PBC-40-based questionnaire [30] Chol-43 revealed an overall moderate quality of life assessed on baseline and first follow up 5 months later. For analysis, answering options were counted with a 1-point to 5-point scale, where higher scores indicate a poorer quality of life with the following results for the different domains at baseline and follow up, respectively: the highest impact on quality of life was found in the symptoms, cognition, and fatigue domains (symptoms: 2.9–3.1; cognition 3.3 and fatigue 3.1–3.2); while social/emotional and itch domains were less affected. Interestingly, ABCB4/MDR3 disease can be misdiagnosed as WD and cognitive impairment has been reported in patients with ABCB4/MDR3 disease [13].
The site of the p.(Asn489Tyr) substitution is located in the RecA-like domain of the nucleotide binding domain (NBD) of MDR3 (Figure 2A) that includes the C-loop, which is involved in nucleotide binding together with the Walker A and Walker B motif of the opposing NBD. The substitution p.(Asn489Tyr) is more space-filling than the wild-type residue (Figure 2B), which might impair close-by C-loop dynamics and, hence, nucleotide binding or positioning and thus MDR3 function. A prediction of the effect of this substitution with variant assessor of MDR3 (Vasor), a machine learning-based classification tool trained on an MDR3-specific dataset of known benign and pathogenic variants, yielded a probability of pathogenicity of 0.89, indicating a high certainty for the substitution to be pathogenic as no false predictions within the data set were observed at such high values [31].
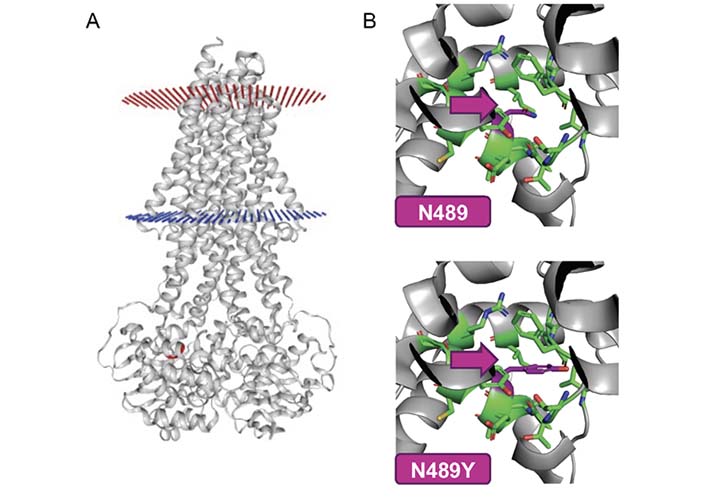
Vasor result for patient-specific MDR3 missense variant. The effect of the MDR3 missense variant p.(Asn489Tyr) (N489Y) was predicted as pathogenic (probability of pathogenicity 0.89, values from 0 = likely benign to 1 = likely pathogenic) using Vasor, https://cpclab.uni-duesseldorf.de/mdr3_predictor/, UniProt ID: P21439-1) [31]. A. Visualization of the MDR3 protein and the respective amino acid position (red) within the RecA-like subdomain of one of the NBDs. The membrane surface towards the canalicular site is marked in red, the cytosolic membrane surface is marked in blue; B. close-up view of the location of the reference amino acid asparagine (N, upper panel) and the variant with tyrosine (Y, lower panel) at position 489 marked in magenta, showing how much more space-filling the substitution is than the wild-type residue. This figure was generated with the Vasor webserver, Panel B was optimized using PyMol (Schrödinger) based on the downloadable PyMol script from the Vasor webserver
This case presentation illustrates the importance of genetic analysis in adult patients with recurrent symptoms of hereditary cholestasis (Figure 3). LPAC affects about 1% of adult patients with symptomatic gallstone disease and thus needs to be considered especially in young patients presenting with recurrent biliary symptoms [17]. Not only for final diagnosis but also for surveillance and therapeutic strategy, genetic analysis is needed in patients fulfilling LPAC criteria (2 out of 3 of the following): (1) symptomatic cholelithiasis before 40th year of life, (2) suspected intrahepatic microlithiasis or sludge on ultrasound, (3) recurrent biliary pain after cholecystectomy due to intrahepatic or bile duct stones as illustrated in the national and European clinical practice guidelines [19, 32–36]. Our patient represents a typical LPAC case with a long time period from disease onset to definite clinical and also genetic diagnosis, female gender fulfilling clinical LPAC criteria with cholelithiasis requiring cholecystectomy before the age of 40 and recurrence of intrahepatic bile duct stones. Therefore, UDCA therapy was initiated immediately. In parallel, other reasons for cholestasis were excluded according to the European guidelines [37]. In our patient, there was no need for a liver biopsy for diagnostic workup.
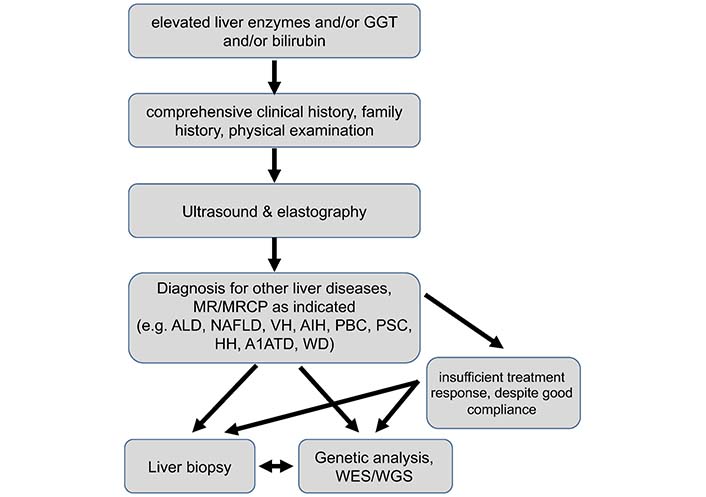
Proposed diagnostic algorithm in adults with elevated liver biochemistry. Genetic testing in most adult patients with elevated liver biochemistry is downstream from clinical history taking, physical examination, ultrasound, and exclusion of common adult liver diseases. It may also be considered in those with insufficient therapy response despite good patient compliance similar to liver (re-)biopsy. In patients with clear clinical presentation of hereditary cholestasis syndromes, such as LPAC, genetic testing may be considered earlier in the diagnostic workup. A1ATD: alpha1 antitrypsin deficiency; ALD: alcoholic liver disease; HH: hereditary hemochromatosis; MR: magnetic resonance; MRCP: magnetic resonance cholangiopancreaticography; NAFLD: non-alcoholic fatty liver disease; WGS: whole genome sequencing
So far, our patient benefits from UDCA therapy but as GGT remains elevated (last value: 1.2-times ULN), lifelong UDCA therapy is recommended, especially for LPAC patients.
Although in the cohort described by de Vries et al. [24], over 90% of ABCB4 variant carriers had liver transplant-free survival under UDCA therapy, three patients underwent liver transplantation due to decompensated cirrhosis or relevant comorbidity. Moreover, as a carrier of a heterozygous ABCB4/MDR3 variant, our patient has an increased CCA risk, therefore, she needs to be monitored at least once per year via ultrasound or MR to detect focal dilated bile ducts indicating either obliteration by intrahepatic stones or early tumor development [17, 23, 24, 37]. In the well-characterized LPAC cohort by Dong et al. [17], over 300 patients showed no progression to fibrosis but, nevertheless, a 5% CCA risk in line with findings from de Vries et al. [24] where two patients died because of CCA.
The initially identified FXR/NR1H4 variant c.-1G>T is known to result in a reduced expression of FXR target genes like ABCB11. The variant is described in ICP patients or cases of drug-induced cholestasis [38, 39]. Besides the cholestasis-associated variants in MDR3/ABCB4 and FXR/NR1H4, further common polymorphisms were identified in BSEP/ABCB11 and FIC1/ATP8B1 gene, which may contribute to the overall phenotype of elevated serum BA levels and LPAC syndrome [20, 40–42].
The predominant clinical presentation was recurrent gallstones requiring cholecystectomy and interventions in line with LPAC despite no signs of ICP in her two pregnancies. In cases of MDR3-related ICP, biochemical remission without UDCA intake can be achieved but needs to be monitored one to three years later because UDCA therapy eventually should be resumed in cases where liver biochemistry is abnormal [24].
Since 2021, the IBAT inhibitor odevixibat is approved in Europe for PFIC patients, who are 6 months or older, but not for patients with milder forms of hereditary intrahepatic cholestasis so far [26]. Moreover, norUDCA can improve cholestasis and liver damage in mice deficient for Abcb4, which are often used as PSC models and clinical development has progressed to a phase III trial for PSC (NCT03872921) [43–45]. Despite the improvement of clinical findings, symptom relief by targeting pruritus or fatigue with potential therapeutic options is important to improve the patients’ impaired quality of life. Application of bezafibrate can reduce pruritus and therefore improve quality of life [28]. Nevertheless, there are no promising therapeutic options targeting fatigue, one of the major aspects of reduced quality of life. As the prevalence of fatigue in hereditary intrahepatic cholestasis is so far unknown, the HiChol registry aims not only to collect clinical and genetic data, but also the quality of life data in these patients. Knowing the impact of fatigue in hereditary intrahepatic cholestasis will help to evaluate potential therapeutic options.
The MDR3 missense variant p.(Asn489Tyr) (6 points) has not been characterized in vitro but affects a highly conserved region, the American College of Medical Genetics and Genomics (ACMG) criteria classify this variant at the threshold of unknown significance (0–5 points) to likely pathogenic (6–9 points) [46, 47]. Vasor is an in silico prediction tool, to evaluate the potential impact of specific amino acid substitutions in MDR3 [31]. Vasor results in a value of 0.89 for p.(Asn489Tyr) and therefore predicts the variant to be probably pathogenic (0 = benign, 1 = pathogenic) which is in line with the clinical findings in the presented LPAC patient and another patient from our cohort with gallstone disease and the respective MDR3 variant. Heterozygous variants of unknown significance (VUS) are potentially relevant for a cholestatic phenotype and malignancy risk [12, 23, 48–51].
Since hereditary intrahepatic cholestasis is not based on specific hot spot variants but hundreds of different variants in more than six genes, a broad genetic analysis is preferable to a restricted genetic analysis of specific hot spots. The advantage of WES is not only the broad analysis of genetic variants in known cholestasis-related genes but also the opportunity to identify new potential cholestasis-relevant genes. Once new genes are identified, WES results can be re-analyzed for relevant variants in the respective genes.
ABCB11: ATP binding cassette transporter superfamily, subfamily B, member 11
ATP8B1: adenosine triphosphatase phospholipid transporting, type 8B, member 1
BA: bile acid
BSEP: bile salt export pump
CCA: cholangiocarcinoma
FIC1: familial intrahepatic cholestasis 1
FXR: farnesoid X receptor
GGT: gamma-glutamyl transferase
ICP: intrahepatic cholestasis of pregnancy
LPAC: low phospholipid-associated cholelithiasis
MDR3: multidrug resistance protein 3
MYO5B: myosin 5B
NBD: nucleotide binding domain
NR1H4: nuclear receptor, subfamily 1, group H, member 4
PBC: primary biliary cholangitis
PFIC: progressive familial intrahepatic cholestasis
PSC: primary sclerosing cholangitis
TJP2: tight junction protein 2
UDCA: ursodeoxycholic acid
ULN: upper limit of normal
Vasor: variant assessor of multidrug resistance protein 3
VH: viral hepatitis
WD: Wilson’s disease
WES: whole exome sequencing
CD: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Visualization, Writing—original draft, Writing—review & editing. TG: Data curation, Formal analysis. AB: Data curation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing—original draft, Writing—review & editing. HG: Data curation, Formal analysis, Funding acquisition, Methodology, Resources, Software, Supervision, Writing—original draft, Writing—review & editing. VK: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
VK has received speaker’s honoraria from Albireo, Falk, Intercept, CSL, Gilead, Sanofi, AbbVie. VK has participated in an advisory board for AstraZeneca. All other authors declare that there are no conflicts of interest.
This study was performed according to the guidelines of the declaration of Helsinki. The HiChol registry was approved by the local ethics committee of the Otto von Guericke University Magdeburg (reference number 160/21, Registry for hereditary intrahepatic cholestasis).
Informed consent to participate in the HiChol registry was obtained from relevant participant.
Informed consent to publication was obtained from relevant participant.
Not applicable.
This study was supported by the Federal Ministry of Education and Research (BMBF) through HiChol [01GM1904A, 01GM2204A, and 01GM2204B] to CD, HG, and VK. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Maitane Asensio ... Jose J. G. Marin
Ricardo Espinosa-Escudero ... Maria J. Monte
Grégory Merlen ... Thierry Tordjmann
Beatriz Sanchez de Blas ... Marta R. Romero
Vasiliy Ivanovich Reshetnyak, Igor Veniaminovich Maev
Jose M. Pinazo-Bandera ... Miren García-Cortés
Elias Kouroumalis ... Argyro Voumvouraki
