 Review
Review

Affiliation:
1Department of Cardiology, Hitit University Erol Olçok Education and Research Hospital, 19040 Corum, Turkey
ORCID: https://orcid.org/0000-0002-6496-7849
Affiliation:
2Department of Cardiology, Faculty of Medicine, Hitit University, 19040 Corum, Turkey
Email: macitkalcik@yahoo.com
ORCID: https://orcid.org/0000-0002-8791-4475
Affiliation:
1Department of Cardiology, Hitit University Erol Olçok Education and Research Hospital, 19040 Corum, Turkey
ORCID: https://orcid.org/0009-0008-9864-4715
Affiliation:
2Department of Cardiology, Faculty of Medicine, Hitit University, 19040 Corum, Turkey
ORCID: https://orcid.org/0000-0002-2444-7523
Affiliation:
2Department of Cardiology, Faculty of Medicine, Hitit University, 19040 Corum, Turkey
ORCID: https://orcid.org/0000-0002-3920-1382
Affiliation:
2Department of Cardiology, Faculty of Medicine, Hitit University, 19040 Corum, Turkey
ORCID: https://orcid.org/0000-0002-2544-1975
Explor Cardiol. 2025;3:101279 DOI: https://doi.org/10.37349/ec.2025.101279
Received: September 19, 2025 Accepted: October 31, 2025 Published: November 06, 2025
Academic Editor: Dimitrios Tousoulis, Athens University Medical School, Greece
The article belongs to the special issue Molecular Mechanisms of Cardiovascular Aging
Cardiovascular aging is characterized by progressive endothelial dysfunction and arterial stiffening, two interrelated processes underlying the increased risk of hypertension, coronary artery disease, heart failure, and atrial fibrillation in older individuals. Endothelial dysfunction results from reduced nitric oxide bioavailability, increased oxidative stress, chronic low-grade inflammation, and accumulation of senescent endothelial cells that secrete pro-inflammatory mediators. In parallel, structural alterations of the vascular wall, including elastin fragmentation, collagen deposition, cross-linking by advanced glycation end products, vascular smooth muscle cell phenotypic switching, and calcification, lead to increased stiffness and impaired vascular compliance. These maladaptive changes reinforce one another, creating a vicious cycle in which dysfunctional endothelium accelerates stiffening, while mechanical alterations in turn amplify endothelial injury. Molecular pathways involving NADPH oxidases, mitochondrial dysfunction, NF-κB, JAK/STAT, AMPK, mTOR, sirtuins, and epigenetic regulators integrate oxidative, inflammatory, and metabolic signals that shape vascular aging. Clinically, endothelial dysfunction and vascular stiffness predict cardiovascular events independent of traditional risk factors and serve as emerging biomarkers of biological vascular age. Established therapies such as statins, renin-angiotensin system blockade, structured exercise, and dietary interventions improve vascular function, while novel approaches targeting senescence and redox imbalance are under investigation. Understanding these mechanisms provides opportunities to mitigate vascular aging and extend cardiovascular health span.
Cardiovascular aging is a major contributor to morbidity and mortality in older populations worldwide. As individuals age, structural and functional changes in the vascular system increase the risk of hypertension, coronary artery disease, heart failure, stroke, and other cardiovascular pathologies [1]. Among these changes, endothelial dysfunction and increased vascular stiffness are central processes that drive deterioration of cardiovascular health [2]. Endothelial cells progressively lose their capacity to maintain vasodilator and vasoconstrictor balance, control inflammation, modulate thrombosis, and respond adequately to mechanical stimuli. Concurrently, the arterial wall undergoes modifications in the extracellular matrix composition, vascular smooth muscle cell (VSMCs) behavior, and mechanical properties, resulting in increased stiffness and reduced compliance [1, 3].
Understanding the molecular mechanisms that underlie these processes is essential for developing interventions aimed at slowing or reversing vascular aging. Key factors involved include oxidative stress, decreased nitric oxide (NO) bioavailability, inflammatory signaling, cellular senescence, alterations in extracellular matrix remodeling, and epigenetic regulation [3, 4]. These mechanisms do not operate in isolation but are highly interconnected. Oxidative stress impairs NO synthase activity and promotes inflammation. Inflammation accelerates cellular senescence, and senescent cells alter extracellular matrix composition [5].
This review will focus on endothelial dysfunction and vascular stiffness as the two most important hallmarks of cardiovascular aging. It will examine normal endothelial functions and vascular elasticity, discuss the pathophysiological mechanisms of endothelial dysfunction in aging, describe the molecular drivers of vascular stiffness, and explain how dysfunction and stiffness interact. Finally, clinical implications will be considered along with emerging therapeutic strategies designed to mitigate or reverse these processes.
The endothelium is a dynamic endocrine surface that maintains vascular homeostasis through continuous synthesis of NO, prostacyclin, and endothelium-derived hyperpolarizing pathways, while constrictor influences such as endothelin-1 are restrained under physiological conditions. Endothelial nitric oxide synthase (eNOS) generates NO, which diffuses to VSMCs to activate soluble guanylate cyclase and reduce intracellular calcium, producing vasodilation. NO also limits leukocyte adhesion, platelet activation, and smooth-muscle proliferation, establishing a tonic antithrombotic and anti-inflammatory state in the vessel wall [6]. eNOS activity is regulated by shear stress, substrate and cofactor availability, phosphorylation, and protein-protein interactions, allowing moment-to-moment adjustment of vascular tone [6].
Hemodynamic shear stress is the dominant physiological stimulus for endothelial phenotype. Laminar shear aligns endothelial cells, augments eNOS expression and NO bioavailability, stabilizes junctions, and suppresses pro-inflammatory signaling, whereas disturbed or oscillatory shear reduces NO signaling and favors atheroprone phenotypes [7]. This mechanoresponsive behavior integrates signals from the glycocalyx, primary cilia, ion channels, and adhesion complexes to tune endothelial output to local flow patterns [7].
The endothelial glycocalyx forms the luminal macromolecular interface that participates in mechanotransduction and regulates transvascular exchange. As a structured network of glycosaminoglycans and proteoglycans tethered to the membrane, it transmits shear forces to the cytoskeleton, modulates access of circulating molecules to endothelial receptors, and contributes to solute and water flux control across the microvasculature [8]. Preservation of glycocalyx integrity supports low vascular permeability and efficient shear sensing, which together sustain normal vascular reactivity [8].
Vascular elasticity emerges from the composite mechanics of the arterial wall. In large elastic arteries, elastin provides reversible extensibility and energy storage, while collagen constrains excessive stretch at higher pressures; smooth muscle tone and extracellular matrix organization adjust the operating point across the pressure range. These layered contributions produce compliance that damps pulsatile flow and reduces ventricular afterload under normal conditions [9, 10]. With physiological levels of distension, load is borne primarily by elastin lamellae, and recruitment of collagen fibers occurs progressively as pressure rises, shaping the nonlinearity of the pressure-diameter relation [10].
Measurement frameworks reflect these material and geometric properties. Carotid-femoral pulse wave velocity captures the stiffness of central elastic arteries and serves as the gold-standard index of aortic wall elasticity in clinical and research settings [11]. In parallel, endothelial-dependent vasodilation assessed by flow-mediated dilation quantifies the NO-dependent component of endothelial function, linking local mechanotransduction to conduit artery behavior in vivo [7, 11].
Beyond NO and prostacyclin, endothelium-derived hyperpolarization (EDH) constitutes a third vasodilator mechanism that is especially prominent in resistance arteries. EDH signaling involves activation of endothelial small- and intermediate-conductance Ca2+-activated K+ channels, myoendothelial coupling through gap junctions, and in some beds cytochrome P450 epoxides or extracellular K+ acting on smooth-muscle inward-rectifier channels and Na+/K+-ATPase. EDH complements NO to maintain microvascular tone and tissue perfusion, with relative dominance varying by vessel size and species [12]. In intact physiology, NO and EDH provide partially redundant, bed-specific control of vasomotor tone that preserves flow despite fluctuations in metabolic demand and pressure [6, 12].
In summary, normal endothelial function depends on intact mechano-chemical transduction of laminar shear, preserved glycocalyx architecture, regulated eNOS activity with adequate NO bioavailability, and cooperative EDH signaling. Vascular elasticity derives from elastin-dominated load sharing at physiological pressures, timely recruitment of collagen at higher strains, and smooth-muscle tone. Together, these systems minimize pulsatile load on the heart, maintain perfusion, and protect against thrombo-inflammatory activation [6–12].
Aging exerts profound effects on endothelial biology, progressively diminishing its vasoprotective functions and predisposing to cardiovascular disease. One of the earliest and most consistent changes is a reduction in NO bioavailability. Decreased eNOS expression and activity, as well as limited availability of its cofactors such as tetrahydrobiopterin, impair NO generation [13]. In parallel, accumulation of endogenous inhibitors such as asymmetric dimethylarginine (ADMA) further compromises NO signaling. These alterations blunt endothelium-dependent vasodilation and predispose to increased vascular tone and thrombogenicity [14] (Table 1).
Key molecular mechanisms of endothelial dysfunction in aging.
| Mechanism | Molecular features | Functional consequence | References |
|---|---|---|---|
| Reduced NO bioavailability | ↓ eNOS activity, ↑ ADMA, tetrahydrobiopterin deficiency | Impaired vasodilation, pro-thrombotic state | [6, 13–15] |
| Oxidative stress | ↑ NADPH oxidase, mitochondrial ROS, eNOS uncoupling | NO degradation, peroxynitrite formation | [15–17, 31–33] |
| Inflammation | NF-κB activation, ↑ IL-6, ↑ TNF-α, adhesion molecules | Endothelial activation, leukocyte adhesion | [18, 33, 46] |
| Senescence | p53/p21, p16/Rb, SASP secretion | Irreversible growth arrest, pro-inflammatory milieu | [17, 18, 29, 30, 40] |
ADMA: asymmetric dimethylarginine; eNOS: endothelial nitric oxide synthase; IL-6: interleukin-6; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NO: nitric oxide; ROS: reactive oxygen species; SASP: senescence-associated secretory phenotype; TNF-α: tumor necrosis factor-alpha.
Oxidative stress is a central driver of endothelial dysfunction in aging. Increased activity of NADPH oxidases, uncoupled eNOS, and mitochondrial sources of reactive oxygen species (ROS) elevate vascular oxidative burden. ROS directly degrade NO, generating peroxynitrite, which not only reduces vasodilator capacity but also modifies lipids, proteins, and DNA, exacerbating cellular injury [15]. This imbalance between oxidant generation and antioxidant defenses establishes a persistent state of redox stress that accelerates endothelial senescence and dysfunction [16, 17].
Chronic low-grade inflammation, often described as “inflammaging,” is another hallmark of vascular aging. Activation of NF-κB and related signaling pathways enhances the production of cytokines such as interleukin-6 and tumor necrosis factor-α, which amplify endothelial activation, leukocyte adhesion, and vascular remodeling. The endothelium adopts a pro-inflammatory phenotype characterized by increased expression of adhesion molecules and reduced barrier integrity, perpetuating vascular injury [18].
Cellular senescence contributes to the progressive decline in endothelial function. Senescent endothelial cells accumulate with age, displaying irreversible growth arrest and adopting a senescence-associated secretory phenotype (SASP). This phenotype is characterized by release of pro-inflammatory cytokines, chemokines, and proteases that propagate inflammation and tissue remodeling. Key molecular regulators of senescence include activation of the p53–p21 and p16–Rb pathways in response to telomere attrition, DNA damage, and oxidative stress [18]. Through SASP signaling, senescent cells amplify vascular inflammation and stiffness, creating a vicious cycle of endothelial deterioration [18].
Together, diminished NO bioavailability, oxidative stress, chronic inflammation, and senescence represent interconnected processes that underlie age-related endothelial dysfunction (Figure 1). These mechanisms shift the endothelium from a vasoprotective, anti-inflammatory, and anticoagulant surface to one that promotes vasoconstriction, leukocyte adhesion, thrombosis, and vascular remodeling. Such dysfunction not only contributes to vascular stiffness but also serves as a pivotal mediator linking aging to the clinical burden of cardiovascular disease [14–18].
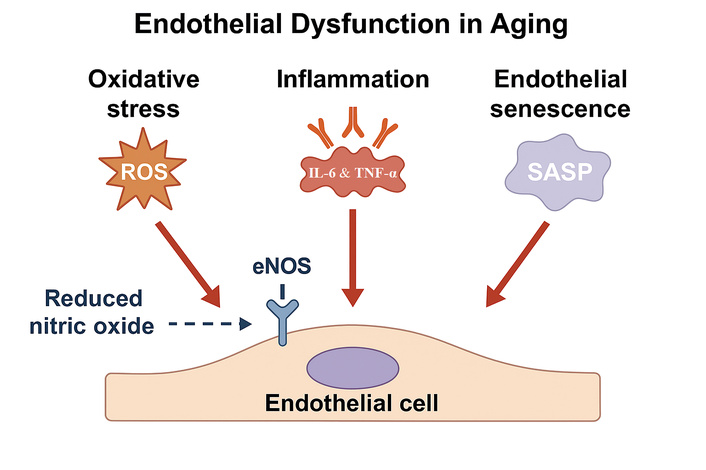
Main molecular mechanisms of endothelial dysfunction. Schematic illustration including reduced nitric oxide (NO) bioavailability due to decreased eNOS activity, increased production of reactive oxygen species (ROS), enhanced secretion of inflammatory cytokines such as IL-6 and TNF-α, and accumulation of senescent endothelial cells releasing SASP factors.
Vascular stiffness is a defining feature of vascular aging and a powerful predictor of cardiovascular morbidity and mortality. It results from cumulative structural, cellular, and molecular alterations within the vascular wall. At the structural level, elastin degradation and fragmentation represent central changes. Elastin is produced mainly during development and early life; with aging, elastin fibers undergo progressive fragmentation due to enzymatic activity of matrix metalloproteinases (MMPs) and mechanical fatigue. The resulting loss of elastic recoil diminishes arterial compliance. Simultaneously, collagen content within the arterial wall increases, providing rigidity and limiting distensibility. These processes are reinforced by the accumulation of advanced glycation end products (AGEs), which cross-link collagen fibers, making them resistant to enzymatic degradation and further stiffening the extracellular matrix [19, 20] (Table 2).
Structural and cellular drivers of vascular stiffness.
| Driver | Molecular/cellular basis | Effect on arterial wall | References |
|---|---|---|---|
| Elastin degradation | ↑ MMP activity, mechanical fatigue | Loss of elasticity, ↑ stiffness | [19, 20, 24, 25] |
| Collagen accumulation | ↑ Synthesis, AGE-mediated cross-linking | Increased rigidity, ↓ compliance | [19, 20, 24–26] |
| VSMC phenotypic switch | Contractile → synthetic phenotype | ↑ ECM production, fibrosis | [21, 22, 27, 30] |
| Vascular calcification | ↑ Phosphate, osteogenic VSMC differentiation | Medial calcification, ↑ pulse wave velocity | [21, 22, 25, 27] |
AGE: advanced glycation end product; ECM: extracellular matrix; MMP: matrix metalloproteinase; VSMC: vascular smooth muscle cell.
At the cellular level, VSMCs play a critical role in the stiffening process. With aging, VSMCs undergo phenotypic switching from a contractile to a synthetic state, characterized by increased proliferation, migration, and production of extracellular matrix components such as collagen and fibronectin [21]. This phenotypic modulation enhances deposition of stiff matrix proteins and contributes to calcification within the vascular wall. VSMCs also exhibit increased sensitivity to pro-calcific stimuli, including phosphate and inflammatory mediators, leading to deposition of hydroxyapatite crystals and medial arterial calcification, which markedly elevate vascular stiffness [22].
Mechanically, these changes translate into reduced arterial compliance and increased pulse wave velocity, the most widely used noninvasive measure of vascular stiffness. The stiffer arterial tree accelerates the propagation of pressure waves, causing earlier return of reflected waves during systole rather than diastole, which augments systolic blood pressure and increases left ventricular afterload. This hemodynamic burden contributes to left ventricular hypertrophy, impaired coronary perfusion, and progression toward heart failure [23].
Matrix remodeling plays an additional role in stiffness development. Dysregulated activity of MMPs and their tissue inhibitors alters the balance between elastin degradation and collagen deposition. Elevated MMP activity accelerates elastin breakdown, while reduced regulation fosters abnormal extracellular matrix turnover. Together, these processes reinforce the shift toward a rigid vascular wall architecture [24].
Inflammation and oxidative stress further aggravate stiffness by promoting endothelial dysfunction, VSMC senescence, and extracellular matrix remodeling. ROS stimulate collagen synthesis and cross-linking, while pro-inflammatory cytokines amplify VSMC phenotypic switching and calcification. Consequently, vascular stiffness is not simply a passive structural alteration of aging but an active, dynamic process driven by molecular and cellular pathways that interact with systemic hemodynamic forces [25].
In summary, vascular stiffness in aging arises from degradation of elastin, increased collagen deposition and cross-linking, VSMC phenotypic modulation and calcification, and dysregulated extracellular matrix remodeling (Figure 2). These structural and cellular alterations translate into mechanical changes that accelerate pulse wave velocity and augment systolic load, thereby establishing vascular stiffness as a central mediator linking aging to cardiovascular disease [19–25].
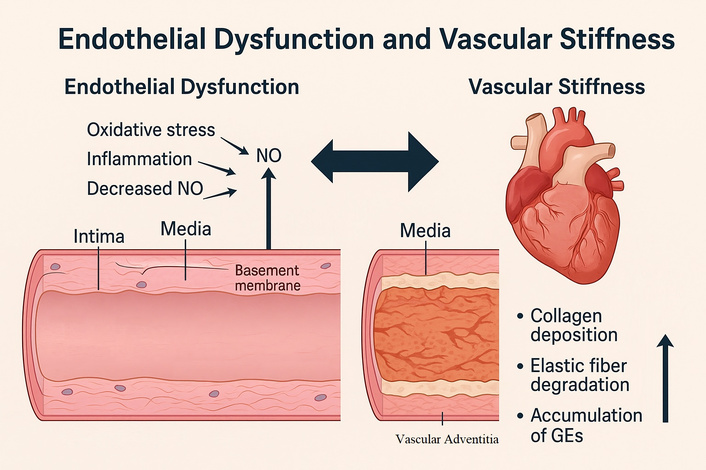
Cross-sectional representation of the arterial wall highlighting age-related structural alterations. Schematic illustration including elastin fragmentation and degradation, increased collagen deposition, cross-linking by advanced glycation end products (AGEs), vascular smooth muscle cell (VSMC) phenotypic switching, and medial calcification, all contributing to increased vascular stiffness.
Endothelial dysfunction and vascular stiffness are not isolated processes but mutually reinforcing phenomena that accelerate vascular aging. Loss of NO bioavailability plays a central role in this interaction. Under physiological conditions, NO maintains VSMCs in a relaxed state and inhibits collagen synthesis. In aging, reduced NO production promotes VSMC contraction and favors extracellular matrix deposition, both of which increase stiffness. Conversely, stiffened arteries impair endothelial mechanosensing of shear stress, further diminishing eNOS activation and NO generation, thereby amplifying dysfunction [26].
Chronic inflammation provides another point of convergence. Endothelial dysfunction increases expression of adhesion molecules and cytokines that recruit leukocytes and stimulate adventitial fibroblasts, fostering extracellular matrix remodeling. The pro-inflammatory environment accelerates elastin degradation, collagen cross-linking, and medial calcification, which all stiffen the arterial wall. In turn, stiffened vessels expose endothelial cells to altered hemodynamic forces, including elevated pulse pressure and oscillatory shear stress, which promote endothelial activation and perpetuate inflammatory signaling [27].
Oxidative stress links the two processes in a feed-forward cycle. Excessive production of ROS reduces NO bioavailability and enhances vascular inflammation, thereby driving dysfunction. At the same time, oxidative modification of elastin and collagen facilitates stiffening. The resulting stiffened arterial wall transmits higher cyclic strain to endothelial cells, increasing their oxidative stress burden and propagating endothelial damage [28].
Senescent cells are another critical mediator of the crosstalk. Senescent endothelial cells secrete a SASP rich in cytokines, proteases, and growth factors [29]. These mediators stimulate VSMC phenotypic switching, extracellular matrix deposition, and calcification, directly promoting stiffness. Likewise, VSMCs that undergo senescence contribute to extracellular matrix remodeling and inflammatory activation of adjacent endothelial cells, creating a vicious cycle of dysfunction and rigidity [30].
The interplay of endothelial dysfunction and stiffness establishes a self-perpetuating system of vascular aging. Impaired endothelial signaling fosters structural and mechanical changes that stiffen the arterial wall, while stiffened vessels feed back to worsen endothelial impairment. This dynamic interaction underscores why both processes should be considered together when evaluating mechanisms of cardiovascular aging and when designing therapeutic interventions [26–30].
Endothelial dysfunction and vascular stiffness are underpinned by a complex network of signaling pathways that regulate redox balance, inflammation, metabolism, and gene expression. These pathways interact dynamically, producing the maladaptive vascular phenotype characteristic of aging.
Oxidative stress-related pathways are central. Activation of NADPH oxidases (particularly NOX2 and NOX4) increases ROS production, while mitochondrial dysfunction enhances superoxide release. ROS reduce NO bioavailability, promote peroxynitrite formation, and damage vascular proteins and lipids. Normally, antioxidant responses mediated by nuclear factor erythroid 2-related factor 2 (Nrf2) counterbalance oxidants by upregulating detoxifying enzymes such as heme oxygenase-1 and superoxide dismutase. However, aging impairs Nrf2 signaling, resulting in inadequate antioxidant defense and sustained oxidative stress [31].
Mitochondrial dysfunction is another key contributor to vascular aging. Aging impairs mitochondrial respiratory chain efficiency, leading to excessive superoxide production and reduced ATP availability. Damaged mitochondria release mitochondrial DNA and danger-associated molecular patterns (DAMPs) that activate inflammatory pathways and inflammasomes, further promoting endothelial injury. Impaired mitophagy exacerbates the accumulation of dysfunctional mitochondria, sustaining oxidative and inflammatory stress within vascular cells [32].
Inflammatory pathways also play a major role. The NF-κB transcription factor is persistently activated in the aged vasculature, driving expression of adhesion molecules, chemokines, and cytokines. This creates a chronic pro-inflammatory state that accelerates endothelial dysfunction and matrix remodeling. In parallel, the JAK/STAT pathway transduces signals from circulating cytokines such as interleukin-6, amplifying inflammatory gene expression and vascular injury. Activation of inflammasomes, particularly NLRP3, has been implicated in promoting IL-1β release and further propagating vascular inflammation and calcification [33].
Metabolic signaling is tightly linked to vascular aging. AMP-activated protein kinase (AMPK) is an energy sensor that enhances eNOS activity, reduces oxidative stress, and limits inflammation. With aging, AMPK activity declines, contributing to impaired endothelial function. Conversely, overactivation of the mechanistic target of rapamycin (mTOR) pathway promotes cellular growth, senescence, and pro-inflammatory signaling in vascular cells. Sirtuins, particularly SIRT1, act as metabolic regulators that deacetylate transcription factors to suppress NF-κB activity, enhance endothelial NO production, and protect mitochondrial function. Age-related decline in SIRT1 activity contributes to loss of vascular resilience [34].
Autophagy, the cellular process responsible for degradation and recycling of damaged organelles and proteins, declines with age. Reduced autophagic flux leads to accumulation of dysfunctional mitochondria and misfolded proteins, aggravating oxidative stress and endothelial senescence. AMPK activation and mTOR inhibition both promote autophagy, highlighting its role as a protective mechanism in maintaining vascular homeostasis [35].
These signaling cascades are functionally interconnected. Persistent activation of NF-κB and JAK/STAT promotes transcription of pro-inflammatory and oxidative genes, which in turn stimulate mTOR activity and suppress SIRT1 signaling. The decline in SIRT1 removes a key inhibitory control over NF-κB, reinforcing inflammatory and senescent phenotypes. At the same time, AMPK and SIRT1 normally act in concert to counterbalance mTOR-driven growth and redox stress. Loss of this regulatory axis with aging establishes a feed-forward loop linking metabolic dysregulation, chronic inflammation, and endothelial senescence, thereby amplifying vascular stiffness and dysfunction [36].
Epigenetic regulation further integrates environmental and metabolic influences on vascular function. DNA methylation changes in endothelial and smooth muscle cells alter gene expression related to inflammation and extracellular matrix remodeling. Histone modifications, such as acetylation and methylation, influence transcription of genes governing oxidative stress responses and cell cycle regulation. Non-coding RNAs, including microRNAs and long non-coding RNAs (lncRNAs), also modulate vascular aging. For instance, miR-34a promotes endothelial senescence by targeting SIRT1, while miR-21 and miR-146a regulate inflammatory signaling in vascular cells [37].
In addition to DNA methylation and histone modification, other epigenetic mechanisms critically influence vascular aging. Global hypomethylation with site-specific promoter hypermethylation alters endothelial gene expression profiles related to oxidative defense and NO synthesis. Age-associated reduction in histone acetylation limits the transcription of antioxidant enzymes such as SOD2 and eNOS, while increased histone methylation at pro-inflammatory loci enhances vascular inflammation. Histone deacetylases (HDACs) and sirtuins, particularly SIRT1 and SIRT6, act as key modulators by linking energy metabolism to chromatin remodeling [38].
Epigenetic drift during aging leads to persistent activation of NF-κB-dependent genes, promoting a pro-inflammatory and pro-senescent phenotype. Pharmacological or lifestyle interventions that restore proper DNA and histone acetylation patterns have been shown to improve endothelial function in experimental models. These findings underscore that epigenetic remodeling is not merely a passive consequence of aging but an active driver of vascular dysfunction [39].
LncRNAs such as MALAT1 and MEG3 influence endothelial cell survival and smooth muscle phenotype, while circRNAs act as molecular sponges regulating microRNA availability. Dysregulation of these RNA networks contributes to impaired endothelial repair, inflammation, and extracellular matrix remodeling [40].
Together, these molecular pathways form a tightly interconnected system. Oxidative stress triggers NF-κB and inflammasome activation, inflammation enhances ROS production, and metabolic regulators such as AMPK, mTOR, and SIRT1 determine the balance between protective and deleterious signaling. Epigenetic modifications stabilize these maladaptive phenotypes, perpetuating endothelial dysfunction and vascular stiffness. Collectively, mitochondrial dysfunction, impaired autophagy, and dysregulated non-coding RNA networks act in concert with oxidative stress, inflammation, and senescence to drive endothelial deterioration and vascular stiffening during aging. Understanding these interactions is essential for identifying therapeutic targets capable of disrupting the cycle of vascular aging [31–37] (Table 3).
Major signaling pathways in vascular aging.
| Pathway | Role in endothelial dysfunction | Role in vascular stiffness | References |
|---|---|---|---|
| NF-κB | Induces pro-inflammatory cytokines | Promotes ECM remodeling | [18, 33, 37, 38] |
| JAK/STAT | Cytokine-driven vascular inflammation | VSMC activation, calcification | [33, 34] |
| Nrf2 | Antioxidant defense, ↓ with aging | Failure of redox balance | [31–33] |
| AMPK | Enhances eNOS, energy sensor | Loss reduces vascular resilience | [34, 35] |
| mTOR | Promotes growth, senescence | ECM deposition, calcification | [34, 36] |
| Sirtuins (SIRT1) | Suppresses NF-κB, ↑ NO | Protects mitochondria, anti-fibrotic | [34, 37, 38, 40] |
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; JAK/STAT: Janus kinase/signal transducer and activator of transcription; Nrf2: nuclear factor erythroid 2-related factor 2; AMPK: AMP-activated protein kinase; mTOR: mechanistic target of rapamycin; SIRT1: sirtuin 1; ECM: extracellular matrix.
The interplay between endothelial dysfunction and vascular stiffness has direct clinical consequences, as both are independently associated with adverse cardiovascular outcomes. Endothelial dysfunction is considered an early marker of atherosclerosis and precedes the development of overt structural vascular disease. Impairment in flow-mediated dilation of the brachial artery has been shown to predict future cardiovascular events, including myocardial infarction and stroke, even in individuals without established coronary artery disease [41]. Similarly, increased carotid-femoral pulse wave velocity, the reference measure of vascular stiffness, strongly predicts all-cause and cardiovascular mortality in both hypertensive and general populations [42].
Hypertension is one of the most common manifestations of the interaction between endothelial dysfunction and vascular stiffness. Reduced NO bioavailability and impaired vasodilator capacity contribute to increased peripheral resistance, while stiffening of central arteries augments systolic blood pressure and pulse pressure. These hemodynamic changes increase left ventricular afterload, promote left ventricular hypertrophy, and predispose to heart failure with preserved ejection fraction (HFpEF), a syndrome highly prevalent among older adults [43].
Coronary artery disease is also closely linked to vascular aging. Endothelial dysfunction fosters atherogenesis by permitting increased leukocyte adhesion, lipid infiltration, and smooth muscle cell proliferation. In parallel, vascular stiffness compromises coronary perfusion by altering the timing of reflected pressure waves and reducing diastolic coronary filling. Together, these mechanisms contribute to myocardial ischemia and adverse outcomes in patients with coronary artery disease [44].
Atrial fibrillation (AF) represents another clinical correlate. Vascular stiffness and endothelial dysfunction create conditions of elevated left atrial pressure, atrial stretch, and oxidative stress, which promote atrial remodeling and arrhythmogenesis. Observational studies have shown that higher pulse wave velocity is independently associated with greater AF risk, suggesting that vascular stiffness contributes to the pathophysiological substrate for arrhythmia in aging populations [45].
Translational relevance is underscored by the fact that the molecular alterations described—oxidative stress, inflammation, mitochondrial dysfunction, and senescence—manifest as measurable vascular phenotypes. Endothelial dysfunction can be quantified by flow-mediated dilation and reactive hyperemia-peripheral arterial tonometry, whereas arterial stiffness is evaluated by carotid-femoral pulse wave velocity. Circulating biomarkers, including ADMA, inflammatory cytokines, and microRNAs such as miR-34a and miR-126, serve as potential early indicators of vascular aging. These clinical parameters provide a direct bridge between molecular mechanisms and patient outcomes, enabling early detection and therapeutic monitoring [46].
In summary, endothelial dysfunction and vascular stiffness not only describe molecular and structural features of vascular aging but also serve as measurable clinical entities that predict cardiovascular outcomes. Their assessment in clinical settings provides prognostic information beyond traditional risk factors, and they are increasingly considered therapeutic targets in the prevention and management of cardiovascular disease [37–45].
Therapeutic approaches target two linked goals: restoration of endothelial signaling and reduction of vascular stiffness. Lipid lowering with statins improves NO-dependent vasodilation across diverse populations. A recent meta-analysis confirms significant improvement in flow-mediated dilation with statin therapy, consistent with anti-inflammatory and pleiotropic actions on endothelial cells [47]. Mechanistic and clinical data indicate statins reduce leukocyte-endothelium interactions and improve vascular reactivity beyond lipid effects [48]. These translational insights have spurred the development of targeted therapies. Experimental and early clinical studies using senolytics, Nrf2 activators, SIRT1 enhancers, and SGLT2 inhibitors demonstrate potential to modulate the same molecular pathways implicated in vascular aging, offering mechanistic continuity between bench and bedside [49] (Table 4).
Therapeutic strategies targeting endothelial dysfunction and vascular stiffness.
| Strategy | Mechanism of action | Evidence/effect | References |
|---|---|---|---|
| Statins | ↑ NO bioavailability, anti-inflammatory | Improve FMD, reduce leukocyte adhesion | [47, 48] |
| ACE inhibitors/ARBs | RAAS blockade | Reduce PWV, improve compliance | [50, 51] |
| Exercise | ↑ eNOS activity, ↓ oxidative stress | Improves FMD, reduces stiffness | [52, 53] |
| Mediterranean diet | Polyphenols, unsaturated fats | Enhances endothelial function | [54] |
| Nitrate supplementation | Exogenous NO donor | Improves FMD, lowers BP | [55] |
| Senolytics | Eliminate senescent cells | Early trials show ↓ SASP mediators | [57–59] |
ACE: angiotensin-converting enzyme; ARB: angiotensin receptor blocker; RAAS: renin-angiotensin-aldosterone system; PWV: pulse wave velocity; FMD: flow-mediated dilation; BP: blood pressure; NO: nitric oxide; SASP: senescence-associated secretory phenotype.
Renin-angiotensin system blockade favorably modifies stiffness and endothelial tone. Meta-analyses of randomized trials show that angiotensin-converting enzyme inhibitors reduce pulse wave velocity and augmentation index, effects that are partly pressure independent and align with improved arterial compliance [50]. Comparative data indicate ACE inhibitors and angiotensin receptor blockers can similarly reduce pulse wave velocity, supporting RAAS inhibition as a stiffness-modifying strategy [51].
Exercise training is a high-yield nonpharmacological intervention [52]. Evidence shows that continuous aerobic exercise meaningfully increases flow-mediated dilation, with benefits observed at moderate and vigorous intensities and with programs of sufficient duration [53]. Diet patterns that emphasize unsaturated fats, polyphenols, and fiber also aid endothelial health. A meta-analysis shows Mediterranean diet interventions improve flow-mediated dilation, indicating measurable endothelial benefit beyond traditional risk factor control [54].
NO donor strategies provide adjunctive endothelial support. Inorganic nitrate from beetroot juice can acutely and subacutely improve flow-mediated dilation and lower blood pressure in selected cohorts, though responses vary with dose, timing, and baseline endothelial status [55]. Glucose-lowering agents with vascular pleiotropy are under evaluation. For sodium-glucose cotransporter-2 inhibitors, pooled evidence suggests neutral average effects on flow-mediated dilation and pulse wave velocity to date, highlighting heterogeneity by population and study design and the need for targeted trials focused on vascular endpoints [56].
Energy restriction improves vascular function by enhancing NO bioavailability and reducing oxidative stress. Human and translational data show that caloric restriction and weight loss enhance endothelial function with aging, supporting lifestyle-first strategies in vascular risk modification [17]. Emerging geroscience approaches seek to interrupt cellular senescence-driven vascular inflammation and remodeling. Early human studies of intermittent senolytic therapy with dasatinib plus quercetin demonstrate reduction of senescence markers, providing proof-of-concept for targeting the senescence program that contributes to endothelial dysfunction and stiffness, though definitive vascular outcome data are pending [57, 58].
In practice, an integrated program combines RAAS blockade and statins when indicated, structured aerobic exercise, Mediterranean-style nutrition with weight control, and selective use of adjuncts such as inorganic nitrate in appropriate patients. Future work should define precision phenotypes that predict responsiveness to specific therapies and test senescence-targeted strategies against hard vascular endpoints [47–57] (Figure 3).
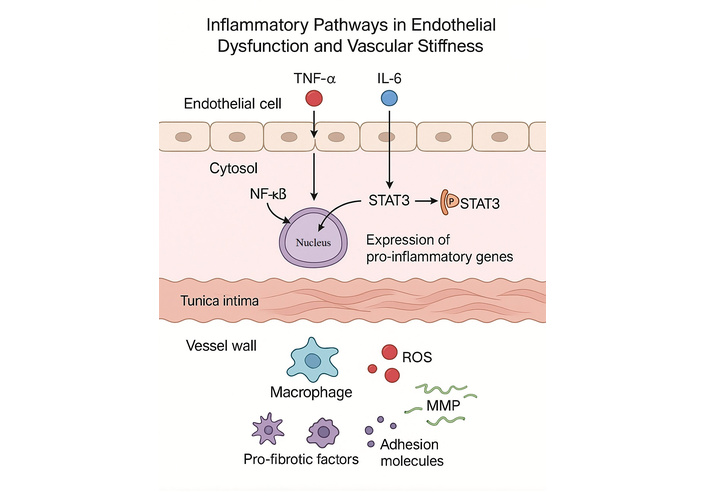
Inflammatory pathways in endothelial dysfunction and vascular stiffness. Schematic representation of key inflammatory signaling cascades contributing to endothelial dysfunction and vascular stiffening. Tumor necrosis factor-α (TNF-α) activates NF-κB signaling, while interleukin-6 (IL-6) activates STAT3 through phosphorylation. Both pathways promote transcription of pro-inflammatory genes within endothelial cells. The resulting cytokine release and endothelial activation enhance macrophage recruitment, reactive oxygen species (ROS) generation, matrix metalloproteinase (MMP) activity, adhesion molecule expression, and secretion of pro-fibrotic factors, all of which contribute to extracellular matrix remodeling and increased vascular stiffness.
Despite major advances in understanding the molecular and clinical aspects of vascular aging, significant gaps remain. One priority is the translation of experimental findings into precision therapies. Although many molecular pathways have been identified—such as oxidative stress, senescence, and inflammation—effective clinical strategies that selectively target these mechanisms are still limited. Senolytics, Nrf2 activators, and sirtuin modulators are promising, but robust evidence from large, long-term clinical trials is required before they can be integrated into standard care [59].
Another challenge is heterogeneity in the vascular aging phenotype. Individuals of the same chronological age often display markedly different levels of endothelial function and vascular stiffness. Genetic background, epigenetic modifications, comorbidities, and lifestyle factors all influence this variability. Future studies should focus on integrating multi-omics data—including genomics, transcriptomics, proteomics, and metabolomics—to identify molecular signatures that define “biological vascular age” more accurately than chronological age [60].
Advances in biomarker discovery also represent a key area of research. Circulating microRNAs, extracellular vesicles, and metabolites linked to endothelial senescence or arterial remodeling are under investigation as potential early indicators of vascular dysfunction. However, their predictive value and standardization for clinical use remain unresolved. Similarly, improvements in noninvasive imaging and vascular function testing are needed to better capture early subclinical changes and track therapeutic responses [61].
Aging research is moving toward integrative systems biology, where computational models simulate interactions between molecular pathways, hemodynamic forces, and clinical phenotypes. Such models may enable personalized prediction of vascular aging trajectories and response to interventions. Combining these insights with digital health platforms could allow continuous monitoring of vascular health, thereby shifting prevention from reactive to proactive strategies [62].
Finally, global demographic changes make it imperative to evaluate interventions across diverse populations. Most clinical studies on vascular aging have been conducted in Western cohorts, with limited data from low- and middle-income countries where aging populations are expanding rapidly. Addressing these disparities is essential to ensure that emerging therapies and diagnostic tools benefit all at-risk groups [63].
Future studies should aim to include ethnically and geographically diverse populations to capture variations in genetic background, cardiovascular risk profiles, dietary patterns, and environmental exposures. Such diversity is essential to identify population-specific mechanisms of vascular aging and to ensure that preventive and therapeutic strategies are broadly applicable. Collaborative multinational cohorts and consortia integrating genomics, metabolomics, and imaging data would enable comparison of vascular aging trajectories across different regions. Expanding global representation will improve the translational relevance and equity of vascular aging research [64].
In summary, future research must bridge mechanistic discoveries with clinical translation, define molecular signatures of vascular aging, standardize novel biomarkers, and embrace population diversity. Only through this multifaceted approach can the cycle of endothelial dysfunction and vascular stiffness be interrupted, reducing the burden of cardiovascular disease in aging societies [59–63].
Endothelial dysfunction and vascular stiffness represent the two principal molecular and structural processes that drive cardiovascular aging. Endothelial cells progressively lose their vasodilator, anti-inflammatory, and antithrombotic functions, primarily due to oxidative stress, reduced NO bioavailability, chronic low-grade inflammation, and cellular senescence. Simultaneously, the arterial wall undergoes elastin fragmentation, collagen accumulation, calcification, and extracellular matrix remodeling, all of which culminate in stiffening. These maladaptive processes are not independent; rather, they reinforce one another in a self-perpetuating cycle that amplifies vascular injury and accelerates the onset of hypertension, coronary artery disease, heart failure, and AF.
Clinical and experimental evidence shows that both endothelial dysfunction and vascular stiffness are measurable, clinically relevant entities that predict cardiovascular outcomes independent of traditional risk factors. Current therapies—such as statins, renin-angiotensin system blockade, structured exercise, and dietary modification—improve endothelial function and reduce vascular stiffness to varying degrees. Emerging approaches, including senolytics, metabolic regulators, and epigenetic interventions, hold promise for disrupting the molecular drivers of vascular aging.
Despite these advances, substantial gaps remain. There is a pressing need for translational research that bridges molecular insights with personalized therapies. Integration of multi-omics biomarkers, refinement of vascular imaging, and incorporation of systems biology models may provide a framework for individualized risk stratification and treatment. Equally important is ensuring that clinical trials encompass diverse populations to address global aging trends.
In conclusion, targeting the intertwined mechanisms of endothelial dysfunction and vascular stiffness offers a rational pathway to reduce the cardiovascular burden of aging. By advancing mechanistic understanding, refining diagnostic tools, and developing novel therapies, it may be possible to shift the trajectory of vascular aging and extend cardiovascular health span in the growing elderly population.
AF: atrial fibrillation
AMPK: AMP-activated protein kinase
EDH: endothelium-derived hyperpolarization
eNOS: endothelial nitric oxide synthase
lncRNAs: long non-coding RNAs
MMPs: matrix metalloproteinases
mTOR: mechanistic target of rapamycin
NO: nitric oxide
Nrf2: nuclear factor erythroid 2-related factor 2
During the preparation of this work, the authors used ChatGPT (OpenAI) to assist with composing and refining the figures. After using the tool, authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
MCÇ: Conceptualization, Investigation, Writing—original draft. MK: Conceptualization, Writing—review & editing. AB: Conceptualization, Investigation, Writing—original draft. MY: Writing—review & editing. LB: Supervision, Validation. YK: Supervision, Validation. All of the authors contributed to planning, conducting, and reporting of the work. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
No financial funding was received for this study.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Filipa Morgado ... Leonel Pereira
Jonica Campolo ... Maria Grazia Andreassi
Abdullatif Taha Babakr
Michael Li ... Robert M. Lust
