 Systematic Review
Systematic Review

Affiliation:
1Anatomy Department, Atma Jaya Catholic University of Indonesia, Jakarta 14440, Indonesia
Email: poppy.kristina@atmajaya.ac.id
ORCID: https://orcid.org/0000-0002-4859-3402
Affiliation:
2Department of Cardiology and Vascular Medicine, Universitas Indonesia Hospital, Depok 16424, Indonesia
ORCID: https://orcid.org/0000-0003-2579-3089
Affiliation:
3Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada, Bulaksumur, Caturtunggal, Sleman 55281, Indonesia
ORCID: https://orcid.org/0009-0003-8969-4992
Affiliation:
4Faculty of Medicine, Universitas Brawijaya, Malang 65145, Indonesia
ORCID: https://orcid.org/0009-0009-5512-790X
Affiliation:
4Faculty of Medicine, Universitas Brawijaya, Malang 65145, Indonesia
ORCID: https://orcid.org/0009-0002-6629-995X
Affiliation:
5Cardiology Research Office, Department of Cardiology and Vascular Medicine, Dr. Sardjito General Hospital, Yogyakarta 55284, Indonesia
ORCID: https://orcid.org/0000-0002-4560-265X
Explor Neuroprot Ther. 2025;5:1004127 DOI: https://doi.org/10.37349/ent.2025.1004127
Received: July 30, 2025 Accepted: November 25, 2025 Published: December 11, 2025
Academic Editor: Aurel Popa-Wagner, University of Duisburg-Essen, Germany
The article belongs to the special issue Therapeutic Targets for Neuroprotection in Ischemic Stroke
Background: Ischemic stroke is a leading cause of disability, with calcium (Ca2+) dysregulation contributing to neuronal injury and impaired recovery. While early clinical trials targeting calcium signaling showed limited success, growing preclinical evidence supports the potential of calcium modulation for long-term neuroprotection. This systematic review evaluates the long-term effects of calcium modulation in animal models of ischemic stroke.
Methods: A comprehensive search across PubMed, Scopus, Web of Science, and the Cochrane Library up to June 2025 identified studies investigating calcium-targeted interventions (e.g., calcium channel blockers, chelators, antioxidants) with ≥ 30 days of follow-up. Risk of bias was assessed using the Risk of Bias in Non-randomized Studies of Interventions (ROBINS-I).
Results: Nine studies met the inclusion criteria. Interventions like L-type calcium channel blockers, magnesium sulfate, and ischemic preconditioning consistently reduced infarct volume (e.g., 22.4 ± 0.5% with preconditioning vs. 51.6 ± 2.1% with knockout) and improved neurobehavioral outcomes [e.g., epigallocatechin gallate (EGCG)-treated rats scored 2.17 ± 0.05 vs. 3.63 ± 0.06 in controls]. Molecular pathways involved included phosphoinositide 3-kinase (PI3K)/AKT, stromal interaction molecule 1 (STIM1)/ORAI1, and calcium-sensor proteins such as NCKX2.
Discussion: Calcium modulation holds strong promise for neuroprotection in ischemic stroke models. Although clinical gaps remain, these findings support the development of calcium-targeted therapies for stroke recovery, especially when combined with multimodal strategies.
Ischemic stroke is a leading cause of death and long-term disability worldwide. It occurs when a sudden reduction in cerebral blood flow triggers a cascade of biochemical and molecular events that result in neuronal injury. One of the critical factors contributing to neuronal damage is calcium (Ca2+) dysregulation. Under normal physiological conditions, intracellular calcium concentrations are tightly regulated, playing essential roles in various neuronal functions, such as synaptic transmission, plasticity, and cell signaling. However, during ischemia, calcium overload occurs due to the uncontrolled influx through N-methyl-D-aspartate (NMDA) receptors, voltage-gated calcium channels, and the release from intracellular stores. This overload induces mitochondrial dysfunction, oxidative stress, and the activation of calcium-dependent enzymes, ultimately leading to neurotoxicity and cell death [1, 2].
Calcium overload contributes not only to acute neuronal injury but also to long-term processes that are vital for recovery and neuroplasticity. This dual role, both as a driver of neuronal damage and a facilitator of recovery, presents challenges in developing targeted therapies [3]. Early clinical trials using calcium channel blockers, such as nimodipine, aimed at treating acute ischemic stroke, showed limited success. These failures were likely due to factors such as non-specific blockade, poor timing of administration, and adverse hemodynamic effects [4]. However, recent advances in calcium signaling research have led to more selective targeting strategies, which involve modulating specific calcium channels, receptors, and intracellular calcium stores. These approaches show promise for providing long-term neuroprotection, as supported by a growing body of preclinical evidence.
Notably, optogenetic activation of astrocytic calcium signaling has been demonstrated to improve neurovascular coupling and enhance functional recovery after stroke, underscoring the importance of glial calcium dynamics during the post-ischemic phase [5]. Clinical studies have also shown that serum calcium levels, measured several days after stroke onset, are associated with infarct volume and neurological function, suggesting that calcium homeostasis continues to influence outcomes beyond the acute phase [6, 7].
Epidemiological data also support a role for calcium in both stroke prevention and recovery. Higher dietary calcium intake and optimal serum calcium levels have been associated with lower risk of ischemic stroke and improved outcomes, implying both protective and restorative effects [8, 9]. However, the heterogeneity of stroke pathology and the complex interactions between calcium signaling and other molecular pathways present significant challenges for the clinical translation of these findings.
Despite a broad and comprehensive search across multiple databases, only nine studies met the predefined inclusion criteria for this review. These studies included those with long-term follow-up (≥ 30 days) and relevant neuroprotective outcomes. While a larger number of studies were initially identified, many were excluded due to differences in study designs, outcome measures, or short follow-up periods. The studies included in this review represent what we believe to be the best available evidence on the long-term effects of calcium modulation in ischemic stroke.
This systematic review aims to synthesize the available preclinical evidence on the long-term neuroprotective effects of calcium modulation in ischemic stroke. By examining how calcium dysregulation impacts stroke outcomes and evaluating the efficacy of various calcium-targeted interventions in animal models, this review seeks to clarify the therapeutic potential of calcium modulation and inform future translational research in stroke recovery.
This systematic review was conducted according to the protocol of the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.
This systematic review included English-language studies that evaluated the impact of calcium (Ca2+) modulation, for example, calcium channel blockers, calcium chelators, or intracellular regulators of calcium, on long-term outcome in animal models of ischemic stroke. Qualifying studies reported at least one recovery-associated or neuroprotective outcome (e.g., neurological function, infarct size, cognitive or motor recovery, or neuronal survival) with a follow-up duration of at least 30 days.
A systematic review was conducted in PubMed, Scopus, Web of Science, and the Cochrane Library with the pre-specified Medical Subject Headings (MeSH) terms and keywords. Editorials, conference abstracts, meta-analyses, and reviews, as well as studies that failed to report discrete outcome data regarding neuroprotection, were excluded. Studies failing to isolate the effects of modulation of calcium from other interventions administered concomitantly were also excluded.
A comprehensive literature search was conducted across PubMed, Scopus, Web of Science, and the Cochrane Library, covering all records up to June 2025. The search strategy utilized MeSH terms and relevant keywords, including “calcium modulation”, “calcium channel blockers”, “ischemic stroke”, “neuroprotection”, and “functional recovery”. Boolean operators (AND, OR) were used to maximize sensitivity and specificity. Filters were applied to include only peer-reviewed studies published in English.
Two independent reviewers screened all titles and abstracts to identify potentially eligible studies. Full texts of selected articles were then assessed against the inclusion and exclusion criteria. Discrepancies were resolved through discussion or consultation with a third reviewer.
The systematic review was conducted in accordance with the PRISMA guidelines. A comprehensive literature search was performed across the following databases: PubMed, Scopus, Web of Science, and the Cochrane Library (up to June 2025).
Studies were selected based on predefined eligibility criteria, which included animal models of ischemic stroke with at least 30 days of follow-up. The data from the selected studies were extracted using a standardized form, which included study design, intervention details, outcome measures, and key findings.
Risk of bias was assessed using the Risk of Bias in Non-randomized Studies of Interventions (ROBINS-I), which evaluates seven domains:
Bias due to confounding.
Bias in selection of participants.
Bias in classification of interventions.
Bias due to deviations from intended interventions.
Bias due to missing data.
Bias in measurement of outcomes.
Bias in selection of the reported result.
Each domain was judged as Low, Moderate, Serious, Critical, or No information risk of bias.
Interventions:
The systematic review included studies using the following interventions:
Calcium-modulating agents, including:
Traditional calcium channel blockers (e.g., L-type blockers).
Magnesium sulfate (acts as a calcium antagonist).
Calcium chelators [e.g., epigallocatechin gallate (EGCG)].
Ischemic preconditioning.
These interventions were tested in various animal models of ischemic stroke to assess their long-term neuroprotective effects.
A total of 3,120 records were identified through database searching: PubMed (n = 1,230), Scopus (n = 980), Web of Science (n = 720), and the Cochrane Library (n = 190). After removing 820 duplicates and 100 irrelevant publication types (e.g., non-English articles, reviews, or editorials), 2,200 records remained for title and abstract screening. Of these, 2,100 records were excluded due to irrelevance.
The full texts of 100 articles were retrieved and assessed for eligibility. After detailed review, 91 studies were excluded due to: irrelevant outcomes (n = 35), wrong study design (n = 28), or other reasons such as incomplete data or absence of a comparator group (n = 28). Ultimately, 9 studies met the inclusion criteria and were included in the final analysis.
Due to the heterogeneity across the included studies, including differences in study designs, animal models, interventions, outcome measures, and follow-up periods, a meta-analysis was not feasible. The differences in study designs and outcome reporting made it difficult to combine the data in a meaningful way. Therefore, a qualitative systematic review approach was used to synthesize the findings from the selected studies.
The full study selection process is summarized in the PRISMA flow diagram (Figure 1).
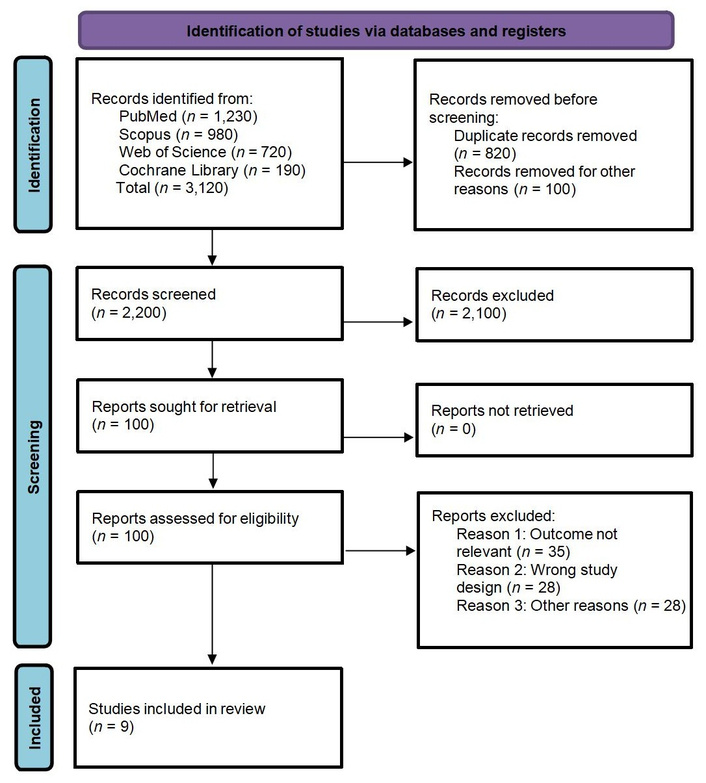
PRISMA Flow Diagram showing study selection process. Adapted from “The PRISMA 2020 statement: an updated guideline for reporting systematic reviews” by Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. BMJ. 2021;372:n71 (https://www.bmj.com/content/372/bmj.n71). CC BY 4.0.
The 9 included studies were published between 2015 and 2025 and involved both in vivo and in vitro experimental models of ischemic stroke. Most studies employed middle cerebral artery occlusion (MCAO) models in rodents. Calcium-targeted interventions included L-type calcium channel blockers, magnesium sulfate, EGCG, ischemic preconditioning, small interfering RNA (siRNA) targeting stromal interaction molecule 1 (STIM1), and other novel approaches.
Outcomes assessed included infarct volume, neuronal apoptosis, neurobehavioral scores, intracellular calcium regulation, and molecular pathway activity. A summary of key study characteristics and findings is provided in Table 1.
Summary of included studies and main findings.
| Study ID | Study design | Participants | Summary of findings |
|---|---|---|---|
| Cuomo et al., 2022 [1] | Experimental in vivo study | Male Sprague-Dawley rats subjected to middle cerebral artery occlusion (MCAO) treated with ischemic preconditioning (IPC) | IPC increased the expression of NCKX2, K+-dependent Na+/Ca2+ exchanger, following MCAO, mediated by the p-AKT signaling pathway. NCKX2 knockout yielded lower neuroprotective capacity, indicated by the greater infarct size (51.6 ± 2.1% with NCKX2 knockout + preconditioning vs. 22.4 ± 0.5% with preconditioning). |
| Dhyani et al., 2023 [2] | In vitro study | HMC3 microglial cell line, subjected to a hypoxic gradient and treated with an L-type calcium channel blocker (CCB) | L-type CCB improved the hypoxic conditions, similar to reoxygenation, indicated by the alleviation of cytosolic calcium level and preservation of cell viability within one hour of treatment, in cell lines with a hypoxic gradient. CCB administration also inhibited immune response by decreasing oxidative stress markers, namely HIF1A and OXR1. |
| Park et al., 2024 [3] | Experimental in vivo study | Male Sprague-Dawley rats subjected to MCAO treated with epigallocatechin gallate (EGCG; 50 mg/kg) | MCAO resulted in diminished neurobehavioral functions and increased infarct size as well as decreased hippocalcin expression, a neuronal calcium sensor protein. Administration of EGCG improved neurological deficits (2.17 ± 0.05 in EGCG vs. 3.63 ± 0.06 in controls), prevented cell death and intracellular calcium overload in glutamate-exposed neurons, and alleviated the decreased hippocalcin expression (0.52 ± 0.05 in EGCG vs. 0.11 ± 0.02 in controls). Additionally, EGCG reduced caspase-3 (1.43 ± 0.02 vs. 1.69 ± 0.02) and cleaved caspase-3 (5.32 ± 0.21 vs. 6.15 ± 0.35) expression levels associated with glutamate exposure. |
| Secondo et al., 2019 [4] | Experimental in vivo study | Sprague-Dawley rats MCAO treated with IPC | IPC stimulated store-operated calcium entry (SOCE) and Ca2+ release, which then inhibited the downregulation of stromal interaction molecule 1 (STIM1) and a structural component of the CRAC calcium channel (ORAI1) associated with oxygen and glucose deprivation. In contrast, STIM1 and ORAI1 silencing resulted in endoplasmic reticulum stress, marked by the increased activity of 78-kDa glucose-regulated protein (GRP78) and caspase-3. |
| Tang et al., 2015 [5] | Experimental in vivo and in vitro study | Male C57BL/6 mice subjected to permanent MCAO and treated with pertussis toxin (PTx) | PTx exhibited maximal reduction of infarct size when a 40–60% reduction of relative CBF was observed following permanent MCAO (2.12 ± 1.58 mm3 vs. 12.3 ± 3.8 mm3, p < 0.01). PTx also inhibited calcium influx into the neurons, which resulted in increased neuronal survival (1.1 ± 0.12, p < 0.01) or apoptosis prevention by reducing caspase-3-positive cell density (1.36 ± 0.05 vs. 1.89 ± 0.2, p < 0.01) and salvage of ischemic penumbra. |
| Wang et al., 2025 [6] | Experimental in vivo study | Male ICR mice subjected to MCAO and treated with the traditional Chinese medicine borneol | The nanoformulation of the traditional Chinese medicine borneol restored intracellular Ca2+ level and redox homeostasis, improved CBF (increased to 93.9 ± 2.4% after 24 hours), alleviated cerebral histopathology, and prevented apoptosis mediated by PI3K/Akt/Bcl-2/Bax/Cyto-C/Caspase-3,9 signaling pathway 3 hours after its administration, following MCAO. Its efficacy on oxidative stress was indicated by superoxide level reduction, both in the middle (by 56.1%) and high (77.4%) doses. Additionally, this formulation also inhibited astrocyte overactivation, microglia polarization towards pro-inflammatory phenotype, and NF-κB/TNF-α/IL-6 signaling pathway. Borneol treatment also alleviated the neurobehavioral score, similar to that of Edaravone, a commonly used pharmacological agent for ischemic stroke. |
| Zhang et al., 2023 [7] | Experimental in vivo and in vitro study | Male C57BL/6J mice subjected to transient MCAO (tMCAO) and treated with small interfering ribonucleic acid (siRNA) and STIM1 knockout | tMCAO induced the deterioration of autophagic flux in the neurons through STIM1 protein level increase (1.83 ± 0.13 vs. sham 1.00 ± 0.02, p < 0.05) by stimulating SOCE and inhibiting AKT/mTOR pathway. STIM1 downregulation through siRNA administration inhibited autophagic activity, and additionally, STIM1 knockdown specimens exhibited greater improvement of neurological deficits following tMCAO. To evaluate the role of the AKT/mTOR pathway, STIM1-knockout mice were treated with AKT/mTOR inhibitor, AZD5363, which exacerbated infarct volume (39.33 ± 4.04% vs. Cap 22.33 ± 5.51%, p < 0.05) and brain edema (23.58 ± 2.43% vs. Cap 15.06 ± 1.53%, p < 0.05). |
| Zhang et al., 2025 [8] | Experimental in vivo and in vitro study | Sprague-Dawley rats subjected to MCAO and treated with intra-arterial hypothermia infusion solution containing magnesium sulfate (MgSO4; IA-SCMI) | IA-SCMI conferred better infarct reduction and the greatest CBF following MCAO compared to intra-arterial selective cooling saline infusion (IA-SCSI). In vitro analysis revealed that MgSO4 and hypothermia resulted in greater cell viability after oxygen and glucose deprivation through inhibition of calcium overload and reactive oxygen species (ROS) production. Additionally, MgSO4 and hypothermia alone exhibited benefits on cell viability. |
| Zhao et al., 2022 [9] | Experimental in vivo study | Adult Sprague-Dawley rats subjected to MCAO and ischemia/reperfusion, treated with moderate ethanol-preconditioning (EtOH-PC) | EtOH-PC 24 hours prior to I/R conferred neuroprotection and increased the calcium-sensitive potassium (BKCa) channels both in the penumbra and infarct core. EtOH-PC also inhibited apoptosis, reduced infarct size, and improved neurological function. |
CBF: cerebral blood flow; HIF1A: hypoxia-inducible factor 1-alpha; OXR1: oxidation resistance 1; IA-SCMI: intra-arterial selective cooling magnesium infusion.
Across all studies, calcium modulation strategies demonstrated consistent neuroprotective effects. These included reduced infarct volume, decreased expression of apoptosis markers (e.g., cleaved caspase-3), improvement in neurological scores, and restoration of intracellular calcium balance.
Key molecular pathways implicated in these effects included phosphoinositide 3-kinase (PI3K)/AKT signaling, caspase-dependent apoptosis, STIM1/ORAI1-mediated store-operated calcium entry (SOCE), and calcium-sensor proteins such as NCKX2 and hippocalcin.
Combination therapies, such as magnesium sulfate with intra-arterial hypothermia or nanomedicine-based delivery, further enhanced neuroprotection in some models.
The methodological quality of the included studies was generally moderate. Risk of bias was assessed using the ROBINS-I. Most studies had low risk in domains such as outcome measurement and classification of interventions. However, some studies showed concerns related to selection bias, lack of randomization details, or potential confounding. Each domain was classified as Low, Moderate, Serious, Critical, or No information risk of bias.
A full summary of the risk of bias evaluation is provided in Table 2.
ROBINS-I risk of bias assessment for included studies.
| Study | Bias due to confounding | Bias in selection of participants | Bias in classification of interventions | Bias due to deviations from intended interventions | Bias due to missing data | Bias in measurement of outcomes | Bias in selection of the reported result | Overall risk of bias |
|---|---|---|---|---|---|---|---|---|
| Cuomo et al., 2022 [1] | Moderate | Low | Low | Low | Low | Low | Moderate | Moderate |
| Dhyani et al., 2023 [2] | Serious | Moderate | Moderate | Low | Moderate | Low | Moderate | Serious |
| Park et al., 2024 [3] | Moderate | Low | Low | Low | Low | Low | Low | Moderate |
| Secondo et al., 2019 [4] | Moderate | Low | Low | Low | Low | Low | Moderate | Moderate |
| Tang et al., 2015 [5] | Moderate | Moderate | Low | Moderate | Moderate | Low | Moderate | Serious |
| Wang et al., 2025 [6] | Serious | Low | Moderate | Low | Low | Moderate | Serious | Serious |
| Zhang et al., 2023 [7] | Moderate | Low | Low | Low | Low | Moderate | Moderate | Moderate |
| Zhang et al., 2025 [8] | Serious | Low | Moderate | Moderate | Low | Moderate | Serious | Serious |
| Zhao et al., 2022 [9] | Moderate | Low | Low | Low | Low | Low | Moderate | Moderate |
This systematic review provides robust preclinical evidence that calcium (Ca2+) dysregulation plays a pivotal role in the pathophysiology of ischemic stroke. Targeted calcium modulation strategies, including interventions such as ischemic preconditioning, L-type calcium channel blockers, antioxidants (e.g., EGCG, borneol), and magnesium sulfate, have demonstrated significant neuroprotective benefits. These interventions mitigate intracellular calcium overload, oxidative stress, infarct volume, and neuronal apoptosis [10], leading to improved neurobehavioral outcomes. The underlying mechanisms involve key molecular pathways, including PI3K/AKT signaling, caspase-dependent apoptosis, STIM1/ORAI1-mediated SOCE, and calcium-sensor proteins like NCKX2 and hippocalcin [11]. Genetic silencing of key calcium-regulatory pathways, such as STIM1 and NCKX2, has been shown to exacerbate neurological damage, reinforcing their causal role in stroke pathophysiology. Furthermore, combination therapies and novel delivery approaches, including nanomedicine formulations, have demonstrated enhanced therapeutic efficacy in experimental models [1–7].
Our findings align with established literature indicating that calcium overload is a major contributor to secondary neuronal injury following cerebral ischemia [2, 3, 10, 11]. Early clinical studies investigating calcium channel blockers like nimodipine yielded limited success in treating acute ischemic stroke, likely due to non-specific blockade, poor timing of administration, and adverse hemodynamic effects [12]. However, recent preclinical research has demonstrated more selective and effective calcium modulation strategies [4].
Notably, newer studies have incorporated the role of calcium signaling in glial cells and immune responses, including factors such as hypoxia-inducible factor 1-alpha (HIF1A) and pathways of endoplasmic reticulum stress [e.g., GRP78 (78-kDa glucose-regulated protein)], adding mechanistic depth to the understanding of ischemia-induced calcium toxicity [13, 14]. The use of dual-target or combinatory strategies, such as magnesium sulfate with hypothermia or siRNA with pathway inhibitors, reflects current trends in translational neuroprotection research, which increasingly favors multimodal approaches over single-agent therapies [6, 9].
A major strength of this review is its focus on long-term outcomes and mechanistic insights from recent preclinical models. The inclusion of both in vitro and in vivo studies provides a comprehensive view of calciumʼs neurobiological role across different experimental contexts. The use of a rigorous methodology, including standardized bias assessment, enhances the reliability of the findings.
However, several limitations exist. The heterogeneity of stroke models, intervention protocols, and outcome measures complicates direct comparisons across studies. Due to these variations, a meta-analysis was not feasible, as differences in study designs, animal models, interventions, and follow-up periods made it difficult to combine the data in a meaningful way. As a result, a qualitative systematic review approach was used to synthesize the findings.
Moreover, despite focusing on long-term outcomes, many included studies still emphasized acute-phase results without extended behavioral follow-up. The exclusive reliance on preclinical data also limits the generalizability of the findings to human stroke patients. Lastly, potential publication bias and incomplete reporting in some studies cannot be excluded.
The preclinical evidence presented herein suggests that targeting calcium signaling pathways holds promise as an adjunctive approach for ischemic stroke management. Interventions such as calcium channel blockers, magnesium sulfate, and natural antioxidants may complement existing reperfusion therapies by mitigating delayed neuronal damage [7, 12]. However, translating these findings into clinical practice necessitates careful consideration of several factors:
Timing and dosage: Determining the optimal timing and dosage of calcium-targeted interventions is crucial to maximize neuroprotective effects while minimizing potential adverse outcomes.
Patient stratification: Identifying patient populations that would benefit most from calcium modulation therapies requires further research into biomarkers and individualized treatment strategies.
Safety and efficacy: Comprehensive clinical trials are needed to assess the safety and efficacy of calcium-targeted therapies in human subjects, considering the variability in stroke subtypes and individual patient characteristics.
Future studies should prioritize clinical validation of calcium-targeted therapies, particularly in combination with anti-inflammatory or antioxidant strategies. Longitudinal studies assessing functional outcomes beyond the acute phase are urgently needed. Research should also investigate calcium’s role in glial activation, immune modulation, and synaptic remodeling to capture the full spectrum of post-stroke neurobiology. Lastly, the development of calcium-related biomarkers may enable early diagnosis, patient stratification, and personalized treatment strategies [15–17].
Calcium (Ca2+) dysregulation is a central mechanism underlying neuronal injury in ischemic stroke, contributing to excitotoxicity, oxidative stress, and apoptosis. This systematic review underscores consistent preclinical evidence demonstrating that calcium modulation, through agents such as L-type calcium channel blockers, magnesium sulfate, antioxidants (e.g., EGCG, borneol), ischemic preconditioning, and genetic modulation, can confer long-term neuroprotective effects in experimental stroke models.
These interventions appear to reduce infarct volume, preserve neuronal integrity, and improve functional recovery by modulating key molecular pathways, including PI3K/AKT, STIM1/ORAI1-mediated SOCE, caspase-dependent apoptosis, and calcium-sensor proteins like NCKX2 and hippocalcin. Moreover, combination therapies and nanomedicine-based delivery approaches have shown potential to further enhance treatment efficacy, offering promising avenues for improving neuroprotective strategies [14].
Despite these promising findings, clinical translation remains limited due to several factors, including a lack of standardized outcome measures, heterogeneous experimental designs, and a general lack of human data. Moreover, variations in animal models, intervention protocols, outcome measures, and follow-up periods prevented the conduct of a meta-analysis. As a result, we used a qualitative systematic review approach to synthesize the available evidence and present key trends in calcium modulation for ischemic stroke recovery.
Future research should prioritize clinical validation of calcium-targeted therapies, focusing on standardized protocols and the exploration of calcium-based biomarkers [18]. These efforts could guide the development of personalized stroke recovery therapies. Longitudinal human studies, including large-scale clinical trials, are urgently needed to assess the safety, efficacy, and optimal treatment windows for these therapies.
In conclusion, calcium modulation represents a promising strategy for adjunctive neuroprotection in ischemic stroke. As the field transitions towards multimodal and personalized stroke management, calcium-targeted therapies deserve increased attention in both translational and clinical research, offering hope for improving stroke recovery outcomes and patient quality of life.
EGCG: epigallocatechin gallate
MCAO: middle cerebral artery occlusion
PI3K: phosphoinositide 3-kinase
ROBINS-I: Risk of Bias in Non-randomized Studies of Interventions
SOCE: store-operated calcium entry
STIM1: stromal interaction molecule 1
We express our sincere gratitude to our supervisors for their invaluable guidance during the preparation of this systematic review. We also thank the library staff for facilitating access to relevant databases and literature. Lastly, we appreciate the support of our colleagues and families throughout this process.
PKS: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. RMJ: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. SAS: Validation, Writing—review & editing, Supervision. AD: Data curation, Methodology, Visualization. ADG: Formal analysis, Writing—review & editing, Project administration. BBB: Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
The datasets that support the findings of this study are available from the corresponding author upon reasonable request.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Said Hachimi-Idrissi
Shafiq Dexter B. Abou Zaki, Johnny K. Lokin
Yang Yang ... Yi Li
Lidija Radenovic
Renata Murguiondo-Pérez ... Antonio Ibarra
