 Review
Review

Affiliation:
1Strathclyde Institute of Pharmacy & Biomedical Sciences, University of Strathclyde, G4 0RE Glasgow, United Kingdom
Email: marta.ruano-aldea@strath.ac.uk
ORCID: https://orcid.org/0000-0003-1087-4919
Affiliation:
1Strathclyde Institute of Pharmacy & Biomedical Sciences, University of Strathclyde, G4 0RE Glasgow, United Kingdom
Affiliation:
2AB Vista, SN8 4AN Wiltshire, United Kingdom
Affiliation:
1Strathclyde Institute of Pharmacy & Biomedical Sciences, University of Strathclyde, G4 0RE Glasgow, United Kingdom
Affiliation:
3ABITEC Corporation, Columbus, OH 43215, United States
Affiliation:
1Strathclyde Institute of Pharmacy & Biomedical Sciences, University of Strathclyde, G4 0RE Glasgow, United Kingdom
ORCID: https://orcid.org/0000-0003-1967-3603
Explor BioMat-X. 2026;3:101362 DOI: https://doi.org/10.37349/ebmx.2026.101362
Received: November 14, 2025 Accepted: February 24, 2026 Published: March 08, 2026
Academic Editor: Laichang Zhang, Edith Cowan University’s School of Engineering, Australia
Antibiotic resistance is a global threat, driven by limited new antimicrobials and rising multidrug-resistant infections. Lipid nanoparticles (LNPs) combine tunable material properties with antimicrobial functionality, providing biocompatibility, controlled release, and biofilm penetration. LNPs provide key advantages over metallic and polymeric nanocarriers, including high biocompatibility, the ability to encapsulate both hydrophilic and hydrophobic agents, controlled release profiles, and reduced cytotoxicity and immune activation. These features enhance drug stability and bioavailability and may help circumvent bacterial defences such as biofilms and efflux pumps. Robust preclinical evaluation platform of antimicrobial biomaterials requires platforms that capture biologically relevant interactions while remaining ethically and economically feasible. The chick embryo model (CEM) has emerged as a versatile platform for infection studies, bridging conventional in vitro assays and mammalian in vivo models. Its vascularized and developing tissue environment enables assessment of nanoparticle biodistribution, local toxicity, and antimicrobial efficacy within a dynamic biological context. This review critically examines the application of the CEM for evaluating LNP-based antimicrobial systems, highlighting current methodological variability and limitations in experimental standardization. By identifying gaps in protocol harmonisation and comparative assessment, this work outlines opportunities to improve reproducibility and translational relevance. Overall, integrating rationally designed LNP systems with optimised CEMs may accelerate the development of next-generation antimicrobial biomaterials to combat antibiotic-resistant infections.
The rapid emergence and global dissemination of antibiotic resistance (AR) constitute one of the most critical public health challenges of the 21st century. The World Health Organization (WHO) has identified AR as a global priority, warning that without urgent and coordinated intervention, infections that were previously treatable may once again become fatal [1]. Extensive overuse and misuse of antibiotics across clinical medicine, veterinary practice, and agriculture have accelerated the selection and spread of resistant microbial strains, undermining the effectiveness of existing therapies and placing a growing burden on healthcare systems worldwide [2]. Current estimates indicate that drug-resistant infections are responsible for approximately 700,000 deaths annually, with projections suggesting this figure could rise to 10 million per year by 2050 in the absence of effective countermeasures [3].
Global surveillance initiatives, including the WHO Global Antimicrobial Resistance Surveillance System (GLASS), consistently report increasing resistance rates across multiple bacterial species, with multidrug-resistant Gram-negative pathogens posing particular concern in both community- and healthcare-associated infections [1, 4]. Beyond clinical settings, environmental reservoirs play a substantial role in the persistence and dissemination of resistance determinants. Agricultural runoff, wastewater effluents, and improper disposal of pharmaceutical compounds contribute to environmental antibiotic contamination, facilitating the horizontal transfer of resistance genes and further amplifying the global AR burden [5, 6].
Despite the escalating clinical need, the antibiotic discovery pipeline has stagnated over recent decades. Most antibacterial agents approved in the past 20 years are derivatives or reformulations of existing drug classes, providing incremental improvements rather than fundamentally new mechanisms of action [7, 8]. Economic disincentives, prolonged development timelines, and high attrition rates have constrained investment in antibacterial research, prompting increased interest in alternative and complementary strategies to conventional antibiotics [9].
Among these strategies, biomaterials-based approaches—particularly nanotechnology-enabled delivery systems—have attracted growing attention for their ability to modulate nano–bio interactions and overcome biological barriers associated with AR. LNPs are of particular interest due to their intrinsic biocompatibility, tunable physicochemical properties, capacity to encapsulate both hydrophilic and hydrophobic agents, and potential for controlled or localized delivery [10, 11]. In antimicrobial applications, LNPs can enhance stability, pharmacokinetics, and site-specific delivery, potentially bypassing resistance mechanisms such as biofilm formation and efflux activity while reducing systemic toxicity [12].
The rational development of LNP-based antimicrobial biomaterials requires robust and predictive preclinical evaluation platforms. While mammalian models provide high physiological relevance, they are often costly, time-intensive, and associated with significant ethical constraints. In this context, the chicken embryo model (CEM) has emerged as a versatile and ethically favorable alternative that bridges the gap between in vitro assays and mammalian in vivo systems [13]. The CEM provides a vascularized and dynamically developing biological environment, supports localized infection and material administration, and aligns with the principles of Replacement, Reduction, and Refinement (3Rs) in animal research [14].
Despite increasing adoption, the application of the CEM in antimicrobial and biomaterials research remains highly variable. Variations in embryo age, inoculation routes, administration volumes, incubation conditions, and experimental endpoints limit reproducibility and complicate direct comparisons between studies [15]. Similarly, while LNP-based antimicrobials are often extensively characterised in terms of physicochemical properties and in vitro activity, fewer studies systematically evaluate their performance in standardized, biologically relevant in vivo-like models or critically compare their advantages and limitations relative to alternative nanocarrier platforms.
This review critically examines the current literature on the use of the CEM for evaluating lipid nanoparticle-based antimicrobial biomaterials. By identifying methodological inconsistencies, highlighting experimental limitations, and discussing opportunities for protocol standardization, this work aims to improve reproducibility and translational relevance, thereby supporting the rational design and evaluation of nanoparticle-enabled strategies to combat antibiotic-resistant infections.
AR represents one of the most pressing global health challenges of the 21st century. The well-established relationship between antibiotic exposure and the emergence of resistant bacterial populations has prompted WHO to warn that, without urgent intervention, drug-resistant infections will increasingly undermine modern healthcare systems worldwide [16]. From a biomaterials and environmental perspective, AR emerges not only as a clinical problem but also as a consequence of complex interactions between microorganisms, anthropogenic activity, and chemical agents distributed across biological and ecological interfaces.
Historically, resistance has followed closely behind antibiotic discovery. The first documented case of AR occurred in Escherichia coli in 1940, shortly after the introduction of sulfonamides, followed by penicillin-resistant Staphylococcus aureus in 1942 [17, 18]. Although the development of new antibiotic classes temporarily restored therapeutic efficacy, resistance rapidly re-emerged, ultimately giving rise to multidrug-resistant (MDR), extensively drug-resistant (XDR), and pan-drug-resistant (PDR) bacterial phenotypes. This recurring pattern highlights the evolutionary adaptability of bacteria and the inherent limitations of conventional small-molecule antibiotics.
Figure 1 illustrates the parallel timeline of major antibiotic introductions and the subsequent emergence of resistant strains. The rapid appearance of resistance following clinical deployment underscores the persistent “arms race” between antimicrobial development and microbial adaptation. Currently, resistant infections are estimated to cause approximately 700,000 deaths annually worldwide, a figure projected to rise to 10 million deaths per year by 2050 in the absence of effective countermeasures [1, 3, 19].

Timeline illustrating the parallel development of major antibiotic classes and the subsequent emergence of resistant bacterial phenotypes. Penicillinase resistance first appeared in 1947 (early clinical reports of penicillin-resistant Staphylococcus aureus) and later in hospitals (~1967). Vancomycin resistance is indicated twice: 1988 for vancomycin-resistant enterococci (VRE) and 2002 for vancomycin-resistant Staphylococcus aureus (VRSA). Notes clarify the species and timeline distinctions. Resistant (R), multidrug-resistant (MDR), extensively drug-resistant (XDR), and pan-drug-resistant (PDR) organisms are shown with approximate detection dates, highlighting the rapid onset of resistance following antibiotic introduction. The figure was created in BioRender. Ruano, M. (https://BioRender.com/e6xuc2s) is licensed under CC BY 4.0.
In addition to clinical misuse, subtherapeutic environmental antibiotic exposure is a major driver of AR. Widespread antibiotic application in medicine, veterinary practice, agriculture, and aquaculture, combined with insufficient wastewater treatment and pharmaceutical manufacturing effluents, has resulted in the continuous release of antimicrobial residues into soil and aquatic environments [6, 20, 21]. These conditions create persistent selective pressure that favors resistant microorganisms and facilitates the horizontal transfer of antibiotic resistance genes (ARGs).
Figure 2 summarizes the main anthropogenic and natural sources of environmental antibiotic contamination. Agricultural runoff, livestock and aquaculture practices, improper medical-waste disposal, and pharmaceutical production introduce antibiotics into soil and water, where they interact with diverse microbial communities. Environmental compartments—soil, water, and biofilms—act as reservoirs and transfer media, facilitating the spread of resistance genes across ecosystems and their eventual re-entry into clinical settings.
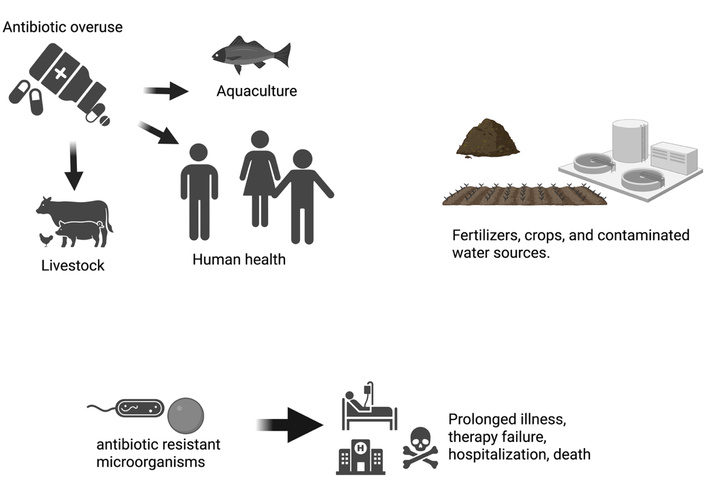
Environmental and anthropogenic drivers of antibiotic resistance. Key sources—including agriculture, pharmaceutical production, human and animal antibiotic overuse, aquaculture, and contaminated water—create selective pressure, promoting bacterial adaptation, horizontal gene transfer, and consequences such as therapy failure, prolonged illness, hospitalization, and occasionally death. The figure was created in BioRender. Ruano, M. (https://BioRender.com/30itgss) is licensed under CC BY 4.0.
Bacteria employ multiple, often synergistic mechanisms to evade antibiotic action [1]. These include:
Enzymatic inactivation or modification, such as β-lactamase-mediated hydrolysis of β-lactam antibiotics;
Target modification, involving mutations or chemical alterations that reduce drug binding to ribosomes, enzymes, or cell wall components;
Active efflux, where membrane-embedded transporters expel antibiotics and reduce intracellular concentrations;
Reduced permeability, resulting from alterations in membrane porins or cell envelope composition; and
Horizontal gene transfer, via conjugation, transformation, or transduction, enables rapid dissemination of resistance traits across species.
These mechanisms are often enhanced by chronic exposure to low antibiotic concentrations in environmental matrices, accelerating the emergence of MDR and XDR phenotypes. Understanding these resistance pathways is critical for the rational design of advanced biomaterials and delivery systems that can bypass or suppress conventional resistance mechanisms.
Despite advances in microbiology and medicinal chemistry, antibiotic discovery has stagnated over the past three decades. No fundamentally new antibiotic classes have reached the market in this period; most recently approved agents are structural derivatives of existing drugs [22, 23]. As a result, monotherapy has become increasingly ineffective for severe infections, necessitating combination regimens, as exemplified by multidrug-resistant tuberculosis [24].
In response, a range of complementary strategies is under investigation, including antimicrobial stewardship to reduce selective pressure, development of antibiotics with novel molecular targets, bacteriophage and probiotic therapies, and gene-editing approaches such as CRISPR-based interventions. Advanced drug delivery systems (DDSs), including nanocarriers, lipid nanoparticles, and bio-inspired materials—have emerged as powerful tools to enhance antibiotic efficacy, improve bioavailability, reduce off-target toxicity, and overcome bacterial defence mechanisms [17, 25]. These material-enabled strategies shift the paradigm from purely pharmacological solutions toward integrated bio-material–microbe interface engineering.
The rising prevalence of bacterial infections, compounded by AR, has intensified the search for novel therapeutic strategies. NPs represent a versatile alternative or adjunct to conventional antibiotics, providing mechanisms of action distinct from traditional small-molecule antimicrobials, which typically target single biochemical pathways. At the bio–nano interface, NPs interact with bacterial membranes, intracellular components, and local microenvironments, providing a multimodal antibacterial effect that can limit resistance development and restore efficacy against MDR pathogens [25].
A broad spectrum of NPs classes—including metallic, metal oxide, polymeric, carbon-based, and lipid-based systems—has demonstrated potent antibacterial activity in vitro and in vivo. For example, oleic acid–coated iron oxide nanoparticles showed nearly two-fold higher antibacterial activity against ampicillin- and kanamycin-resistant Escherichia coli compared with free antibiotics [26]. Similarly, silver nanoparticles (AgNPs) exhibited strong, dose-dependent antibacterial effects against MDR Vibrio parahaemolyticus improving survival rates and reducing infection-related pathology [27]. AgNPs have also synergised with conventional antibiotics against resistant Pseudomonas aeruginosa, enabling reduced antibiotic dosing without loss of efficacy [28].
Metal oxide NPs such as cerium oxide (CeO2) act primarily as antibiotic adjuvants, enhancing antibacterial efficacy through redox cycling and modulation of reactive oxygen species (ROS) rather than direct bactericidal action [29]. These examples underscore the versatility of NP platforms in addressing AR through complementary, multimodal mechanisms [30].
NPs exert antibacterial effects via multiple, often concurrent mechanisms, reducing the likelihood of resistance compared with conventional antibiotics [31]. The known antibacterial mechanisms include:
Physical membrane disruption: Direct interactions of sharp NP edges with bacterial cell walls compromise membrane integrity, leading to leakage of cytoplasmic contents [32].
Generation of ROS: Oxidative stress overwhelms bacterial defences, even in dark conditions [33, 34].
Bacterial trapping/immobilization: NP aggregation can physically trap bacteria, limiting nutrient uptake and proliferation [35].
Redox imbalance: NPs disrupt cellular redox homeostasis, damaging proteins, lipids, and DNA [36].
Metabolic interference: NPs may inhibit glycolysis or energy production, impairing bacterial survival [37].
DNA damage: Interaction with genetic material can induce strand breaks and replication errors [38].
Electric charge transfer: Electron exchange between NPs and bacteria can disrupt membrane potential and enzymatic function [39].
Metal-ion release: Release of ions (e.g., Ag+) interferes with enzymes and membrane integrity [40].
Nanobubble formation/explosion: Localized cavitation events can mechanically damage bacterial structures [41].
Charge polarity effects: Surface charge polarity of NPs influences bacterial adhesion and membrane disruption [42].
The multimodal action of NPs underpins potent activity against multidrug-resistant pathogens and biofilms, where single-target antibiotics often fail (Figure 3).
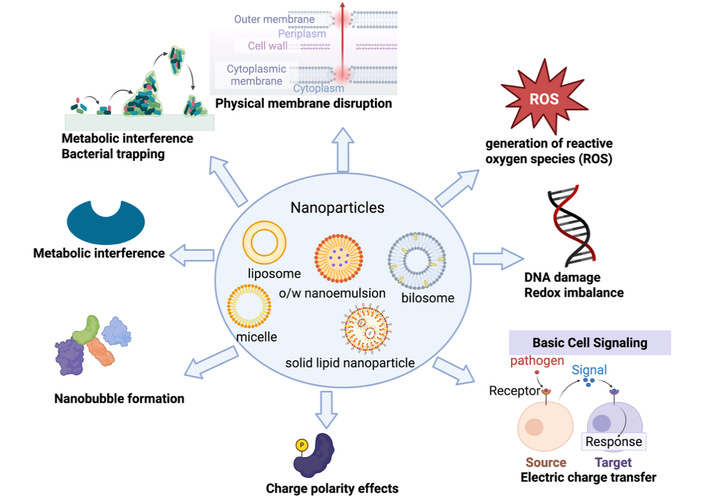
Multimodal antibacterial mechanisms of nanoparticles (NPs). NPs disrupt bacterial membranes, induce ROS and redox stress, impair metabolism, damage DNA, release toxic ions, and alter membrane potential. These combined actions enhance bactericidal efficacy and limit the development of conventional resistance [43]. The figure was created in BioRender. Ruano, M. (https://BioRender.com/yx7ilj1) is licensed under CC BY 4.0.
Various nanocarrier systems differ in biocompatibility, toxicity, stability, and translational potential (Table 1). Metallic NPs (Ag, Au, Fe3O4) show strong antibacterial activity but risk oxidative toxicity and accumulation [44]. Metal oxide NPs (ZnO, TiO2, CeO2) act as potent antibiotic enhancers, though biodegradability can be limited [12]. Polymeric NPs (PLGA, chitosan) provide controlled release but may present monomer toxicity and batch variability [45]. Carbon-based nanomaterials are effective due to high surface area and membrane interactions but face regulatory hurdles [46]. LNPs combine high biocompatibility, biodegradability, and scalability, making them highly translational for antimicrobial applications [10].
Comparative overview of nanocarrier systems for antimicrobial applications.
| Nanocarrier type | Key advantages | Key limitations | Relevance to AR |
|---|---|---|---|
| Metallic NPs (Ag, Au, Fe3O4) [31] | Strong intrinsic antibacterial activity; ROS generation | Long-term accumulation; oxidative toxicity | Effective against MDR bacteria; mainly topical/local use |
| Metal-oxide NPs (ZnO, TiO2, CeO2) [12] | Potent antibacterial and adjuvant effects | Cytotoxicity; limited biodegradability | Useful as antibiotic enhancers |
| Polymeric NPs (PLGA, chitosan) [47, 48] | Controlled release; tunable degradation | Monomer toxicity; batch variability | Sustained antibiotic delivery |
| Carbon-based nanomaterials [49, 50] | High surface area; membrane disruption | Safety and regulatory concerns | Promising but limited translation |
| LNPs [10, 51, 52] | High biocompatibility; biodegradability; scalable | Physical instability in some formulations | Strong translational potential |
AR: antibiotic resistance; LNPs: lipid nanoparticles; NP: nanoparticle.
LNPs uniquely balance antibacterial efficacy with biocompatibility, offering minimal systemic toxicity, low immunogenicity, and limited long-term accumulation. Composed of physiologically compatible lipids, they exhibit minimal systemic toxicity, low immunogenicity, and limited long-term accumulation [51, 53, 54]. LNPs encapsulate both hydrophilic and hydrophobic antimicrobials, protecting labile agents from enzymatic degradation and enabling controlled or stimuli-responsive release [52, 54, 55].
LNP systems include non-vesicular platforms (solid lipid nanoparticles, nanostructured lipid carriers, cubosomes, lipid–drug conjugates) and vesicular systems (liposomes, lipid–polymer hybrids, niosomes, bilosomes) [56–58]. Cubosomes, for instance, enhance antimicrobial peptide stability and achieve rapid bactericidal activity, outperforming free peptides [59]. Similarly, cubosome-encapsulated norfloxacin improved local deposition by ~150% in otitis externa models [60]. Clinically, lipid-based nanocarriers such as AmBisome®, Abelcet®, and Epaxal® exemplify their translational readiness [61].
LNPs exhibit high biocompatibility, superior drug loading, robust colloidal stability, and scalable manufacturing, outperforming many polymeric and inorganic systems (Table 2) [10, 57]. Their surfaces can be functionalized for targeted delivery, enhancing biodistribution for personalized medicine [62].
Comparative advantages of LNPs over other nanocarriers.
| Feature | LNPs | Liposomes | Polymeric NPs | Inorganic NPs |
|---|---|---|---|---|
| Biocompatibility | High | Moderate | Moderate | Variable |
| Drug Loading | High | Moderate | Moderate | Variable |
| Stability | High | Moderate | Moderate | Often high, may aggregate |
| Targeted delivery | Easily functionalized | Possible, less efficient | Possible | Possible |
| Scalability & GMP | Easy | Moderate | Moderate | Difficult |
| Clinical translation | Validated in mRNA vaccines | Some approved drugs | Limited | Mostly preclinical |
LNPs: lipid nanoparticles; NP: nanoparticle.
These attributes position LNPs as a leading platform for targeted antimicrobial delivery, reduction of systemic toxicity, facilitation of combination therapies, and support of personalized medicine approaches.
The CEM provides a vascularized and cost-effective system. It is ethically favorable for evaluating nanoparticle biodistribution, toxicity, and therapeutic efficacy. By enabling injection into the chorioallantoic membrane (CAM) or yolk sac, the CEM allows assessment of NP–host and NP–pathogen interactions, infection progression, and mid-throughput screening prior to mammalian studies [63, 64].
The distinctive physicochemical properties of NPs—including nanoscale size, high surface area, and tunable surface chemistry—necessitate tailored preclinical evaluation strategies to assess therapeutic efficacy, mechanisms of action, and potential toxicity. LNPs are particularly sensitive to composition, size distribution, surface charge, and structural organization, making thorough evaluation essential [65].
Preclinical evaluation typically follows a tiered approach, integrating in vitro, ex vivo, and in vivo models to assess NP behavior and efficacy [66]. While in vitro assays are indispensable for early screening, they cannot fully capture systemic interactions, biodistribution, and host responses. The CEM provides a physiologically relevant intermediate platform bridging in vitro assays and mammalian studies.
In vitro assays provide the first step in NP evaluation, offering controlled conditions, high throughput, and cost efficiency. They are commonly used to assess cytotoxicity (e.g., MTT or Alamar Blue™), cell viability, metabolic activity [67], and mechanistic endpoints such as cytokine release, oxidative stress, and gene expression [68]. Antimicrobial activity is typically evaluated using disc diffusion, broth microdilution, or metabolic readouts, enabling direct comparison with conventional antibiotics [44, 68, 69].
The main advantages of in vitro screening are experimental control, scalability, and rapid identification of lead candidates (Table 3). These assays also align with the 3Rs principle, reducing reliance on animal testing while providing mechanistic insight into NP–cell and NP–pathogen interactions. Limitations include the absence of systemic factors (immune response, metabolism, multicellular interactions), oversimplified tissue models, and limited predictive power for long-term toxicity, chronic exposure, or in vivo efficacy. Advanced commercial models, such as organotypic intestinal systems, developed by Alimetrics, improve physiological relevance but cannot fully replace in vivo validation.
Advantages and limitations of in vitro screening.
| Advantages | Limitations |
|---|---|
| Controlled conditions (pH, temperature, medium) | Lack of systemic interactions (immune response, metabolism) |
| Rapid, high-throughput screening | Limited physiological relevance of immortalized or tumor-derived cell lines |
| Ethical alignment with 3R principle | Oversimplified environment, lacking ECM and mechanical forces |
| Mechanistic insights into toxicity and antimicrobial action | Poor prediction of chronic toxicity or long-term effects |
| Cost-effective compared with animal studies | Short experimental duration limits assessment of sustained effects |
In vitro assays are indispensable for early-stage NP screening but must be complemented by intermediate and mammalian models to assess systemic safety and therapeutic performance.
In vivo studies capture complex organism-level interactions, including pharmacokinetics, tissue distribution, immune responses, efficacy, and toxicity. Ex vivo models complement these studies by preserving tissue or organ physiology under controlled conditions, enabling mechanistic investigation of NP–host interactions.
Model selection balances ethical considerations, cost, throughput, and translational relevance. Mammalian models (mice, rabbits, non-human primates) provide detailed pharmacokinetic, immunological, and toxicological data [70] but are constrained by high cost, ethical requirements, and low throughput (Table 4). Non-mammalian models, such as Caenorhabditis elegans [45, 71–73], Drosophila melanogaster [74], Galleria mellonella [75–78] and zebrafish [79–81] provide cost-effective, ethically favorable alternatives for higher-throughput screening, though species differences limit predictive power for humans.
Advantages and limitations of in vivo models.
| Advantages | Disadvantages |
|---|---|
| Replicate complex organismal interactions | Ethical concerns; regulatory approval required |
| Capture physiological complexity (immune response, metabolism) | High maintenance costs and specialized facilities |
| Enable comprehensive toxicity evaluation | Inter-species variability limits translational accuracy |
| Support long-term studies | Time-consuming |
| Multi-endpoint assessment (behavioral, histopathological, biochemical) | Limited throughput for large-scale screening |
Ex vivo platforms bridge the gap between in vitro and in vivo studies, maintaining tissue architecture and mechanistic insight while lacking systemic integration.
Overall, in vivo models remain indispensable for comprehensive safety and efficacy evaluation, while non-mammalian and ex vivo systems support early decision-making and mechanistic insight. A tiered screening strategy combining these models is therefore essential to balance translational relevance, ethical responsibility, and development efficiency.
The CEM provides an effective intermediate platform between in vitro assays and mammalian studies, balancing medium-to-high throughput, physiological relevance, and ethical feasibility for preclinical NP screening. The CEM provides a vascularized, dynamically developing, and ethically favorable platform for nanoparticle evaluation, supporting infection modeling, immune assessment, and biodistribution studies.
Compared with other non-mammalian models, such as Caenorhabditis elegans, Drosophila melanogaster, Galleria mellonella, and zebrafish, the CEM provides greater physiological complexity and clinical relevance, while maintaining manageable costs and throughput (Table 5). Simpler invertebrate models allow very high-throughput screening but lack vascularization and adaptive immunity, whereas zebrafish provide partial organ and immune complexity yet remain limited in modeling later-stage immune responses [80]. The CEM therefore occupies a unique position, combining vascularized tissues, immune development, and experimental accessibility through in ovo or ex ovo approaches [82].
Comparison with other non-mammalian models.
| Feature | CEM | Caenorhabditis elegans[45, 71] | Zebrafish[81] | Drosophila melanogaster[83] | Galleria mellonella[76, 77, 84, 85] |
|---|---|---|---|---|---|
| Cost | Low–moderate | Very low | Low | Very low | Low |
| Throughput | Moderate–high | Very high | High | Very high | Moderate–high |
| Physiologic complexity | Organ growth, blood vessels, immune progress | Simple anatomy, dynamic | Organ growth, immune progress | Simple anatomy, no adaptive immunity | Innate immunity only |
| Immune system | Innate + adaptive develop | Innate only | Innate + partial adaptive | Innate only | Innate only |
| Ease of manipulation | Moderate (in ovo/ex ovo, CAM access) | Easy | Moderate | Easy | Moderate |
| Clinical relevance | Moderate–high | Low | Moderate | Low | Low–moderate |
| Ethical issues | Less restriction before ED15 | Minimal | Low | Minimal | Low |
Overall, the CEM provides a robust compromise between complexity and scalability, supporting mechanistic insight and early translational decision-making. As such, it represents an ideal intermediate model for nanoparticle evaluation prior to mammalian testing.
The CEM exhibits a rapidly maturing immune system with structural and functional parallels to humans, enabling translationally relevant studies of host–pathogen interactions and immunomodulatory interventions (Figure 4).
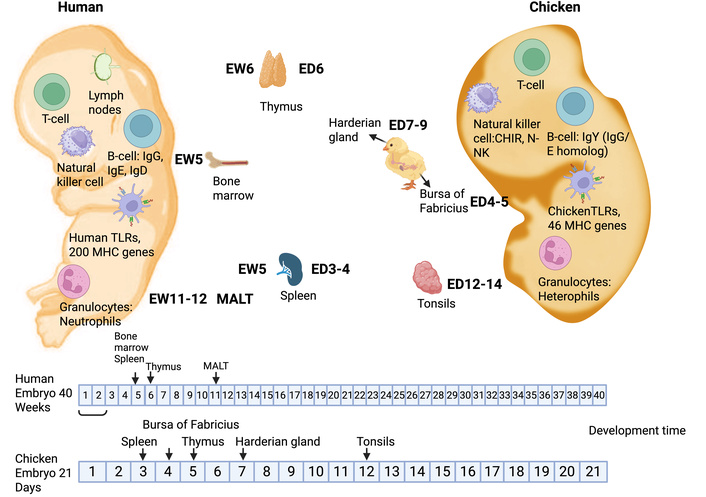
Comparative immune development in humans and chickens. Human immune maturation occurs over ~40 gestational weeks (embryonic weeks, EW), whereas chicken immune development is completed within 21 embryonic days (ED). Key primary and secondary lymphoid organs are shown, including the thymus, spleen, bone marrow (humans), and the bursa of Fabricius (chickens), as well as mucosa-associated lymphoid tissue (MALT) in humans and cecal tonsils in chickens, highlighting conserved functional immunity despite differences in developmental timing and MHC diversity. The figure was created in BioRender. Ruano, M. (https://BioRender.com/d52c6jx) is licensed under CC BY 4.0.
Primary lymphoid organs develop in a defined sequence: the thymus supports T-cell differentiation (γδ T cells precede αβ T cells), the bursa of Fabricius mediates B-cell maturation (functionally analogous to human bone marrow), and the spleen acts as a secondary lymphoid organ in both species. Innate immunity is highly conserved; Toll-like receptors (TLRs) detect pathogen-associated molecular patterns, triggering comparable inflammatory signaling in chickens and humans.
Antigen presentation occurs through the major histocompatibility complex (MHC), which, despite lower genetic diversity in chickens (~46 genes versus > 200 in humans), enables effective T-cell activation. Early emergence of macrophages, dendritic cells, and NK cells, together with embryonic cytokine expression (IL-1β, IL-8, IFN-γ), allows functional evaluation of immune responses pre-hatch. The rapid, functional immune maturation makes the CEM a versatile platform for screening nanocarriers and immunomodulatory therapies, bridging mechanistic insight with potential clinical translation.
The CEM enables multiple experimental configurations for nanomaterial evaluation (Figure 5). In ovo cultivation preserves physiological conditions and is used for toxicity, survival, and biodistribution studies [86]. Ex ovo cultivation allows direct access to the CAM for angiogenesis assays and imaging-based analyses, with higher risk of dehydration and contamination [87].
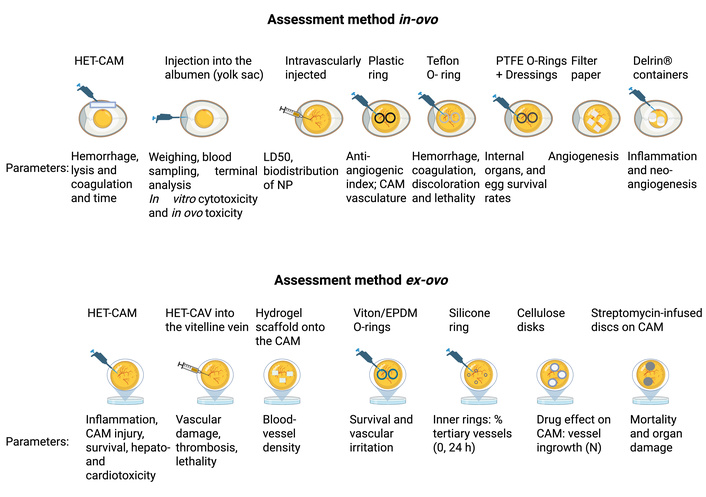
Experimental workflow of the CEM. Overview of in ovo and ex ovo cultivation, nanoparticle delivery routes (CAM injection, yolk sac injection, topical CAM application), and analytical readouts, including CAM angiogenesis assays, fluorescence/bioluminescence imaging, histological analysis, and toxicity assessment, for integrated evaluation of nanocarrier biodistribution and efficacy. CAM: chorioallantoic membrane; CEM: chick embryo model; HET-CAM: Hen’s Egg Test – Chorioallantoic Membrane; HET-CAV: Hen’s Egg Test – Chorioallantoic Vasculature. The figure was created in BioRender. Ruano, M. (https://BioRender.com/v51f276) is licensed under CC BY 4.0.
NP administration is performed via CAM injection [87], yolk sac injection [88], or topical CAM application [89], depending on the intended exposure profile. CAM injection enables localized assessment of vascular interactions, angiogenic responses [90], and antimicrobial efficacy. Yolk sac injection provides systemic exposure, supporting evaluation of NP circulation and organ distribution. Topical CAM application allows confined delivery to the membrane surface and is particularly suited for localized antibacterial or anti-angiogenic studies. These standardized routes, combined with multimodal readouts [82, 91], support reproducible and translational screening of LNPs in the CEM.
The CEM is widely applied for infection modeling and antimicrobial evaluation, leveraging CAM accessibility, high vascularization and favorable ethical status. The model supports controlled investigation of pathogen invasion, host responses, and nanocarrier-mediated therapeutic efficacy across multiple microbial classes.
CEM-based studies encompass bacterial, fungal, parasitic, and probiotic systems, including clinically and agriculturally relevant pathogens. Bacterial models include Klebsiella pneumoniae, Escherichia coli K-12, Salmonella Typhimurium, Staphylococcus aureus, Pseudomonas aeruginosa, Clostridium perfringens, Enterococcus cecorum, Chlamydia psittaci, and Chlamydia abortus [92]. Fungal infections have been established using Candida albicans [93], Aspergillus fumigatus [94], and Cryptococcus gattii [90], while parasitic infections such as Neospora caninum [95] have also been reported. In parallel, probiotic interventions (e.g., Enterococcus faecium, Bacillus subtilis) have been evaluated against enteric pathogens including Campylobacter jejuni [96].
Antimicrobial assessment in the CEM typically follows a tiered screening strategy. Initial in vitro assays (disc diffusion, broth microdilution, metabolic viability assays) define minimal inhibitory concentrations and baseline antimicrobial activity. Subsequent in vivo/ex vivo infection studies involve pathogen inoculation at defined embryonic stages (commonly ED5–ED10) via CAM, yolk sac, allantoic cavity, or amniotic routes, followed by quantification of embryo survival, pathogen burden, histopathology, and immune markers. Topical CAM application further enables localized therapeutic evaluation while limiting systemic exposure.
Pathogen-dependent outcomes are consistently observed in the CEM. Highly invasive extracellular bacteria induce rapid vascular dissemination and embryo lethality, whereas intracellular pathogens display delayed progression and tissue-restricted infection. Fungal pathogens show species-specific virulence profiles reflected in survival kinetics, inflammatory responses, and CAM vascular remodeling.
Methodological heterogeneity limits reproducibility. Variability in inoculation route, embryonic day, pathogen dose, cultivation mode, and relative humidity (RH) significantly affects infection outcomes and complicates cross-study comparison. Ex ovo cultivation is associated with increased baseline mortality, while RH fluctuations markedly influence embryonic susceptibility (Table 6), underscoring the need for standardized infection protocols.
Summary of microorganisms investigated using the chicken embryo model (CEM).
| Name | Day | T/RH°C/% | ED | Dose | Key findings | Ref |
|---|---|---|---|---|---|---|
| Klebsiella pneumoniae, Escherichia coli K-12, Salmonella Typhimurium | 13 | 37°C/45% | 11 | 4 × 106 – 2 × 107 CFU/100 μL | Dose-dependent embryo mortality and immune activation | [86] |
| Chlamydia psittaci, C. abortus | 12 | 37.8°C/60% | 10 | 5 × 104 IFU/egg | Successful infection and tissue colonization | [92] |
| Cryptococcus gattii | 18 | ND | 10 | 107 CFU/mL | Fungal dissemination | [97] |
| Clostridium perfringens, Eimeria tenella | 15 | 37.6°C/55% | 10, 15 | 104 CFU/100 μL | Gut lesions resembling avian intestinal disease | [98] |
| E. coli DH5α bioluminescent | 20 | 37.2–37.7°C/ND | 18 | 103 CFU/μL | Enabled in vivo tracking of bacterial distribution | [91] |
| Staphylococcus aureus | 12 | 38°C/80–90% | 9 | 102 CFU/μL | Embryo mortality; inflammatory response | [87] |
| Salmonella enterica serovar Typhimurium | 17 | 37°C/40–60% | 6, 10, 13, 16 | 106 CFU/μL | Dose- and time-dependent systemic infection | [99] |
| Enterococcus cecorum | 18 | 37°C/55% | 11 | 3.4 × 107 CFU/egg | Embryo mortality; liver/gut colonization | [100] |
| Pseudomonas aeruginosa | 6 | 37°C/80% | 5 | 105 CFU/μL (10 μL) | Rapid infection; high embryo lethality | [101] |
| Probiotic strains (E. faecium, Bacillus subtilis) | 19 | 37.5°C/54% | 17.5 | 5–16 × 109 CFU/egg | No adverse effects; microbiome modulation | [102] |
| Salmonella spp., APEC, Campylobacter jejuni | 10–17 | 37°C/58% | 10 | 3 log CFU/egg | Colonization; pathogen interaction studies | [96] |
| Salmonella pullorum, E. coli | 15 | 37°C/58% | 13 | 2–6 log CFU/embryo | Embryo mortality; pathogen-specific immunity | [103] |
| Neospora caninum | 8–18 | ND | 8 | 10–105 tachyzoites/embryo | Infection and parasite proliferation | [104] |
| Candida albicans | 18 | 37.6°C/60% | 7, 10 | 108 cells/egg | Colonization; embryo lethality | [105, 106] |
| Aspergillus fumigatus | 16 | 37.6°C/50–60% | 14 | 0.1 mL of 107–102 CFU | Respiratory lesions; dose-dependent | [107] |
The CEM provides a rapid, translational platform for NP evaluation: rapid organogenesis (21 days) [13], low ethical constraints before ED15 [108], human-relevant physiology, and highly vascularized CAM for real-time NP tracking. Physiological and immune parallels with humans (Figure 4) support mechanistic insights, while the highly vascularized CAM allows real-time assessment of NP biodistribution and efficacy [109].
The CEM provides a cost-effective, ethically favorable platform for infection and therapeutic studies, but several biological and technical constraints must be considered. Table 7 summarizes the main limitations.
Limitations of the CEM for biomedical and pharmacological applications.
| Limitation | Description | Ref |
|---|---|---|
| Underdeveloped immunity | Adaptive immunity immature pre-hatch, limiting immune and immunotherapy studies | [110] |
| Ethical constraints | Studies after ED14 need approval | [111] |
| Limited systemic evaluation | Unsuitable for chronic or systemic studies, including toxicity, pharmacokinetics, and metabolism | [112] |
| Challenges modelling complex diseases | Immature development limits studies of neurodegeneration and advanced cancers | [13] |
| Ex ovo technical risks | Contamination, dehydration, and high mortality limit long-term reliability | [113, 114] |
CEM: chick embryo model.
Remaining gaps include standardized pathogen inoculation and LNP dosing, limited systemic immune evaluation, and few comparative studies with mammalian models to validate translational relevance. Complex phenomena such as wound healing or scaffold dynamics remain challenging to assess in this model.
To enhance reproducibility and comparability, we propose the following standardized framework in Table 8.
Proposed standardization framework for CEM-based studies.
| Parameter | Recommendation | Notes |
|---|---|---|
| Inoculation timing | ED5–10 for systemic studies; ED12–18 for immune response assessment | Adjust based on pathogen/study goal |
| Microbial dose | Report in CFU/μL or per egg; determine sublethal/lethal ranges via titration | Ensures reproducibility |
| Environmental conditions | Temperature 37–38°C; humidity 40–80% | Maintain consistently |
| Administration route | Specify in ovo vs. ex ovo, injection site, volume, and technique | Impacts survival/outcomes |
| Experimental endpoints | Embryo survival, pathogen colonization, immune markers, histology, imaging | Standardized readouts improve comparison |
| Replicates and controls | Include biological and technical replicates; positive/negative controls | Ensures statistical robustness |
CEM: chick embryo model.
Despite the promise of LNPs and the CEM, the field currently faces key gaps: lack of standardized protocols across infection models, limited comparative studies of LNPs versus other nanocarriers and insufficient translational validation between CEM and mammalian systems.
Future studies should focus on systematic, mechanistically guided, and standardized investigations to maximize the CEM’s predictive power and accelerate clinical translation of nanoparticle-based therapeutics and key areas for further development include:
Standardization of experimental parameters: harmonized SOPs for inoculation, LNP dosing, temperature, and RH to reduce variability.
Integration of advanced readouts: use of fluorescence lifetime imaging, whole-embryo tomography, and transcriptomic profiling to better understand LNP biodistribution and infection mechanisms.
Improved immune modelling: use immune modulators, cell transplants, or immune-reporter pathogens to evaluate host responses.
Pathogen-specific frameworks: tailor inoculation days, RH, and disease markers to major bacterial, fungal, and parasitic species.
Rational LNP optimization: screen LNP formulations varying in lipid composition, surface chemistry, or targeting ligands.
Bridging to mammalian models: benchmark CEM outcomes against rodents to validate translational predictivity.
In conclusion, standardized protocols are critical to ensure reproducible, comparable infection studies using the CEM. Pathogen-specific optimization of inoculation timing, dose, and evaluation parameters is essential to capture diverse infection dynamics. Implementing harmonized frameworks (Table 8) will strengthen reproducibility and translational relevance.
AgNPs: silver nanoparticles
APEC: avian pathogenic Escherichia coli
AR: antibiotic resistance
CAM: chorioallantoic membrane
CEM: chick embryo model
HET-CAM: Hen’s Egg Test – Chorioallantoic Membrane
HET-CAV: Hen’s Egg Test – Chorioallantoic Vasculature
LNPs: lipid nanoparticles
MDR: multidrug-resistance
NPs: nanoparticles
RH: relative humidity
ROS: reactive oxygen species
WHO: World Health Organization
XDR: extensively drug-resistant
M.R. acknowledges that the foundational knowledge underpinning this study was developed during a Knowledge Transfer Partnership (KTP) project between AB Vista, ABITEC Corporation, and the University of Strathclyde.
MR: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. CO: Conceptualization, Investigation, Writing—original draft. MB: Project administration. ABM: Project administration. DK: Project administration. VAF: Supervision, Resources, Project administration, Conceptualization, Investigation, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest. The publication of the article is not subject to any financial or other restrictions.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 2187
Download: 39
Times Cited: 0
