Original Article
Original Article

Affiliation:
1G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch of the Russian Academy of Sciences, 690022 Vladivostok, Russia
Email: sergeimerx@gmail.com
ORCID: https://orcid.org/0000-0001-9961-8350
Affiliation:
1G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch of the Russian Academy of Sciences, 690022 Vladivostok, Russia
ORCID: https://orcid.org/0000-0002-3558-0821
Affiliation:
1G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch of the Russian Academy of Sciences, 690022 Vladivostok, Russia
ORCID: https://orcid.org/0000-0002-4027-9064
Affiliation:
1G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch of the Russian Academy of Sciences, 690022 Vladivostok, Russia
ORCID: https://orcid.org/0000-0003-0093-5757
Affiliation:
1G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch of the Russian Academy of Sciences, 690022 Vladivostok, Russia
ORCID: https://orcid.org/0000-0003-3815-6142
Affiliation:
1G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch of the Russian Academy of Sciences, 690022 Vladivostok, Russia
ORCID: https://orcid.org/0000-0002-5382-8296
Affiliation:
1G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch of the Russian Academy of Sciences, 690022 Vladivostok, Russia
ORCID: https://orcid.org/0000-0002-5587-2610
Affiliation:
1G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch of the Russian Academy of Sciences, 690022 Vladivostok, Russia
2Department of Biomedical Science and Environmental Biology, Kaohsiung Medical University, Kaohsiung City 80708, Taiwan, China
ORCID: https://orcid.org/0000-0002-1073-4994
Explor Neurosci. 2024;3:39–50 DOI: https://doi.org/10.37349/en.2024.00035
Received: October 13, 2023 Accepted: November 29, 2023 Published: February 22, 2024
Academic Editor: Giustino Varrassi, President of the Paolo Procacci Foundation, Italy
The article belongs to the special issue Neuropathic Pain
Aim: The ability of synthetic 1,4-naphthoquinones (1,4-NQs) to prevent adenosine triphosphate (ATP)-induced and purinergic P2X7 receptor (P2X7R) mediated inflammation in macrophage and neurodegeneration of neuronal cells in vitro was previously established. The aim of the present study was to investigate analgesic-like and anti-inflammatory activity of 1,4-NQs thioglucoside derivatives, compounds U-286 and U-548, in in vivo experiments.
Methods: Spectrofluorimetry approach and YO-PRO-1 fluorescent dye uptake determination were applied to study the effect of 1,4-NQs upon ATP-induced P2X7R mediated macropore formation in mouse neuroblastoma Neuro-2a cells and macrophages RAW 264.7 cells. An acetic acid-induced writhing test, hot plate test, and carrageenan-induced paw edema test were used as an in vivo mouse models to study the ability of 1,4-NQs to inhibit pain and inflammation. In the in vivo experiments, compounds were administered to mice intraperitoneally at dosages of 0.1 mg/kg, 1.0 mg/kg and 10.0 mg/kg. A group of animals that received injections of sterile water was used as a control. Each dosage group and the control group consisted of 6 mice.
Results: In the present work the analgesic-like and anti-inflammatory activity of 1,4-NQs, U-286 and U-548, was demonstrated. Compound U-548 showed a significant inhibitory effect in antinociceptive tests reducing the number of mouse writhings and eliminating the latent time of mouse hind paw licking, correspondingly. Selected compounds were able to almost completely reduce the size of carrageenan-induced paw edema 24 h after injection and had a potent anti-inflammatory activity. Observed effects were accompanied with aptitude of studied 1,4-NQs to inhibit the formation of purinergic P2X7R macropore associated with inflammation and nociceptive pain.
Conclusions: The results obtained allow to consider compounds U-286 and U-548 and as a pharmacological basis for the development of new analgesic-like and anti-inflammatory drugs.
Chronic and acute nociceptive pain is caused by cellular damage with an accompanying inflammation. Multiple factors contribute to the development of pain, and it is widely acknowledged that inflammatory cytokines play a crucial role in this process. The role of adenosine triphosphate (ATP) and P2X7 purine receptors in the release of inflammatory cytokines during pain has been consistently demonstrated by numerous studies [1]. In response to damage of the plasma membrane of surrounding cells or the entry of an infectious pathogen, high concentrations of ATP enter the extracellular space through breaks in the cell membrane or by diffusion through pannexin-1 hemichannels. A rapid increase in the extracellular level of ATP leads to the activation of P2X7 receptor (P2X7R) localized in immune and neuronal cells of the peripheral and central nervous systems [2].
P2X7R are ionotropic receptors, one of the subtypes of membrane ion channels of the P2X family [3]. When activated by high concentrations of ATP, receptors provide the influx of calcium ions (Ca2+) and sodium ions (Na+), as well as the outflow of potassium ions (K+), which leads to cell membrane depolarization [4]. Long-term exposure of the agonist to the receptor causes the opening of a macropore, permeable to molecules weighing up to 900 Da, as well as an apoptotic or necrotic pathway of cell lysis, depending on the type of tissue [5].
Microglial cells contain special receptors that can recognize hazardous exogenous molecules [5]. Such molecules include primarily bacterial lipopolysaccharides (LPS) and danger-associated molecular patterns, for example, ATP. The combined effects of such agents on cells can lead to severe inflammation [6]. Activation of P2X7R by ATP causes a cascade of events resulting mainly in the assembly of the nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome and the proinflammatory interleukin 1β (IL-1β) release [7].
ATP can act as a sensory neurotransmitter at synapses. In the central nerve terminals of neurons in the dorsal root group (DRG), this neurotransmitter transmits pain signals to the thalamus. DRG satellite glial cells as well as dorsal root astrocytes express P2X7R, which can be released from lysosomes when activated by IL-1β as well as gliotransmitters such as ATP [6]. Activation of P2X7R leads to the initiation of macrophages, microglia and astrocytes, as well as the secretion of cytokines, prostaglandins and pro-inflammatory proteases into surrounding tissues [7]. In addition, the opening of the P2X7R channel causes the production of reactive oxygen species (ROS) and nitrogen monoxide (NO), and the release of excitotoxic glutamate, which causes hyperexcitation of neurons [8]. In this regard, P2X7R are one of the main participants in the development of chronic inflammation pain, and are also involved in the transmission of acute nociceptive pain signals [9].
Several researches have demonstrated that P2X7R plays a significant role in modulating behavioral responses to painful stimuli [10, 11]. The phenotype of P2X7 knockout mice, characterized by the absence of both isoforms of IL-1β, has been observed in studies [12]. It has been proposed that the release of IL-1β resulting from nerve injury or prolonged inflammation may contribute to an elevation in the phosphorylation of the NMDA receptor subunit 1 (NR1) and N-methyl-d-aspartate receptor subunit 2B (NR2B) subunits of the N-methyl-D-aspartate receptor, potentially leading to a phenomenon similar to long-term potentiation (LTP), causing hypersensitivity and pain [13]. It has been demonstrated that the administration of the P2X7 antagonist A438079 via intrathecal route can effectively prevent mechanical hypersensitivity [14].
1,4-Naphthoquinones (1,4-NQs) are a class of compounds that have a wide range of biological activities [15], including antimicrobial [16], anti-ischemic [17], cardioprotective [18] and anti-inflammatory properties [19]. It was previously discovered that 2-hydroxy-3-iodo-1,4-NQs (AN-03) and 2-hydroxy-3-phenylnaphthalene-1,4-dione (AN-04) exhibited higher potency compared to brilliant blue G (BBG) and A740003 in inhibiting dye uptake, release of IL-1β, and have anti-inflammatory effect in carrageenan-induced paw edema in vivo [20]. Based on these findings, it can be inferred that 1,4-NQs hold promise as novel blockers of P2X7R and potential anti-inflammatory drugs. In previous study [21], it was demonstrated that the synthesized acyclic thioglucoside U-548 and its tetracyclic derivative U-286 can inhibit ATP-induced calcium influx in RAW 264.7 and Neuro-2a cells. The tested compounds also reduced the production of ROS and NO in these cell lines caused by high concentrations of ATP. Compound U-286 was shown to inhibit the production of the mature form of IL-1β, as well as the proinflammatory cytokine tumor necrosis factor α (TNF-α) in RAW 264.7 macrophages. In addition, these compounds significantly reduced the activity of the proinflammatory enzyme cyclooxygenase 2 (COX-2) [22].
This study is a continuation of the investigation of the synthesized 1,4-NQs effects on the functioning of purinergic P2X7R in macrophage and neuronal cells. The novelty of the work lies in new data on the compounds U-286 and U-548 biological activity in the experimental animal models of pain and inflammation. The antinociceptive effect of selected 1,4-NQs was established on the hot plate test model of thermal hyperalgesia, as well as on the “Acetic acid-induced writhing test” model of acid-induced muscle pain in vivo. The anti-inflammatory effect of the studied compounds was assessed in an in vivo model of carrageenan-induced mouse paw edema. The results obtained indicate that compounds U-286 and U-548 can be considered as potential blockers of P2X7R and can be used for the development of new types of analgesic-like and anti-inflammatory drugs.
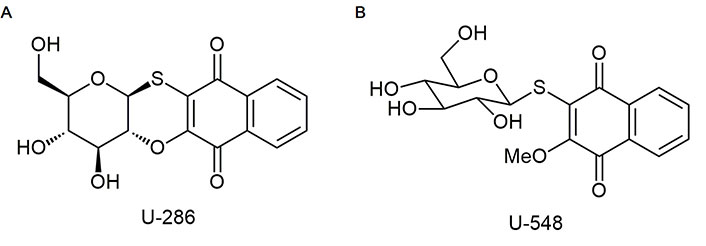
Studied compounds U-286 [(2R,3R,4S,4aR,12aS)-2-hydroxymethyl-3,4-dihydroxy-3,4,4a,12a-tetrahydro-2H-naphtho(2, 3-b) pyrano(2,3-e)(1,4)-oxathiine-6, 11-dione] and U-548 [3-(β-D-glucopyranosyl-1-thio)-2-methoxynaphthalene-1,4-dione] were synthesized in accordance with early described protocols [22]. Stocks of compounds were prepared in dimethyl sulfoxide (DMSO, Biolot, Russia) as 10 mmol/L solutions. For experiments, substances were dissolved in double distilled water (ddH2O) so that the concentration of DMSO in the solution did not exceed 1%.
Mice of two lines, C57BL/6 and CD-1 (females, two months old, 20–22 g), were purchased from the Russian National Center for Genetic Resources of Laboratory Animals (ICiG SB RAS, Novosibirsk, Russia). The animals were divided into experimental groups of 6 mice in each group and kept in a vivarium at a 12 h light/dark cycle and a temperature of 22°C ± 1°C with free access to the food and water. All experiments were performed in obedience to the International Recommendations for Biomedical Research Using Animals, adopted by the International Council of Medical Scientific Societies (CIOMS) and the Order of the Ministry of Health and Social Development of Russia dated 23.08.2010 No. 708n “On Approval of Laboratory practice Rules”.
Mouse neuroblastoma Neuro-2a cells [American Type Culture Collection (ATCC)® CCL-131™] and mouse macrophages RAW 264.7 (ATCC® TIB-71™) were obtained from ATCC (Manassas, VA, USA). Cells were cultivated in CO2-incubator in 75 cm2 cultural flask (Corning, Phoenix, USA) in Dulbecco’s modified Eagle’s medium (DMEM) culture medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin antibiotics (Biolot, St. Petersburg, Russia).
Neuro-2a (3 × 104 cells/well) or RAW 264.7 (4 × 104 cells/well) cells were seeded in 96-well plates and cultivated at 37°C in a CO2-incubator for 24 h. Before experiments culture medium was replaced with a Hanks’ balanced salt solution (HBSS) solution (140 mmol/L NaCl, 5 mmol/L KCl, 0.8 mmol/L MgCl2, 2 mmol/L CaCl2, 10 mmol/L glucose, 10 mmol/L HEPES, pH 7.4) and test compounds were added to the cells at the final concentrations 0.1 μmol/L, 1 μmol/L and 10 μmol/L. After incubation for 1 h, HBSS solution was replaced with a HBSS containing the fluorescent dye YO-PRO-1 (Sigma, USA, 5 μmol/L for Neuro-2a or 2.5 μmol/L for RAW 264.7 cells, final concentration). Then, 15 min later 2 mmol/L ATP (Sigma, USA, A26209) was added to each plate well and the cells were incubated for an additional 10 min followed by washing with buffer solution without dye. The final fluorescence intensity of the cell penetrating dye was assessed using a PHERAstar FS plate reader (BMG Labtech, Germany) at λex = 480 nm and λem = 520 nm. A selective blocker of the P2X7R, A438079 (10 μmol/L, Sigma, USA, A9736), was used as a standard antagonist [23].
Hot plate analgesia meter (Columbus Instruments, Columbus, OH, USA) was used to examine the mouse thermal hyperalgesia. For this purpose, the animals were placed individually on a 47°C preheated device plate surface and exposed to heat until a specific nociceptive reaction was recorded. The time of the first withdrawal or licking of the hind paw was taken as a reaction latency period. If there was no reaction in the selected time period, the latent time was taken to be 80 s. Compounds U-286 and U-548 at dosages of 0.1 mg/kg, 1.0 mg/kg or 10.0 mg/kg were administered to mice intraperitoneally (100 µL/20 g body weight) 60 min before the start of the experiment. A group of animals that received injections of sterile water was used as a control. A separate group of mice was used for each experiment.
Substances U-286 and U-548 at dosages of 0.1 mg/kg, 1.0 mg/kg or 10.0 mg/kg were administered to mice intraperitoneally (100 µL/20 g body weight) 60 min before the experiment. Then the animals were given an intraperitoneal injection of 0.7% acetic acid (CH3COOH, 100 µL/10 g body weight) solution. Next, the animal was placed in a closed box and video was filmed using a camera SONY HDR-CX280 (Virio Tessar, 50×, Japan). Observation was carried out for 15 min, immediately after the injection of acetic acid. As a result of the experiment, the number of specific pain reactions (writhing) accompanied by characteristic movements of the animals (contraction of the abdominal muscles followed by relaxation, stretching of the hind limbs and arching of the back) was calculated, in the form of characteristic stretching of the animal’s two hind limbs. The number of latent reactions was counted manually while watching the video recording of the experiment. Animals that received injections of sterile water were used as a control group. A separate group of mice was used for each experiment.
Mouse paw edema was caused by subplantar injection of carrageenan-δ (Sigma Aldrich, St. Louis, MO, USA) in phosphate buffered saline (1.5 mg/mL, 20 µL). Test compounds at dosages of 0.1 mg/kg, 1.0 mg/kg or 10.0 mg/kg were introduced to mice intraperitoneally 1 h before carrageenan application. Acute local inflammation parameters (size of the inflamed edema, the volume of the area of the animal’s paws from the toes to the hock joint) were inspected with a plethysmometer (Ugo Basile, Gemonio, VA, Italy) 1 h, 2 h, 4 h and 24 h post carrageenan administration. A separate group of mice was used for each experiment. The control group of mice that received a saline injection were used as a baseline for all behavioral tests.
Statistical analysis was performed using SigmaPlot 14.0 software (Systat Software Inc., San Jose, CA, USA). One-way and two-way analyses of variance (ANOVA) with a Tukey post-hoc test were used for multiple comparisons, and Student’s t-test was used for two-group comparisons. All results are presented as means ± standard error of mean (SEM), and differences with P < 0.05 were considered significant.
It is known that P2X7R, when exposed to long-term high concentrations of ATP (hundreds of micromoles), is capable of opening a macropore, allowing charged molecules up to 900 Da to pass through [5]. The ability of antagonists to block macropore formation when exposed to ATP is a distinguishing marker in the screening of potential P2X7R blockers. In present work, we investigated the capability of synthetic 1,4-NQs, compounds U-286 and U-548 (Figure 1), to inhibit ATP-induced entry of the YO-PRO-1 dye in mouse Neuro-2a neuroblastoma cells and mouse RAW 264.7 macrophage cells that mimic mouse brain neurons and microglia.
In Neuro-2a neuronal cells, 1,4-NQs derivative U-286 inhibited fluorescent dye entry at concentrations of 0.1 μmol/L, 1 μmol/L, and 10 μmol/L by 12.7% ± 8.8%, 25.4% ± 10.4%, and 46.3% ± 2.1%, respectively, demonstrating a dose-dependent effect (Figure 2A). Compound U-548 turned out to be more effective, inhibiting the entry of fluorescent dye at concentrations of 0.1 μmol/L, 1 μmol/L and 10 μmol/L by 35.1% ± 12%, 18.6% ± 2.1% and 61% ± 4.2%, respectively. A standard P2X7R blocker A438079 at a concentration of 10 μmol/L showed a comparable effect, inhibiting dye uptake by 32.8% ± 3.6% (Figure 2A).
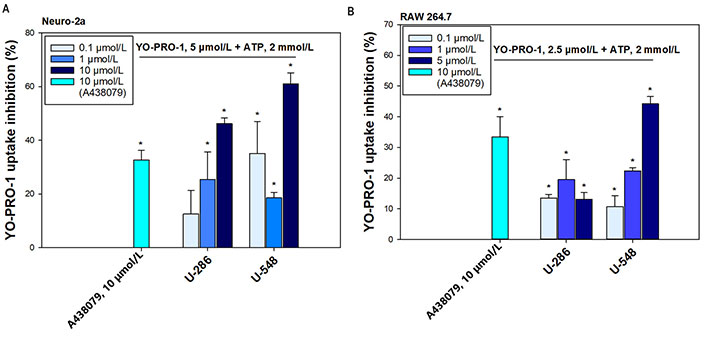
Synthetic 1,4-NQs U-286 and U-548 inhibit ATP-induced uptake of the fluorescent dye YO-PRO-1. (A) In Neuro-2a mouse neuroblastoma cells; and (B) in RAW 264.7 macrophage cells. A438079 (10 μmol/L) was used as a standard P2X7R antagonist. Data are presented as percent inhibition of dye entry relative to the ATP positive control. Values are presented as mean ± SEM (n = 3); * P < 0.05 compared with the effect of ATP alone
These 1,4-NQs also showed their effectiveness in RAW 264.7 macrophage cells (Figure 2B). Compound U-286 exhibited a bell-shaped concentration dependence, inhibiting YO-PRO-1 uptake at concentrations of 0.1 μmol/L, 1 μmol/L, and 5 μmol/L by 13.6% ± 1.2%, 19.6% ± 6.5%, and 13.1% ± 2.3%, respectively. Compound U-548 in this cell line also showed significant reduction of dye inflow in a pronounced dose-dependent manner at concentrations of 0.1 μmol/L, 1 μmol/L and 5 μmol/L by 10.7% ± 3.7%, 22.4% ± 1.1% and 44.2% ± 2.5%. The standard blocker A438079 at a concentration of 10 μmol/L inhibited YO-PRO-1 uptake by 33.4% ± 6.7% (Figure 2B).
Antinociceptive activity of the tested compounds was inspected in the acetic acid-induced writhing test. Compound U-286 significantly diminished the number of writhing only at the maximum dosage (10 mg/kg) by 39.2% ± 10.1%, relative to the positive control. The other two dosages were ineffective (Figure 3A). Compound U-548 at dosages of 0.1 mg/kg, 1 mg/kg and 10 mg/kg significantly and strongly eliminated the number of latent reactions by 72.9% ± 7.8%, 66.1% ± 14.5% and 88.1% ± 5.7%, respectively (Figure 3A).
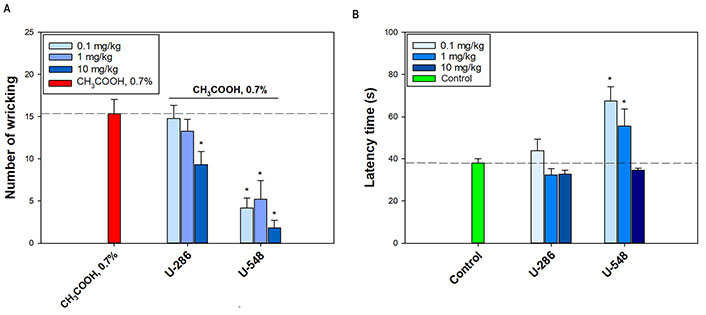
Analgesic-like effect of 1,4-NQs. (A) Influence of compounds U-286 and U-548 on number of writhings of CD-1 mice in a model of acid-induced muscle pain; (B) on latent time of hind paw licking of C57BL/6 mice in hot plate test. The dotted line indicates the levels of positive control (A, CH3COOH, 0.7%) or negative control (B, Control). Results are presented as mean ± SEM (n = 6). The significance of the differences is presented in comparison with the group that received only acetic acid alone (A) or only saline (B). * P < 0.05
Thermal hyperalgesia was studied using the hot plate test (Figure 3B). Compound U-548 demonstrated an inverse dose-dependent effect, raising latency reaction time by 77.5% ± 18.1% and 46.3% ± 21.5% at dosages of 0.1 mg/kg and 1 mg/kg, respectively. Compound U-286 did not exhibit a significant analgesic-like effect in this experiment, however, at a dosage of 0.1 mg/kg, a slight elevation of the animal’s latent reaction time was observed (Figure 3B).
The anti-inflammatory activity of U-286 and U-548 was studied in a model of acute local inflammation induced by carrageenan (Figure 4A and B). In a paw edema test, injection of carrageenan into the hind footpads of mice showed a huge increase in paw edema as a function of time after administration of the inductor. This model is well established for assessing the anti-inflammatory effect of potential anti-inflammatory and antinociceptive drugs.
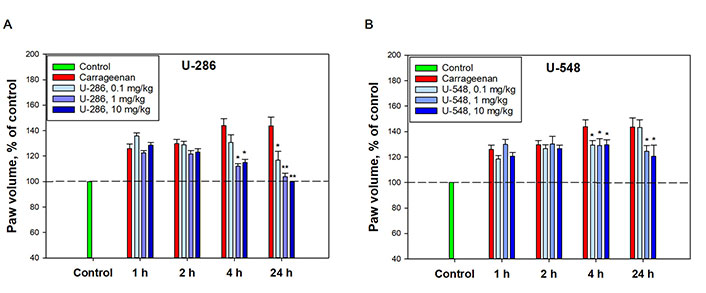
Anti-inflammatory effect of compounds U-286 and U-548 in a model of carrageenan-induced paw edema of CD-1 mice. (A) U-286; (B) U-548. The dotted line indicates the level of mouse paw volume before carrageenan administration. A solution of 1,4-NQs was administered intraperitoneally 60 min before the induction of inflammation. Paw volume was measured 1 h, 2 h, 4 h and 24 h after intraplantar injection of carrageenan at a concentration of 1.5 mg/mL in a volume of 20 μL. The results obtained are presented as mean ± SEM (n = 6); * P < 0.05, ** P < 0.01 compared to carrageenan
Compound U-286 did not have a significant effect on paw volume and inflammatory edema growth index after 1 h and 2 h. However, 4 h after administration of carrageenan, it significantly diminished edema by 72.7% ± 4.6% and 65.9% ± 6% at dosages 1 mg/kg and 10 mg/kg, respectively. After 24 h, U-286 at dosages of 0.1 mg/kg and 1 mg/kg reduced paw volume relative to carrageenan by 61.7% ± 15.8% and 91.3% ± 6.1%, respectively. Notably, the highest dosage of this compound (10 mg/kg) completely eliminated the swelling caused by carrageenan, returning the paw volume to its initial size (Figure 4A).
Compound U-548 also did not demonstrate an anti-inflammatory effect 1 h and 2 h after carrageenan administration. After 4 h, the compound inhibited the growth of inflammatory edema at dosages of 0.1 mg/kg, 1 mg/kg and 10 mg/kg by 32.7% ± 8.1%, 34.1% ± 12.5% and 32.4% ± 9%, respectively. After 24 h, U-548 did not exhibit a significant anti-inflammatory effect at a dosage of 0.1 mg/kg, but reduced inflammation at dosages of 1 mg/kg and 10 mg/kg by 44% ± 10.7% and 53.1% ± 20.1%, respectively (Figure 4B).
This work is a continuation of the study of the anti-inflammatory activity of the acyclic thioglucoside U-548 and its tetracyclic derivative U-286. In earlier research these compounds were found to exhibit antagonistic properties associated with blocking reaction pathways mediated by P2X7R in macrophage and neuronal cells in vitro [21, 22]. Activation of P2X7R induces large-scale release of ATP into the extracellular space of tissues, due to its ability to form membrane macropores, as well as activate pannexin-1 hemichannels, thereby enhancing purinergic signaling and the inflammatory process [24]. The release of high concentrations of ATP is a natural danger signal indicating tissue damage or cell penetration by infectious pathogens [7]. ATP, which activates the P2X7R, is a second messenger to stimulate the assembly of the NLRP3 inflammasome, which induces both the maturation and release of proinflammatory cytokines such as IL-1β, IL-6 and IL-18 [25]. The P2X7R also induces the production of ROS and nitrogen species, and is also involved in the activation of caspases and the initiation of necrosis and apoptosis processes. In addition, the P2X7R is involved in adaptive immunity and regulates the balance between the production of T helper type 17 (Th17) and T regulatory (Treg) lymphocytes [14]. In this regard, the P2X7R is participated in many pathological conditions associated with chronic inflammation and tissue degradation, such as arthritis, Crohn’s disease, asthma and many others [25]. A large number of studies prove that P2X7R is closely related to neuronal excitation, neurosensitization, neuroinflammation, pain transmission and chronic pain [1].
Previously, the anti-inflammatory activity of 1,4-naphthoquines was repeatedly emphasized, but only in recent years have studies appeared demonstrating the potential of this class of compounds to block P2X7 type purinergic receptors [20, 26]. One of the distinctive features of this receptor subtype is its capability to form a large macropore in the cell membrane, permeable to large charged molecules [3]. In this work, we showed that synthetic 1,4-naphthoqunone derivatives, U-286 and U-548, abolished the entry of the fluorescent dye YO-PRO-1 caused by the application of a high concentration of ATP to cells. In the case of neuronal cells, a stable dose-dependent inhibitory effect of both compounds was observed, exceeding the effect of the standard P2X7R blocker, A438079. On macrophage and neuronal cells, compound U-286 had a lesser effect than U-548, but consistently inhibited dye entry at all concentrations tested. The experimental results indicate a direct blocking effect of selected 1,4-NQs on ATP-induced formation of the P2X7R macropore.
The analgesic-like activity of the studied 1,4-NQs was assessed using the hot plate test and acetic acid-induced writhing test models in vivo. The hot plate test is a well-established method for studying the pain response of animals to a thermal stimulus. The method is more often used to study the pharmacological activity of centrally acting analgesics, while the acetic acid-induced writhing test is more sensitive to peripherally acting drugs. Compound U-548 significantly increased the latent reaction time of the animal to a thermal stimulus, exhibiting a pronounced dose-dependent effect at dosages of 0.1 mg/kg and 1 mg/kg, while a dosage of 10 mg/kg did not show a sufficiently significant difference from the control level. Compound U-286 did not exhibit pronounced analgesic-like activity in this test; only at a concentration of 0.1 mg/kg there was a tendency to increase the latent time of the reaction. At the same time the observed increase in latency during this test suggests that the investigated compounds have the potential to modify heat perception, which is essential for protective purposes. This possible side effect requires further investigation.
In the acetic acid-induced writhing test for acid-induced muscle pain, compound U-286 demonstrated a dose-dependent effect at dosages of 1 mg/kg and 10 mg/kg, while a dosage of 0.1 mg/kg was ineffective. Compound U-548, on the contrary, showed a significant suppressing effect, reducing the number of latent reactions to the stimulus at all concentrations tested. Generally, obtained results indicate that studied 1,4-NQs alleviated pain caused by thermal or acid burn.
The modest P2X7R inhibitory and analgesic-like effects of compound U-286 in comparison with U-548 in these tests can be explained by their different chemical structures. Unlike substance U-286 compound U-548 has a sugar moiety in the molecule conjugated with quinone, since this compound is a quinone-thioglucoside conjugate (Figure 1) and therefore has a greater aqueous solubility. This chemical feature may influence on its pharmacokinetic behavior. Increased bioavailability, due to better aqueous solubility, can be considered as the reason why compound U-548 has greater effect over short periods of time, while U-286 has greater potential over time.
Acute inflammation is a process that involves the production of free radicals, the activation of a complex of enzymes, and the release of several inflammatory and proinflammatory cytokines. Carrageenan-induced paw edema is a well-established model of acute inflammation that has been widely used to screen new anti-inflammatory compounds. Acute inflammation caused by subcutaneous administration of a proinflammatory adjuvant is a process that develops over time and is divided into two phases [27]. During the acute inflammation phase, in response to carrageenan entering the tissue, biogenic amines such as histamine, serotonin and bradykinin are produced. During the second phase, several hours after the induction of inflammation, the development of a chronic inflammatory response occurs, initiated primarily by the release of ATP into the surrounding tissues and the mobilization of P2X7R to the surface of cell membranes. Activation of P2X7R leads to the maturation of the NLRP3 inflammasome and the production of mature forms of various pro-inflammatory cytokines, such as IL-1β, IL-1α, IL-18 and TNF-α, followed by activation of the COX-2 enzyme responsible for the production of prostaglandin E2 and the production of ROS, hydroxyl radicals and nitric oxide [27]. As shown in present investigation, the studied compounds did not affect the size of the paw edema 1 h and 2 h after carrageenan administration. However, after 4 h and 24 h, a clear trend towards a decrease in the inflammatory response was visible. The results of this study are in excellent agreement with earlier studies of these compounds in cellular models, where compounds U-286 and U-548 were shown to abrogate ATP/LPS-induced production of the mature form of IL-1β and TNF-a, and also reduce the activity of pro-inflammatory enzyme COX-2 [11]. We did not find much difference in the anti-inflammatory activity of the two 1,4-NQs studied. This is probably due to the ability of both naphthoquinones not only to block the P2X7R, but also to have direct antioxidant activity [11], which may also remarkably contribute to their anti-inflammatory effect. It is known that intraperitoneal pretreatment with clinically used indomethacin (10 mg/kg) after carrageenan subcutaneous injection significantly reduces the increase in paw thickness and edema in mice by approximately 46% [28] that is comparable with that of 1,4-NQs.
It was discovered that, the subcutaneous administration of 300 mg/paw of A438079, a P2X7R antagonist, coinjected with carrageenan, resulted in a notable decrease in hyperalgesia and the levels of pro-inflammatory markers TNF-a, IL-6, and cytokine-induced neutrophil chemoattractant (CINC) in the inflamed paw of rats. [29]. The administration of BBG, which blocks P2X7R, has been confirmed to effectively reduce behavioral deficits, pain mediators, and microglial activation. By inhibiting the activation of the P2X7R related NLRP3 inflammasome, BBG prevents the release of ILs IL-1 and IL-18, which are responsible for nociceptors activation. These studies have demonstrated that BBG increases reaction latency in rats during hot plate and tail-flick warm water tests [30]. The data presented indicate the similarity between the anti-inflammatory effects of the compound BBG and the investigated 1,4-NQs, suggesting the involvement of P2X7R blockers in their anti-inflammatory activity. Furthermore, these findings confirm the association between P2X7R activation and thermal hyperalgesia.
Thus, this work demonstrates that studied 1,4-NQs relieved induced nociceptive pain and inflammation in in vivo models. The data obtained demonstrate a putative relation between the analgesic-like and anti-inflammatory activities of the studied compounds and their ability to block P2X7R in neuronal and macrophage cells. The results allow us to consider compounds U-286 and U-548 as a chemical scaffold for the creation of anti-inflammatory and antinociceptive drugs that suppress inflammatory and nociceptive pain initiated by activation of P2X7R.
The present work has certain limitations associated with a limited set of experimental approaches and methods. Subsequently, our future research aims to extensively investigate the anti-inflammatory and analgesic properties of these compounds. This research will involve employing transgenic cell cultures that stable express the P2X7R and utilizing electrophysiology techniques on both neuronal and microglial cells in vitro. Moreover, we intend to evaluate the effectiveness of these compounds in models of neuropathic pain and neuroinflammation in vivo. Additionally, we plan to assess the pharmacokinetics of 1,4-NQs and their capacity to cross the blood-brain barrier.
1,4-NQs: 1,4-naphthoquinones
ATP: adenosine triphosphate
BBG: brilliant blue G
COX-2: cyclooxygenase 2
HBSS: Hanks’ balanced salt solution
IL-18: interleukin 18
NLRP3: nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3
P2X7R: P2X7 receptor
ROS: reactive oxygen species
SEM: standard error of mean
SK: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. EP: Conceptualization, Writing—review & editing. EM, EC, YS, and SP: Metodology. IA: Investigation. DA: Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
The study was approved by the local ethics committee of the G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Easter Branch of the Russian Academy of Sciences (protocol No. 02/23 of February 6, 2023). All experiments were conducted according to the International Recommendations for Biomedical Research Using Animals, adopted by the International Council of Medical Scientific Societies (CIOMS) and the Order of the Ministry of Health and Social Development of Russia dated 23.08.2010 No. 708n “On Approval of Laboratory practice Rules”.
Not applicable.
Not applicable.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Hannah G. Matejowsky ... Alan D. Kaye
Howan Leung ... Rainer Freynhagen
Bushra Yasin ... Alex Bekker
Tom Berfelo ... Jan R. Buitenweg
Youyi Peng ... Shan Chen
Laura Demartini, Cesare Bonezzi
Giustino Varrassi ... Matteo L.G. Leoni