 Review
Review

Affiliation:
Pain Medicine Unit, Department of Mini-Invasive Surgery, ICS Maugeri IRCCS, 27100 Pavia, Italy
Email: laura.demartini@icsmaugeri.it
ORCID: https://orcid.org/0000-0002-3343-859X
Affiliation:
Pain Medicine Unit, Department of Mini-Invasive Surgery, ICS Maugeri IRCCS, 27100 Pavia, Italy
ORCID: https://orcid.org/0000-0001-9330-2531
Explor Neurosci. 2025;4:100686 DOI: https://doi.org/10.37349/en.2025.100686
Received: January 01, 2025 Accepted: April 09, 2025 Published: April 27, 2025
Academic Editor: Dirk M. Hermann, University of Duisburg-Essen, Germany
The article belongs to the special issue Neuropathic Pain
Neuropathic pain, defined by the International Association for the Study of Pain as “pain caused by a lesion or disease of the somatosensory system”, has an estimated prevalence of 7–9.2% in the general population and is associated with poorer health-related quality of life than other types of pain. Diagnosis can be improved by the use of diagnostic algorithms, but treatment remains rather unsatisfactory, with only 30–40% of patients achieving an acceptable response. Some authors have suggested that the poor results in the treatment of neuropathic pain may be related to the different mechanisms present in each patient and have tried to correlate them with clinical characteristics in order to evaluate possible targeted treatments. This approach has been used in some studies evaluating the response to specific pharmacotherapies in clusters of patients, with encouraging results but still limited applicability to clinical practice. In this narrative review, we attempt to analyse the literature suggesting possible pathogenetic mechanisms manifested along the nociceptive pathway due to a lesion or disease of the nervous system; aware of the limitations of exploring such a wide field, we look for conditions that could be targeted by the available pharmacological or interventional treatment options. Functional changes may occur in the nociceptive system from the periphery to the cerebral cortex, in particular in the nociceptive terminals, along the first-order neuron and the dorsal root ganglion, at the first synapses, or at supraspinal levels. Clinical assessment is the first step in the study of anatomical and functional changes; the diagnostic hypothesis should be confirmed, if possible, by instrumental studies or diagnostic blocks or procedures to guide an individualised therapeutic algorithm from less to more invasive treatments.
Neuropathic pain was defined by the IASP Neuropathic Pain Special Interest Group in 2011 [1] as “pain caused by a lesion or disease of the somatosensory system”, modifying previous definitions from 1994 and 2008. It is important to distinguish neuropathic pain from pain resulting from damage to other tissues of the body, nociceptive pain, or pain due to dysfunction of the pain modulating system, nociplastic pain [2], because although different types of pain may share some form of plastic changes in the functionality of the nociceptive system, neuropathic pain is sustained by specific mechanisms that should be identified and potentially addressed by therapeutic strategies.
Neuropathic pain is included in the WHO International Classification of Diseases and Related Health Problems (ICD-11) [3], where it is classified according to the site of the lesion (peripheral or central), the cause (e.g., nerve lesion, neuropathy, radiculopathy, postherpetic neuralgia, spinal cord injury, trauma, stroke, multiple sclerosis), and more precisely according to intensity, interference, temporal patterns and psychosocial factors. Neuropathic pain has an estimated prevalence of 7–9.2% in the general population and is associated with poorer health-related quality of life than other types of pain [4, 5].
To facilitate the diagnosis of neuropathic pain for clinical and research purposes, the Neuropathic Pain Special Interest Group (NeuPSIG) of the IASP proposed a grading algorithm in 2008 [6], which was revised in 2016 [7]. The advantage of the algorithm is that it outlines the steps towards a definitive diagnosis:
The patient’s clinical history and the distribution of the pain in a neuroanatomically plausible area may suggest possible neuropathic pain;
The presence of sensory changes in the same territory defines probable neuropathic pain;
Instrumental diagnostic tests demonstrating a lesion of the somatosensory system confirm the diagnosis of definite neuropathic pain.
There are a few exceptions where the grading system is not useful: the principal is trigeminal neuralgia, a characteristic neuropathic pain in which it is often impossible to identify sensory deficits, and there is no specific diagnostic test. Other challenging neuropathic pain syndromes include some forms of peripheral nerve entrapment, such as radial tunnel syndrome involving the deep branch of the radial nerve, where electromyography (EMG) and nerve conduction studies (NCS) are often negative and the only sign is deep pressure allodynia, which is also seen in tendinitis [8].
Neuropathic pain may be present in a percentage of patients with neurological diseases, but it is still unknown why patients with the same disease can report no pain or pain with different characteristics: a typical example is herpes zoster with post-herpetic neuralgia. Regardless of the aetiology, neuropathic pain conditions share clinical characteristics, suggesting common underlying mechanisms and different therapeutic options, from drugs to neuromodulation techniques, are generally recommended for neuropathic pain in general [9].
On the other hand, the efficacy of treatments for neuropathic pain is not yet satisfactory; recent meta-analyses of pharmacotherapy have shown that only 30–40% of patients have an acceptable response (30–50% pain reduction) compared with placebo [10]. The number needed to treat (NNT), a measure of drug efficacy, has increased in more recent trials compared with earlier ones because of differences in study design; for example, pregabalin had an NNT of 4 (25% of patients responding to the drug) in a 2004 trial and 9–12 in 2017 trials [10]. Non-pharmacological, non-invasive or interventional treatments, from peripheral nerve blocks to spinal cord or motor cortex stimulation (MCS), have low or inconclusive recommendations for neuropathic pain, mainly due to the lack of placebo-controlled trials [11].
The poor efficacy of proposed treatments for neuropathic pain may be related to the different underlying mechanisms present in each patient, and some authors have suggested hypothesising the different mechanisms based on the clinical characteristics of patients (phenotypes) and evaluating possible targeted treatments [12]. In particular, according to quantitative sensory test data, patients can be grouped into three main phenotypes [13]:
Thermal hyperalgesia: relatively preserved large and small fibre sensory function combined with heat and cold hyperalgesia and only low intensity dynamic mechanical allodynia (DMA). Persistent hyperactivity in surviving nociceptors may be responsible for persistent pain and may lead to some degree of central sensitisation in the dorsal horn.
Mechanical hyperalgesia: predominant loss of cold and heat sensitive small fibre function in combination with blunt pressure hyperalgesia, pinprick hyperalgesia, and marked and more frequent DMA. Central sensitisation to mechanical stimuli is prominent. Persistent pain in this subgroup indicates spontaneous activity of the nociceptive system, which may originate from the peripheral and/or central nervous system.
Sensory loss: loss of small and large fibre function, possible presence of paradoxical heat sensation. Spontaneous pain is likely to be due to ectopic action potentials generated at proximal sites of injured nociceptors, such as the dorsal root ganglion (DRG) or deafferented central nociceptive neurons.
This approach has been used in some studies to evaluate the response to specific pharmacotherapies in groups of patients with different sensory profiles; the results are encouraging, but at present they have limited implications for clinical practice [14].
In this narrative review, we attempt to analyse the possible mechanisms present in different neuropathic pain conditions in order to suggest rational, if not evidence-based, treatments.
Clinical assessment is the first step in diagnosing the origin and pathophysiological mechanisms of pain. The medical history can provide information about specific events that precipitated the symptoms, such as trauma or surgery, systemic diseases that may be related to nerve damage (e.g., diabetes, hypothyroidism), or known neurological pathologies [6].
Pain distribution is important to suggest the possibility of neuropathic pain, but also to diagnose other types of pain. It is recommended to draw the pain distribution on the patient’s body or on a body image, taking into account some possible differences in the use of the two methods; the body image representation implies some psychological aspects [15]. The distribution of pain must lead the clinician to hypothesise possible causes of localised, referred, or radiating pain. Patient-reported symptoms, pain descriptors, and other sensitive abnormalities such as paresthesias or dysesthesias are more typical of neuropathic pain but are not specific. Patients’ symptoms can be analysed using standardised questionnaires, which help to suggest a diagnosis of neuropathic pain and monitor response to treatment over time. Useful tools for diagnosing neuropathic pain include the Douleur Neuropathique en 4 questions (DN4) [16] and the Leeds Assessment of Neuropathic Symptoms and Signs (LANSS) [17], which include a brief examination, and the painDETECT [18]. The Neuropathic Pain Scale (NPS) [19] and the Neuropathic Pain Symptom Inventory (NPSI) are helpful in characterising patients and grouping them into phenotypes [20].
Other important information is the temporal pattern of the pain (continuous, intermittent), whether it is spontaneous or evoked, and the provoking stimuli or activities, the interference with daily life, and sleep. Some typical examples are painful attacks induced by mechanical stimuli (washing the face, shaving, brushing the teeth, eating, talking) in trigeminal neuralgia and the disappearance of pain during sleep in central pain and some forms of peripheral neuropathic pain. Radicular pain is often provoked by standing or walking, but is sometimes present or worse when lying down.
A thorough neurological evaluation is fundamental for looking for sensory but also motor abnormalities.
Different sensory modalities are assessed using mechanical and thermal stimuli to determine the functionality of Aβ, Aδ, and C-fibres, the lemniscus pathway, and components of the spinothalamic tract. A gain or loss of function can be documented using simple and inexpensive stimuli in the painful area and a comparable healthy area of the body. Reimer et al. [21] suggest a battery of instruments, including a metal piece, a Q-tip, a cotton swab, a brush, a tuning fork, a 64 mN Von Frey hair, a 0.7 mm/0.4 mm Chicago Medical Supply (CMS) hair, a 10 mL syringe, but also simpler instruments such as a cotton swab or brush for mild mechanical sensation and DMA, the fingertip for deep mechanical allodynia or hyperalgesia, a metal piece (the tuning fork can be used) for cold sensation and cold allodynia, a tube filled with warm water or a hand warmer for warm sensation and allodynia. Pain is carried through Aδ and C-fibres, the function of which can be inferred from sensitivity to thermal stimuli. If the bedside assessment documents a loss of function of specific modalities and the distribution suggests the possible site of the lesion (peripheral symmetric, peripheral asymmetric, radicular, various spinal patterns, trunk or supratentorial lesion), the more appropriate confirmatory tests will be prescribed. As neurophysiological tests are specific to the diagnosis of certain sensory or motor functions, it is important that the clinician visiting pain patients has a sound neurological background. A detailed description of the diagnostic tools to confirm neuropathic pain can be found in the work of De Stefano and Truini [22].
It is essential to understand the aetiology of the neurological lesion or disease in order to determine whether the cause can be removed with possible recovery: typical examples are entrapment syndromes, with a success rate of 75–90% in long-term carpal tunnel release studies [23], and radiculopathy due to herniated discs, which may heal spontaneously or improve after specific decompressive treatments. Even some forms of polyneuropathy may improve over time, such as diabetic polyneuropathy with improvement in glycaemic control or chemotherapy-induced polyneuropathy, which resolves in two-thirds of patients after six months [24].
Clinical assessment should help to stratify patients into phenotypes in order to determine, together with the aetiological diagnosis, the site of the lesion and the pathophysiological mechanisms involved. For example, in radicular pain, the lesion site is close to the DRG, but pathophysiological changes may occur at the DRG level with hyperexcitability to mechanical stimuli and spontaneous activity [25] and at the dorsal horn level [26]. In the case of sensory loss, clinical assessment is not always sufficient to determine whether the lesion is distal or proximal to the DRG, creating completely different conditions: in the first case, the spinal second-order neuron still receives afferences from the DRG, whereas in the second case it is deafferented and its spontaneous activity can cause pain, and this condition should be documented with specific neurophysiological or imaging tests [27].
Neuropathic pain is characterised by abnormal ectopic activity at various sites in the peripheral and central somatosensory pathways, particularly in the spinothalamic pathway. From the periphery, some of the possible pathophysiological mechanisms (Figure 1) are:
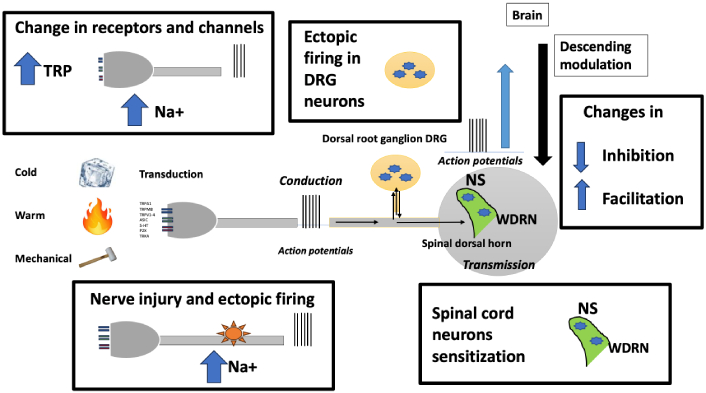
Sites of functional changes in the case of lesions of pain pathways. TRP: transient receptor potential; DRG: dorsal root ganglion; NS: nociceptive specific; WDRN: wide dynamic range neurons
1.Changes in the expression of ion channels and receptors in spared nerve terminals.
2.Ectopic discharges at the site of the lesion in fibres and associated ganglia with chemical and mechanical hypersensitivity.
3.Neuroimmune interactions leading to increased and/or altered production of inflammatory molecules that increase excitability.
4.Sensory-sympathetic coupling and other changes in receptor signalling [28].
5.Functional disruption of inhibitory interneurons and glial activation in the dorsal horn of the spinal cord, leading to reorganisation of neuronal circuits and changes in the excitability of second-order neurons [29].
6.Changes in trunk modulatory activity and thalamocortical networks that develop autonomous pain maintenance processes [30].
Identifying the site of the lesion and the possible sites of functional changes is important for determining therapeutic strategies; clinical assessment and instrumental studies can confirm a possible peripheral or central lesion, but are not able to give us information about the degree of plasticity of the nervous system. For example, in the case of a traumatic peripheral nerve lesion, the lesion site can be documented by conduction velocity study and nerve sonography [31], but specific changes in activity in rostral sites of the pain pathway cannot be documented. Functional neuroimaging studies using positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) have shown various abnormalities in the central nervous system, but no specific biomarker for neuropathic pain [22]. Some functional changes have been shown to be reversible once effective treatment improves pain control [22].
In nociceptors present in the skin (or deep tissues), under pathological conditions, such as peripheral nerve or nerve root lesion, changes may occur in ion channels and receptor expression [32]. Changes have been demonstrated in transient receptor potential vanilloid 1 (TRPV1) channels on the terminal surface, leading to hyperexcitability to heat but also participating in mechanical and cold-induced neuropathic pain [33]. The role of transient receptor potential melastatin 8 (TRPM8) is more ambiguous, as its activation promotes cold hypersensitivity, but decreases mechanical hypersensitivity [33]. In diabetes-related neuropathic pain, TRPV1 channels are stimulated by hyperglycemic and hypoxic conditions, inducing heat and mechanical hypersensitivity. Transient receptor potential ankyrin 1 (TRPA1) receptors are also directly stimulated by a diabetes by-product, methylglyoxal (MG), which leads to mechanical hyperalgesia. Recent in vitro evidence suggests a similar role for TRPM8 [33]. In chemotherapy-induced neuropathic pain, protease-activated receptor 2 (PAR2)-mediated TRPV1 sensitization has been implicated in heat and mechanical hypersensitivity mediated by chemotherapeutic drugs. TRPA1 activation participates in both mechanical and cold hypersensitivity, whereas TRPM8 has a major role in cold hypersensitivity [33].
Na+ channels are located at the coding site [34] (where the action potential is generated), along nerve fibres and on the DRG; their overexpression leads to hyperexcitability to various stimuli [35]. The voltage-gated sodium channel Nav1.7 is expressed selectively in sensory and autonomic neurons; inactivating mutations in sodium voltage-gated channel alpha subunit 9 (SCN9A), which encodes Nav1.7, result in congenital insensitivity to pain, whereas gain-of-function mutations in this gene produce pain syndromes such as erythromelalgia, paroxysmal extreme pain disorder, and small-fibre neuropathy [36]. Nav1.8 is involved in peripheral sensitization due to inflammatory conditions, and it has been demonstrated that an injury-induced redistribution of Nav1.8 to uninjured axons leads to hyperexcitability in animal models [37]. There is evidence of a role for Nav1.9 in inflammatory, diabetic neuropathy, and orofacial neuropathic pain [37].
In case of peripheral nerve lesion, an immune response is activated in order to repair the damage; the inflammatory mediator cytokines such as IL-1b, TNF-α, together with peptides released from the nerve terminals, substance P (SP) and calcitonin gene related peptide (CGRP), sensitize and stimulate nociceptors generating hypersensitivity to stimuli (allodynia) or spontaneous pain [38]. The inflammatory response is functional to nerve repair; therefore, the treatment should modulate and not suppress inflammation [38]. Histamine, released by mast cells, has been shown to contribute to hypersensitivity in peripheral nerve injury models [39].
Clinically, alterations of TRPV and Na+ channels and peripheral neurogenic inflammation present with the “irritable nociceptors” [32] or “thermal hyperalgesia” [12] phenotype, characterized by the prevalence of positive signs of all sensory modalities. This clinical presentation is often present in polyneuropathies and ganglionopathies (e.g., postherpetic neuralgia).
When a peripheral sensitization due to overexpression or overactivity of TRPV1 receptors is hypothesized, topical capsaicin is a treatment option. It can be applied in cream from 0.025% to 0.1% more times a day or in 8% plasters every three months [40]. High-concentration capsaicin induces a long-term defunctionalisation of TRPV1 receptors, through the ablation of TRPV1-expressing axon terminals induced by a calcium-dependent protease, which is followed by regeneration [41]. Capsaicin demonstrated a disease-modifying effect in some forms of neuropathic pain [42].
In order to target Na+ channels, lidocaine 5% patches can be used and have been demonstrated to be effective for pain paroxysms and deep aching pain [43]. Systemic oxcarbazepine, an antiepileptic drug acting on Na+ channels has been demonstrated to be more efficacious for relief of peripheral neuropathic pain in patients with the irritable vs. the non-irritable nociceptor phenotype [44]. Another Na+ channel blocker, lacosamide, failed to demonstrate efficacy in a population of peripheral neuropathic pain patients [45]. An old drug, ambroxol, used for its antitussive or anti-secretory effects, has been demonstrated to block Nav1.7 and Nav1.8 channels and a subtype of Ca channels with anti-inflammatory benefits and some effect on neuropathic pain symptoms in animal models of neuropathic pain and some case reports as a topical formulation [46]. Given the involvement of Na+ channels in pain perception, several blockers are being studied; recently, a new drug, suzetrigine, a potent and selective Nav1.8 inhibitor, has been approved by the FDA for acute pain. Suzetrigine binds to a specific site on the voltage-sensing domain 2 (VSD2) of Nav1.8, stabilizing the channel in the closed state [47]. Clinical studies in models of acute pain demonstrated efficacy compared to placebo, similar to opioids; interestingly, central nervous system or cardiovascular side effects were not observed at effective dosage [47]. The actual indication is acute pain, but the characteristics of the drug induce to considering it promising for neuropathic pain.
The possible role of histamine receptor antagonism in the treatment of neuropathic pain has been assessed in a recent review [39]: the authors present a possible role of different histamine antagonists on the symptoms of neuropathic pain; particularly interesting is the analgesic effectiveness of H3 receptor antagonists/inverse agonists, mainly on mechanical and cold allodynia.
Botulinum toxin (BTX), usually used to reduce spasticity, has been studied for the relief of pain, not only related to muscle hyperactivity, in particular for neuropathic pain. BTX acts by inhibiting the release of Ach at the neuromuscular junction, and also SP, CGRP, and glutamate, which are involved in neurogenic inflammation at the nerve endings [48].
Various forms of electrical stimulation applied to the skin or just beneath the skin have been proposed and are used to reduce pain and improve function [49]. Different electrical parameters can be delivered to the painful anatomical target through the skin, transcutaneous electrical nerve stimulation (TENS), through percutaneous temporary leads, percutaneous electrical nerve stimulation (PENS), or through implantable leads, peripheral nerve field stimulation (PNFS); unfortunately, the evidence for effectiveness is poor mainly due to the lack of standardization for each treatment [49].
Neuromodulation treatments applied to the nerve trunk or on the DRG can also modify the functionality of peripheral nerve endings.
A lesion along the axon can be caused by compression in entrapment syndromes, by trauma, or by surgery. According to the type and quantity of fibers involved, clinical presentation can be grouped in the “thermal hyperalgesia”, “mechanical hyperalgesia” or “sensory loss” phenotypes [13]; it is important to understand that in case of sensory loss we should consider denervation and not deafferentation since an activity in the peripheral axon and DRG is still transmitted to the second order neuron.
Ectopic sites can develop in the areas of axonal lesion, in regenerating sprouts and neuromas (terminal or in continuity), in the site of demyelination, and in DRG cell bodies [50]. An altered distribution of voltage-dependent Na+ channels has been demonstrated in the demyelinated segments of the membrane. The newly expressed channels include Nav1.7, Nav1.3, a channel normally expressed only in sensory neurons during embryonic development, and Nav1.8 [51]. Combined with a decrease in K+ channels and an increase in nonselective hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, these changes can result in lowered depolarization threshold with the possibility of ectopic discharge, spontaneous or evoked by mechanical and chemical stimuli, and enhancement of action potential transmission through the demyelinated segment [52].
Pain is always radiated in the peripheral innervation territory, and it is not perceived at the site of injury. The frequency and type of discharge can be different from case to case: they vary from very slow (< 1 pulse/second) to very high (> 100 pulses/second). Some afferents discharge single pulses (spikes) at a low frequency with an irregular rhythm, others discharge trains of pulses at a regular rhythm in sequences (bursts) [53].
Ectopic sites can be generated on any fiber type, and the sensation varies from fiber to fiber, such as burning or constriction for unmyelinated C-fibers, electric discharge for myelinated Aδ fibers, and paresthesias for myelinated Aβ fibers. In physiological and pathological nociceptive pain, only C fibers and Aδ are involved in the generation of action potentials from tissue terminals, but in neuropathic pain, tactile and proprioceptive Aβ fibers are also involved. In neuropathic pain, the encoding of the message at the site of nociceptive terminals that characterizes nociceptive pain does not occur, therefore, the perceived sensation does not correlate with the type of stimulus but with the type of fibers involved [28]. Ectopic impulses can generate spontaneously and/or be evoked by stimuli of various kinds (e.g., Tinel’s sign).
The spontaneous origin of the potential is a function of the degree of subthreshold high-frequency oscillation of the membrane potential of the ectopic site [52]. Ectopic spikes can also originate from healthy fibers close to those involved by the injury, especially from C-fibers [54].
In some conditions of neuropathic pain, the involved nerve fibers present a rest period after the discharge; this feature has been shown in animals, and is typical in patients with trigeminal neuralgia, where the ectopic site is proximal to the ganglion [55].
In addition to ion channels and receptors, altered expression, accumulation of infiltrating immune cells such as neutrophils, macrophages, and mast cells at the site of nerve injury contribute to axonal hyperexcitability and sustained electrical discharge in the majority of neuropathic conditions [56].
Axonal sensitivity to various types of inflammatory mediators, such as bradykinin (BK), E2 prostaglandins, acid pH, and capsaicin, has been demonstrated [57, 58]. Neuroma sprouts present mechano-sensitivity and thermo-sensitivity [57, 59].
As inflammation and mechanical stimuli contribute to the genesis of pain in peripheral nerve lesions, it is important to diagnose the presence of entrapment and/or inflammatory processes in order to resolve the hyperexcitability phenomenon due to hypoxia and inflammatory mediators.
If we consider carpal tunnel syndrome, a typical entrapment syndrome, the literature on the use of Na+ channel blockers is poor; symptoms may be improved by corticosteroid injections [60], which act on inflammatory mediators and oedema contributing to compression. Symptoms of carpal tunnel syndrome may be relieved by splinting, which limits range of motion and thus mechanical stimulation [61]. Surgery is often curative [23]. Reconstructive surgery has been proposed for neuromas with significant results [62].
If surgery is not an option, the first step is pharmacological therapy targeting altered Na+ channel activity. In addition to the antiepileptic drugs that act on Na+ channels, such as carbamazepine, oxacarbazepine, lacosamide, topiramate, and lamotrigine, some antidepressants have shown inhibitory activity on Na+ channels expressed on nerve fibres, particularly on the DRG, with amitriptyline being the most potent inhibitor [63]. Carbamazepine is the drug of choice for trigeminal neuralgia [10].
TENS has been shown to modulate action potential propagation when applied to a peripheral nerve [64] and may therefore be an option in the treatment of peripheral neuropathic pain [11]. Peripheral nerve stimulation (PNS) with implanted leads and temporary leads (PENS) has shown positive results for postoperative neuropathic pain [65] and may share the same mechanism of action as TENS. Other neuromodulation techniques, such as cryoneurolysis or pulsed radiofrequency, are commonly used for peripheral neuropathic pain, but evidence of their efficacy is currently lacking. Cryoneurolysis, through the application of intense cold to a specific nerve (approximately −70°C), creates a reversible injury to the nerve fibers: the duration and degree of tissue injury depend on the time of application and probe dimension [66]. Pulsed radiofrequency applied proximal to a nerve lesion has been demonstrated to reverse hypersensitivity through an epigenetic mechanism [67].
Dorsal root neurons show altered activity both in diseases directly affecting the ganglion (e.g., radiculopathy due to disc herniation, herpes zoster) and in diseases affecting the axons. Ganglion cells have many important roles in the physiology of nociception: they nourish the axons and are involved in metabolic diseases causing polyneuropathy; they produce ion channels, receptors, and chemical mediators involved in pain transduction, transmission, and inflammatory processes; they regulate action potential propagation [68].
Clinically, DRG dysfunction can present with signs and symptoms of all three phenotypes. In the case of sensory loss due to disease affecting the ganglion, the pathophysiological mechanism is deafferentation [26]. Mechanosensitivity of ganglion cells is well known in herniated disc compression injuries and is characterized by prolonged discharges along all afferent sensory axons [69]. Ganglion cells become hyperexcitable following nerve injury and generate spontaneous and evoked action potentials. After peripheral nerve injury, there is a reorganization of ion channel expression in DRG neurons. Regarding Na+ channels, some are reduced, some are newly expressed, and some are redistributed to different parts of the neuron [70]. Indeed, an increase in tetrodotoxin-sensitive (TTX-s) Nav1.3 (not normally expressed by ganglion cells) and Nav1.7 channels was observed, together with a reduction in TTX-r (resistant) Nav1.8 and Nav1.9 channels [71]. The increase in Nav1.3 leads to hyperexcitability of the cell and the onset of ectopic discharges. A redistribution of Nav1.8 channels from the cell body to the axon may explain the increased sensitivity of neuromas [72].
Peripheral nerve injury also causes increased expression of the Ca2+ channel subunits Cav α2-δ1 in DRGs, BK B1 and capsaicin (TRPV1) receptors, and a reduction in B2 receptors, SP, and opioid receptors [73]. A reduction in K+ channels contributes to neuronal hyperexcitability.
Sprouting of sympathetic neurons contributes to the development of ectopic activity. Under normal conditions, ganglion neurons are insensitive to norepinephrine, but after nerve fiber injury, there is an increase in α-adrenergic receptors and the formation of clusters of sympathetic axons around ganglion neurons [74].
In a recent article, Hanani [75] highlights the importance of satellite glial cells (SGCs) that surround DRGs; a lesion to peripheral sensory neurons increases their firing rate, inducing increased production of nitric oxide in the neurons; nitric oxide diffuses from the injured neurons and activates SGCs that surround them and probably adjacent neurons. SGC activation causes increased gap junctional coupling, increased sensitivity to ATP, upregulation of ERK, and increased release of pro-inflammatory cytokines (IL-1β, TNF, IL-6, fractalkine, and others). The increased gap junctions and sensitivity to ATP will enhance Ca2+ waves, leading to neuronal excitation. Cytokines released from SGCs will also contribute to neuronal sensitization and excitation. The overall effect will be increased firing (spontaneous and/or evoked) of sensory neurons and pain.
Using the spinal nerve ligation (SNL) model in rats, it has been shown that cell bodies in the DRG contribute 3 times more to ectopic nerve activity than the site of nerve injury. The DRG is therefore a fundamental target for pain control [73]. Functional changes involve large, medium, and small neurons in both injured and uninjured afferents [76]. Targeted administration of lidocaine to the DRG at sub-anesthetic concentrations is able to temporarily suppress allodynia [77]. Neurons with axotomized peripheral processes present a diminished filtering of action potential (AP) trains at the T-junction of C-type neurons, enhancing the transmission of impulses originating ectopically in a neuroma, possibly contributing to pain generation [78].
The DRG is a fundamental site to determine pain characteristics: a neural lesion distal to the ganglion can cause peripheral neuropathic pain, while a lesion involving the ganglion or proximal to it, if sufficiently extensive, can cause deafferentation pain, a form of central pain [27].
Direct involvement of the DRG by an inflammatory process, such as radiculopathy secondary to disc herniation, can be treated with local anesthetic and steroid injections [79, 80]. Surgery can release the nerve root in cases of compression due to disc herniation.
There are no selective drugs targeting plastic changes at the DRG level in chronic pain, but many different approaches have been hypothesized and are under investigation, such as compounds acting on transient receptor potential (TRP) channels, monoclonal antibodies targeting CGRP, NGF, TNF already available for other diseases, bacterial products, gene therapy, stem cells, systemically or topically administered [81].
The DRG is also the target of neuromodulation techniques, in particular DRG pulsed radiofrequency and DRG stimulation. DRG pulsed radiofrequency is widely used for radicular pain due to disc herniation and other painful ganglionopathies, such as post-herpetic neuralgia [82, 83]. The quality of evidence is currently low [82]. DRG stimulation has been shown to be effective for lower extremity complex regional pain syndrome (CRPS), but the evidence is considered to be of low quality. Observational studies and case series show promising results for DRG-S in painful diabetic neuropathy (PDN), focal neuropathy, and polyneuropathy [84].
The spinal nociceptive synapse in the dorsal horn is rarely directly involved in diseases that cause neuropathic pain. Dorsal horn cells can be damaged by infectious (e.g., herpes zoster) or inflammatory diseases, by trauma or ischemia; the central terminal of the first-order neuron can be damaged by avulsion, preganglionic or ganglionic radiculopathy. In all these cases, the most likely phenotype is “sensory loss” with deafferentation pain. The perisynaptic area is an important site of plasticity in all cases of subacute or chronic pain. A synaptic dysfunction clinically presents the characteristics of “mechanical hyperalgesia” but also of “thermal hyperalgesia”.
The state of hyperexcitability of spinal neurons has been widely discussed and attributed to functional changes in the mechanisms involved in transmission and inhibition, but also to truly plastic and therefore persistent changes [85]. Many events can cause changes that alter spinal transmission and create a state of altered processing of afferent impulses or produce pain in the absence of afferent impulses. In neuropathic pain, the excitability of the presynaptic element is influenced by the increased synthesis of neurotransmitters and neuromodulators, the density of Ca2+ and voltage-gated Na+ channels expressed on the membrane [85]. HCN channels contribute to the increased excitability and ectopic firing of peripheral sensory neurons, which is further enhanced by the downregulation of K+ channels [85]. At the same time, processing of afferent messages can be altered or spontaneous activity can be generated at the level of the postsynaptic element.
Aβ fibers, which physiologically transmit harmless sensations and have an inhibitory function, can generate pain either by the establishment of postsynaptic sensitization through release of excitatory mediators [SP, brain-derived neurotrophic factor (BDNF), and cofactor for nitric oxide synthase (NOS)] and creating new spinal cord connections through sprouts from deep to superficial laminae [86].
Changes affecting microglia and astrocytes are much more pronounced and lasting than in inflammatory pain. Microglia release both cytokines and other transmitters and trophic factors (BDNF) that contribute to sensitization and lead to a reduction in inhibitory mechanisms [87].
The inhibitory system changes its functionality. There is a loss of GABAergic inhibitory function and a reduction in glycinergic one, partly due to apoptosis of inhibitory interneurons [88]. Noradrenergic inhibitory activity on α2-adrenoreceptors is lost [89], and serotonergic descending activity becomes facilitatory instead of inhibitory [90, 91]. Opioid receptors reduce their expression and sensitivity [92]. Changes in descending control have been shown in humans by quantifying conditioned pain modulation in neuropathic pain patients [93].
The term “deafferentation pain” is generally used to define pain that results from the interruption of afferent impulses between one neuron and the next in the nerve pathway. Peripheral deafferentation occurs when the lesion affects the root or sensory ganglion but not the peripheral nerve. Central deafferentation pain (or simply central pain), which would theoretically include all forms of deafferentation pain, refers to that which occurs after injury to the central nervous system, spinal and supraspinal pain pathways (e.g., post-stroke pain, syringomyelia, spinal lesions) [94]. In patients with brachial plexus avulsion, the incidence of deafferentation pain is reported to be as high as 90% [95].
The majority of analgesic drugs recommended for neuropathic pain [10] act at the first synapse level.
Antidepressants, acting on the bioavailability of inhibitory mediators, noradrenaline and serotonin, represent one of the first classes of drugs used for neuropathic pain. Tricyclic antidepressants (amitriptyline, nortriptyline, imipramine), serotonin-norepinephrine reuptake inhibitor (SNRI) antidepressants (particularly duloxetine and venlafaxine) are recommended as first line for peripheral and central neuropathic pain [10]. Although the amount of literature favors the use of SNRI, they present an NNT of 6.4 compared to 3.6 for tricyclic antidepressants (TCAs). The number needed to harm (NNH) of TCAs for major adverse effects leading to discontinuation of the drug is 28, and 6 for minor adverse effects (sedation, dizziness, dry mouth, constipation, urinary retention, and headache) [96]. SNRIs have an NNH (sedation or insomnia, headache, and/or decreased appetite or libido) of 11.8 [10]. It is important to emphasize that the analgesic action in patients with neuropathic pain is independent of the antidepressant action.
Gabapentin and pregabalin represent a relatively recent class of anti-epileptics [10]. These drugs, which have become the gold standard for neuropathic pain, have inaugurated a new class of antiepileptics, called the α2-δ ligands. Newer α2-δ ligands, such as mirogabalin, available in Japan, or newer gabapentin formulations (gabapentin extended release) have been subsequently developed [97]. Gabapentin has a strong recommendation for use in neuropathic pain (high quality based on new evidence and trials) [10]. In the French revised recommendations, Moisset et al. [11] downgraded pregabalin as a second-line choice because of modest results from recent double-blind trials and reporting of abuse and misuse (higher than gabapentin).
There is no evidence on clonazepam, a benzodiazepine acting on GABA-B receptors that has been reported effective on pain paroxysms and in cases of night awakening due to pain [11].
Tramadol, a weak opioid acting also on noradrenaline and serotonin reuptake, is recommended as a second-line drug [10]. Because of the opioid crisis, particularly in North America, strong opioids are only used as third-line treatment when first and second-line treatments are ineffective for neuropathic pain [97]. The effective dose of these different opioids is generally higher than the doses used in acute or non-neuropathic chronic pain [97]. The main reason for the limitation of long-term use of opioids is the number of side effects they can induce, mainly dizziness, confusion, nausea, constipation, but also respiratory depression, and more importantly, tolerance and a high risk of substance abuse [98].
Other pharmacologic options considered for neuropathic pain are cannabinoids and oral ketamine, for which there is no clear evidence [11].
The main non-pharmacological option for neuropathic pain acting at the spinal cord level is spinal cord stimulation (SCS); it acts with different demonstrated mechanisms that change the balance of nociceptive and antinociceptive inputs through the activation of segmental and supraspinal pathways [99]. SCS has been used for more than 40 years to treat localized chronic medically refractory neuropathic pain involving limb(s) and trunk. The most frequent indications remain CRPS, failed back surgery syndrome (FBSS), and peripheral neuropathy. The risk/benefit ratio is favorable to SCS, considering that chronic pain patients undergoing this procedure are usually resistant to all the other therapies [100]. A recent review demonstrated a weak GRADE recommendation for the use of conventional SCS in neuropathic pain associated with FBSS and diabetic painful neuropathies (moderate final quality of evidence) [11].
Intrathecal delivery of drugs acting at the synaptic level, mainly morphine and ziconotide, can be considered for neuropathic pain patients not responsive to other therapies, but there is no literature evidence [11].
In addition to the peripheral and spinal mechanisms discussed, supraspinal mechanisms have been shown to play an important role in neuropathic pain.
Descending neuronal pathways from midbrain and brainstem nuclei physiologically modulate pain perception at the level of the spinal cord with a predominant inhibitory effect. In chronic pain, both inflammatory and neuropathic, serotonergic-mediated facilitation may prevail over descending noradrenergic and serotonergic inhibition [85].
Functional plasticity of many components of the so-called “pain matrix”, the network of brain regions activated by painful stimuli, has been demonstrated in chronic pain patients in general. Specific cortical areas are involved in nociception and pain localization (mainly posterior operculo-insular areas), whereas conscious pain is elaborated in a second-order network involving posterior parietal, prefrontal, and anterior insular areas [101]. These regions are responsible for conscious perception, attentional modulation, and control of vegetative responses. The experience of pain is influenced by a third network that takes into account past experiences, beliefs, and emotional state. Neuropathic pain is associated with alterations at all levels of integration [101].
Pain can originate directly from the central nervous system in lesions or diseases of the central somatosensory pathways: spinal cord injury, stroke, multiple sclerosis, and other diseases of the central nervous system are often associated with pain [102]. In these cases, sensory and motor deficits can be observed in typical distributions according to the site of the lesion. Pain can result from the spontaneous activity of deafferented neurons in the central nervous system and/or the altered activity of spared neurons in the spinothalamic tract [102].
Unfortunately, there is a paucity of literature on the treatment of central pain, and a number of drugs that have been studied have been shown to have an effect on signal transmission at the first synapses (opioids, gabapentinoids), whereas in these cases, the pain originates more rostrally [103].
The impairment of endogenous analgesia following nerve injury may be the rationale for the use of SNRIs in neuropathic pain [104]. Dopaminergic neurotransmission has been shown to play a central role in the modulation of pain perception and analgesia in supraspinal regions, including the basal ganglia, insula, anterior cingulate cortex, thalamus, and periaqueductal grey, and in descending inhibition [105]. Fluphenazine, a dopaminergic antipsychotic, has been shown to be effective in controlling neuropathic pain symptoms [106]. Second-generation antipsychotics have fewer extrapyramidal side effects and may be useful in the treatment of neuropathic pain; in addition to blocking dopamine (D2) receptors, they also act on adrenergic, acetylcholine, catecholamine, histamine, and serotonin receptors in the central nervous system [107].
Non-invasive neuromodulation techniques [repetitive transcranial magnetic stimulation (rTMS), and transcranial direct current stimulation (tDCS)] targeting cortical areas involved in the affective component of pain (prefrontal cortex, motor cortex) have shown some analgesic effects, but with low-quality evidence [11].
Invasive MCS has been used in cases of resistant central pain, but the number of patients enrolled is very small [11].
As the affective component is one of the main aspects of chronic pain, psychological interventions such as cognitive-behavioral therapy or mindfulness therapy may be recommended along with other treatment modalities [108].
Neuropathic pain is difficult to diagnose and even more difficult to treat. Evidence-based medicine gives us some suggestions, but pharmacotherapy is often unsatisfactory, and the majority of interventional treatments commonly used in clinical practice still lack evidence [9]. As chronic pain affects daily life, patients need a therapeutic algorithm based on a thorough diagnosis of the causes and mechanisms of pain: pharmacological and non-pharmacological treatments should be proposed based on their effects on specific mechanisms that often overlap (e.g., peripheral and central). Different therapeutic modalities acting at different sites should be combined to improve outcomes and reduce side effects.
Research is providing more insight into the functional changes that occur in chronic neuropathic pain, but it remains difficult to understand the wide range of clinical manifestations of nervous system lesions. New drugs are in the pipeline, and old drugs are being studied for new indications, but there is still a long way to go to achieve analgesia.
BDNF: brain-derived neurotrophic factor
BTX: botulinum toxin
CGRP: calcitonin gene related peptide
CRPS: complex regional pain syndrome
DMA: dynamic mechanical allodynia
DRG: dorsal root ganglion
FBSS: failed back surgery syndrome
HCN: hyperpolarization-activated cyclic nucleotide-gated
MCS: motor cortex stimulation
NNT: number needed to treat
PENS: percutaneous electrical nerve stimulation
SCS: spinal cord stimulation
SGCs: satellite glial cells
SNRI: serotonin-norepinephrine reuptake inhibitor
SP: substance P
TCAs: tricyclic antidepressants
TENS: transcutaneous electrical nerve stimulation
TRP: transient receptor potential
TRPA1: transient receptor potential ankyrin 1
TRPM8: transient receptor potential melastatin 8
TRPV1: transient receptor potential vanilloid 1
LD: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. CB: Investigation, Writing—original draft, Writing—review & editing. Both authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Hannah G. Matejowsky ... Alan D. Kaye
Sergei Kozlovskiy ... Dmitry Aminin
Howan Leung ... Rainer Freynhagen
Bushra Yasin ... Alex Bekker
Tom Berfelo ... Jan R. Buitenweg
Youyi Peng ... Shan Chen
Giustino Varrassi ... Matteo L.G. Leoni
