 Original Article
Original Article

Affiliation:
1Centre for Genomics of Non-Communicable Diseases and Personalized Healthcare, University of Lagos, Akoka, Lagos 101017, Nigeria
2Department of Medical Laboratory Science, University of Lagos, Akoka, Lagos 101017, Nigeria
3Department of Medical Laboratory Science, Neuropsychiatric Hospital, Aro, Abeokuta 110101, Nigeria
Email: bimpeadebayo2002@yahoo.com
ORCID: https://orcid.org/0000-0003-3587-9767
Affiliation:
2Department of Medical Laboratory Science, University of Lagos, Akoka, Lagos 101017, Nigeria
ORCID: https://orcid.org/0009-0000-2499-2997
Affiliation:
2Department of Medical Laboratory Science, University of Lagos, Akoka, Lagos 101017, Nigeria
ORCID: https://orcid.org/0009-0008-2364-3910
Affiliation:
2Department of Medical Laboratory Science, University of Lagos, Akoka, Lagos 101017, Nigeria
ORCID: https://orcid.org/0009-0001-7726-6734
Affiliation:
2Department of Medical Laboratory Science, University of Lagos, Akoka, Lagos 101017, Nigeria
ORCID: https://orcid.org/0009-0002-5818-5370
Affiliation:
3Department of Medical Laboratory Science, Neuropsychiatric Hospital, Aro, Abeokuta 110101, Nigeria
4Department of Public Health, Daffodil International University, Dhaka 1216, Bangladesh
ORCID: https://orcid.org/0000-0002-3809-4271
Affiliation:
5Department of Clinical Sciences, Neuropsychiatric Hospital, Aro, Abeokuta 110101, Nigeria
ORCID: https://orcid.org/0000-0002-0660-9103
Affiliation:
6Faculty of Clinical Sciences, University of Lagos, Akoka, Lagos 101017, Nigeria
ORCID: https://orcid.org/0009-0006-6870-3214
Affiliation:
1Centre for Genomics of Non-Communicable Diseases and Personalized Healthcare, University of Lagos, Akoka, Lagos 101017, Nigeria
2Department of Medical Laboratory Science, University of Lagos, Akoka, Lagos 101017, Nigeria
Email: oakinloye@unilag.edu.ng
ORCID: https://orcid.org/0000-0002-7546-8244
Explor Neurosci. 2026;5:1006134 DOI: https://doi.org/10.37349/en.2026.1006134
Received: December 03, 2025 Accepted: March 17, 2026 Published: April 27, 2026
Academic Editor: Xinhua Shu, Glasgow Caledonian University, UK
Aim: Redox-oxidative dysregulation is implicated in the aetiology of several diseases, including schizophrenia, with a possible influence on clinical symptoms. This study investigated the influence of redox, lipid peroxidation, and micronutrient antioxidants on the expression of clinical phenotypes of schizophrenia.
Methods: A total of 220 consenting drug-naïve volunteers, including 120 participants with schizophrenia and 100 apparently healthy controls, were recruited. Schizophrenia symptoms were evaluated using the Positive and Negative Syndrome Scale (PANSS). Lipid peroxidation (malondialdehyde; MDA) was quantified using the thiobarbituric acid reactive substances (TBARS) spectrophotometric method; glutathione (GSH), superoxide dismutase (SOD), and catalase (CAT) were assessed using established enzymatic activity assays; total antioxidant capacity (TAC) was determined by the phosphomolybdenum colorimetric method; vitamins C and E were measured using spectrophotometric biochemical assays; and zinc (Zn) and selenium (Se) concentrations were quantified using atomic absorption spectrophotometry (AAS).
Results: Enzymatic antioxidants, SOD (19.58 ± 0.80; 10.12 ± 0.45 U/L) and CAT (41.73 ± 1.81; 21.33 ± 0.98 U/L), increased in schizophrenia compared with controls (p < 0.05), but decreased non-enzymatic antioxidants; GSH (14.5 ± 0.28; 15.9 ± 1.59 µmol/L, p < 0.05). Furthermore, serum levels of zinc (1.8 ± 0.01; 2.7 ± 0.02 mg/L), selenium (0.08 ± 0.01; 0.10 ± 0.01 mg/L), and vitamin C (12.98 ± 0.49; 15.08 ± 0.37 mg/L) were lowered in schizophrenia compared with controls (p < 0.05). GSH had a negative correlation with positive symptoms (r = –0.285, p = 0.013) while SOD (r = 0.281, p = 0.001) and CAT (r = 0.179, p = 0.034) correlated positively with MDA (p < 0.05). In contrast, GSH (r = –0.247, p = 0.003) and TAC (r = –0.221, p = 0.009) correlated negatively with MDA (p < 0.05).
Conclusions: Drug-naïve Nigerian individuals with schizophrenia appear to exhibit a pattern of redox imbalance, including increased lipid peroxidation, altered antioxidant enzyme activity, and reduced non-enzymatic antioxidants, with lower GSH levels modestly associated with greater positive symptom severity.
Schizophrenia is a disorder that affects social and emotional function [1, 2]. It is a constellation of features which may be characterized as positive, negative, and general psychopathological symptoms [3]. Positive symptoms include hallucinations, delusions, and paranoia, while alogia, anhedonia, avolition, and blunted affect mark the negative domain. General psychopathology tends to refer to impairments such as poor memory and poor executive functioning [4]. It is the most common diagnosis resulting in help-seeking in Nigeria, and affects 1% of the general population worldwide, and has been known to contribute significantly to years lost to disability [5, 6]. Schizophrenia is a disorder with widespread structural and functional alterations, which are not completely understood. However, studies have shown numerous biochemical pathways in the neurobiology of the disorder. Some of which are not directly causative but act as risk factors for the disorder [7, 8].
In the past, much focus and emphasis had been placed on the dopamine system alterations based on indirect evidence from the role most antipsychotic medications play by acting on the dopamine receptor and specifically the D2 subtype [9]. However, efforts have been made to identify other biological processes that may contribute to the process and symptomatology in schizophrenia. One of these is the redox-oxidative stress and micronutrient antioxidants in schizophrenia [10]. Redox dysregulation as a potential pathway in symptomatology manifestation rises when there is increased oxidative damage and decreased capacity of the intracellular antioxidant defense system. This gives rise to oxidative stress, which results from the disequilibrium between pro-oxidant processes and the antioxidant defense system in favor of the former [11, 12].
There are several consequences of oxidative stress due to increased free radical production and inefficient antioxidant systems, which lead to lipid peroxidation [13, 14]. Oxidative stress has been linked to low levels of antioxidant vitamins such as C, E, and beta carotene [15]. People with schizophrenia have also been found to have altered antioxidant levels in the blood and brain and altered antioxidant enzyme activity. Additionally, oxidative stress has been suggested as a potential connection between schizophrenia and vitamin deficiency [16]. Studies have also shown that antioxidant vitamins such as C and E offer protection against cellular damage due to either inflammation or highly reactive oxygen species (ROS) [17]. Redox dysregulation is linked to the reduction of parvalbumin-containing GABA interneurons and volumes of their perineuronal nets, white matter abnormalities, and microglia activation. Activity of transcription factors, kinases, and phosphatases regulating diverse aspects of neurodevelopment and synaptic pathway vary according to cellular redox state [18, 19].
Furthermore, the mechanisms through which nutrition and nutritional deficiencies contribute to the onset or exacerbation of schizophrenia remain incompletely elucidated; however, multiple interacting biological modalities have been proposed. These include inadequate dietary quality and maladaptive eating patterns that result in deficiencies of essential micronutrients involved in redox homeostasis and neurotransmitter synthesis [20]. Nutritional insufficiency has been associated with hyperhomocysteinemia due to impaired one-carbon metabolism, disturbances in oxidant–antioxidant balance, mitochondrial dysfunction, and increased lipid peroxidation [21]. In addition, micronutrient deficits may precipitate immune dysregulation characterized by altered cytokine profiles, microglial activation, and heightened pro-inflammatory signaling. Emerging evidence also implicates disruptions in gut–brain axis signaling, epigenetic modulation, and impaired neurodevelopmental processes as potential pathways linking dietary inadequacy to symptom severity and disease progression in schizophrenia [22, 23] The neuroprogressive hypothesis, which regarded abnormalities in oxidative stress and immune-inflammatory indicators as potential pathways in the pathophysiology of schizophrenia has previously discussed the significance of inflammation and oxidative stress in schizophrenia [24, 25]. According to current theories, the connection between nutrition and schizophrenia may involve the stimulation of neuroinflammation and modification of the gut microbiota, both of which have been linked in some instances to the exacerbation of schizophrenia symptoms [25, 26].
Oxidative stress and redox imbalance can disrupt the normal functioning of neurotransmitter systems in the brain, particularly the glutamate system [27]. Glutamate is an excitatory neurotransmitter that plays a crucial role in synaptic transmission and cognitive processes [27]. Imbalances in glutamate levels or receptor functioning have been implicated in the pathophysiology of schizophrenia and may contribute to symptoms such as cognitive impairments, hallucinations, and disorganized thinking [28]. In addition, increased oxidative stress can lead to damage to neuronal structures, including lipid peroxidation and protein oxidation [29]. This can disrupt neuronal membrane integrity and impair cellular functions [24]. Oxidative stress can also trigger inflammatory processes in the brain, leading to neuroinflammation. Neuroinflammation has been associated with the development and progression of schizophrenia and may contribute to the positive symptoms of the disorder [24]. Furthermore, redox imbalance in schizophrenia may involve a deficiency in the antioxidant defense system. Antioxidants such as glutathione (GSH), superoxide dismutase (SOD), and catalase (CAT) play a crucial role in neutralizing ROS and protecting cells from oxidative damage [30]. Reduced levels or impaired activity of these antioxidants may result in increased oxidative stress and cellular dysfunction [17, 31]. Also, Mitochondria, the cellular powerhouses responsible for energy production, are vulnerable to oxidative damage [32]. Oxidative stress can disrupt mitochondrial function, leading to decreased energy production, impaired cellular metabolism, and increased ROS production [12]. Mitochondrial dysfunction has been implicated in the pathophysiology of schizophrenia and may contribute to symptoms such as fatigue, cognitive impairment, and negative symptoms [33]. More importantly, oxidative stress can induce DNA damage through the formation of DNA adducts and strand breaks [34]. DNA damage can lead to genetic and epigenetic alterations that affect neuronal development, synaptic plasticity, and neurotransmitter signaling, potentially contributing to the emergence of schizophrenia symptoms [35, 36]. Diets low in anti-inflammatory nutrients or high in pro-inflammatory ones (omega-6 fatty acids are pro-inflammatory, whereas omega-3 fatty acids are anti-inflammatory) can activate and exacerbate neuroinflammation, which, if left unchecked, can lead to pathological changes in schizophrenia and worsen its symptoms [37, 38]. Previous studies reporting redox and oxidant parameters have reported changes that have been heterogeneous, with less emphasis on their relationship with the psychopathology of the disorders [39]. Therefore, this study is aimed at investigating the influence of redox activity, lipid peroxidation, and micronutrient antioxidants in the expression of clinical phenotypes of schizophrenia. We hypothesize that individuals with schizophrenia will exhibit significant redox-oxidative imbalance characterized by increased lipid peroxidation and reduced antioxidant micronutrient levels compared to healthy controls, and that these biochemical alterations will correlate with the severity and pattern of positive, negative, and general psychopathological symptoms.
This is a case-control study matched for gender and age, with no family history of mental illness in controls [40]. A total number of 220 consented drug naïve volunteers, including 120 participants with Schizophrenia and 100 apparently healthy controls, were recruited. The schizophrenia group included 65 males and 55 females (mean age 35.22 ± 12.0 years), while the control group included 54 males and 46 females (mean age 38.04 ± 13.9 years). Controls were recruited from hospital staff and community volunteers to improve representativeness and reduce potential healthy worker bias. The study participants were clients visiting the assessment unit of the Neuropsychiatric Hospital, Aro, Abeokuta, Ogun State, Nigeria. The Positive and Negative Syndrome Scale (PANSS) was applied to evaluate psychotic symptoms [41]. The sociodemographic characteristics of the study participants have been described previously [42]. This study employed a frequency-matched case–control design rather than individual (pairwise) matching. Controls were selected to achieve comparable group-level distributions of age and sex relative to cases. Matching was performed using age bands (± 3 years) and sex categories, with an approximate case-to-control ratio of 1.2:1. Because matching was frequency-based and not individual, statistical analyses were conducted using independent-sample comparisons and multivariable regression models.
Diagnosis of schizophrenia was established according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria through structured clinical assessment conducted by a senior psychiatrist, and subsequently confirmed by an independent consultant psychiatrist who was blinded to the initial diagnostic evaluation. “Drug-naïve” was defined as individuals who had never received antipsychotic medication or who had not been exposed to psychotropic treatment for at least four weeks (washout period) prior to enrollment. Participants were predominantly first-episode or early-stage presentations attending the assessment unit, and the duration of illness was obtained from clinical history where available. Inclusion criteria for cases were: (i) diagnosis of schizophrenia according to DSM-5 criteria confirmed by a consultant psychiatrist, (ii) drug-naïve or medication-free for ≥ 4 weeks prior to recruitment, (iii) age 18–60 years, and (iv) willingness to provide informed consent either by the participants or their relative. Exclusion criteria for both groups included: chronic inflammatory disease, liver or renal dysfunction, diabetes mellitus, cardiovascular disease, alcohol or substance abuse, antioxidant supplementation within the last 3 months, pregnancy/lactation, or acute infection at the time of sampling. Controls were screened by a consultant psychiatrist using a structured clinical interview to exclude psychiatric disorders and a first-degree family history of psychosis.
Prior to enrollment, all eligible participants (and, where applicable, their legally authorized representatives) were provided with detailed verbal and written information about the study objectives, procedures, potential risks, and benefits in clear and understandable language. Adequate time was given for questions and clarification before consent was obtained. Written informed consent was obtained from each participant who had decision-making capacity. For participants whose capacity to consent was temporarily impaired due to acute psychotic symptoms, written consent was obtained from a legally authorized relative or guardian, in accordance with institutional ethical guidelines. Participants were assured of confidentiality, voluntary participation, and their right to withdraw at any stage without any impact on their clinical care. All signed consent forms were securely stored, and participant data were anonymized prior to analysis. A priori sample size considerations were based on feasibility and comparable published biomarker studies; the final sample (n = 220) was considered adequate to detect moderate effect sizes between groups. Ethical approval was obtained from the Neuropsychiatric Hospital Aro Health Research Ethics Committee (Approval No: PR003/19), and all procedures complied with the Declaration of Helsinki.
Following an overnight fast (8–10 h), 10 mL of venous blood was collected between 08:00 and 10:00 h to minimize circadian variation. Five millilitres were dispensed into EDTA tubes for antioxidant enzyme assays and trace element analysis, and 5 mL into plain tubes for serum vitamin assays. Samples were centrifuged at 3,000 rpm for 10 min at 4°C within 1 h of collection. Plasma/serum aliquots were stored at −80°C and analysed within 4 weeks. Freeze–thaw cycles were avoided. Haemolysed samples were excluded due to interference with oxidative stress assays. All laboratory analyses were performed blinded to case–control status to reduce measurement bias.
Lipid peroxidation was quantified as malondialdehyde (MDA) using the thiobarbituric acid reactive substances (TBARS) method, and absorbance was measured at 532 nanometers using a UV–visible spectrophotometer (Mindray BA-88A, Mindray Bio-Medical Electronics Co., China). Concentrations were calculated using an MDA standard curve and expressed as µmol/L [43, 44]. CAT activity was determined by monitoring the decomposition of hydrogen peroxide at 240 nm and was recorded spectrophotometrically [45, 46]. SOD was determined by its ability to inhibit autooxidation of epinephrine to adrenochrome at pH 10.2, which was monitored at 480 nm [47, 48]. Total antioxidant capacity (TAC) was estimated by the phosphomolybdenum method with absorbance measured at 695 nm [49, 50]. Zinc and Selenium were analysed by atomic absorption spectrophotometry (AAS; PerkinElmer AAnalyst 200). Calibration standards were prepared from certified reference solutions in accordance with the manufacturer’s instructions, and measurements were performed under standard operating conditions. Samples were digested with nitric acid prior to analysis. Zinc was read at 213.9 nm and selenium at 196.0 nm. Internal quality controls and blanks were run with each batch [51]. Vitamin C was determined using the 2,4-dinitrophenylhydrazine (DNPH) colorimetric method and read at 520 nm. Vitamin E (α-tocopherol) was quantified using the Emmerie–Engel reaction with spectrophotometric detection at 536 nm [52, 53].
Statistical analysis was performed using IBM Statistical Package for the Social Sciences (SPSS v. 25). Continuous variables were evaluated using the Student’s t-test. Pearson correlation was used to determine the association of psychopathology scores with biochemical parameters. Logistic regression analyses were performed to explore which characteristics were related to the risk of schizophrenia. Predictor variables were entered into logistic regression models using their original measurement units without standardization or rescaling. Given the exploratory nature of the study and moderate sample size, regression assumptions were assessed pragmatically through inspection of data distributions and correlation matrices. Statistical significance was set at p < 0.05.
The mean scores for the Positive, Negative, and General Psychopathology scores in the case are 19.76 ± 7.1, 20.14 ± 7.7, and 35.54 ± 12.1, respectively (Figure 1). The serum levels of both enzymatic and non-enzymatic antioxidants in Schizophrenia and Controls were presented in Table 1. There was a significant increase in enzymatic antioxidant levels in individuals with schizophrenia compared to the controls. Specifically, levels of SOD and CAT were elevated in schizophrenia cases (SOD: 19.58 ± 0.80 vs. 10.12 ± 0.45 U/L; CAT: 41.73 ± 1.81 vs. 21.33 ± 0.98 U/L; p < 0.05). In contrast, non-enzymatic antioxidants were significantly reduced in schizophrenia patients compared to controls. A reduction in GSH levels was observed in schizophrenia (GSH: 14.5 ± 0.28 vs. 15.9 ± 1.59 µmol/L). Similarly, micronutrients, non-enzymatic antioxidants, zinc (1.8 ± 0.01 vs. 2.7 ± 0.02 mg/L), selenium (0.08 ± 0.01 vs. 0.10 ± 0.01 mg/L), vitamin C (12.98 ± 0.49 vs. 15.08 ± 0.37 mg/L), and TAC (0.65 ± 0.0157 vs 0.81 ± 0.0148 mmol/L), (p < 0.05) were lower in cases compared to controls. However, vitamin E showed no significant difference between test and control (1.74 ± 0.15 and 1.91 ± 0.12 mg/dL), p > 0.05. In addition, MDA, a product of lipid peroxidation, was significantly higher in patients with schizophrenia compared to controls (2.32 ± 0.19 and 1.17 ± 0.10 µmol/L), p < 0.05. Table 2 showed the logistic regression predictors of schizophrenia. The regression model showed that an increase in MDA levels was associated with higher odds of schizophrenia (OR = 2.017). Reductions in GSH, TAC, zinc, selenium, and vitamin C levels were also associated with increased odds of schizophrenia. Pearson correlation of MDA with antioxidant parameters reveals that MDA was correlated with certain antioxidant parameters (0.281, 0.179, –0.247, and –0.221) (p < 0.05) (Table 3). On the other hand, Zinc, Selenium, vitamin C, and vitamin E did not show any significant correlation (–0.101, –0.139, –0.060, and –0.045) (p > 0.05). Table 4 presents the correlation analysis between oxidative stress parameters and the three symptom domains of the PANSS scale. A negative correlation between GSH and positive symptoms (r = –0.285, p = 0.013). No correlation was found for other variables such as CAT, SOD, vitamin C, vitamin E, and TAC.
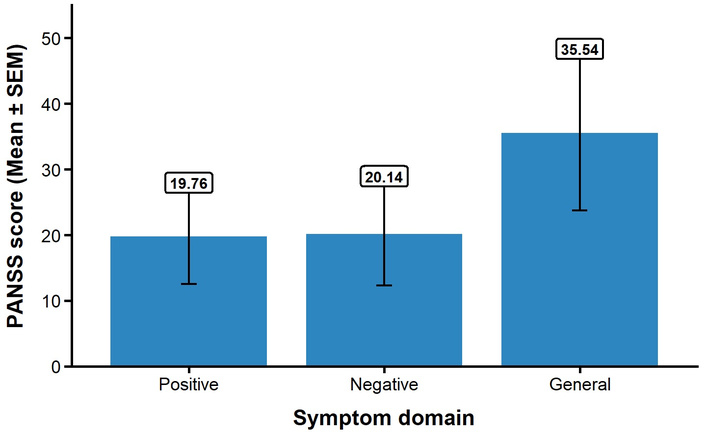
PANSS symptom scores in schizophrenia participants (n = 120). Bars represent mean scores for Positive, Negative, and General symptom domains. Error bars indicate standard error of the mean (SEM). PANSS: Positive and Negative Syndrome Scale.
Serum oxidative stress markers, antioxidant indices, and micronutrient levels in participants with schizophrenia and controls.
| Variables | Schizophrenia = 120 (Mean ± SEM) | Controln = 100 (Mean ± SEM) | t-value | p-value |
|---|---|---|---|---|
| Zinc (mg/L) | 1.8 ± 0.01 | 2.7 ± 0.02 | –3.548 | 0.001* |
| Selenium (mg/L) | 0.08 ± 0.01 | 0.10 ± 0.01 | –2.296 | 0.024* |
| Vitamin C (mg/L) | 12.98 ± 0.49 | 15.08 ± 0.37 | –3.225 | 0.002* |
| Vitamin E (mg/dL) | 1.74 ± 0.15 | 1.91 ± 0.12 | –0.875 | 0.383 |
| TAC (mmol/L) | 0.65 ± 0.02 | 0.81 ± 0.01 | –7.274 | < 0.001* |
| MDA (µmol/L) | 2.32 ± 0.19 | 1.17 ± 0.10 | 4.878 | < 0.001* |
| GSH (µmol/L) | 14.5 ± 0.28 | 15.9 ± 1.59 | –8.647 | < 0.001* |
| CAT (U/L) | 41.73 ± 1.81 | 21.33 ± 0.98 | 9.915 | < 0.001* |
| SOD (U/L) | 19.58 ± 0.80 | 10.12 ± 0.45 | 9.439 | < 0.001* |
*Significant at p < 0.05. Values are presented as Mean ± SEM. SEM was used to reflect the precision of the estimated group mean for inferential comparisons between the schizophrenia and control groups. TAC: total antioxidant capacity; MDA: malondialdehyde; GSH: glutathione; CAT: catalase; SOD: superoxide dismutase.
Logistic regression showing predictors of schizophrenia.
| Variables | Odds ratio | 95% CI | p-value |
|---|---|---|---|
| MDA | 2.017 | 1.466–3.027 | < 0.001 |
| TAC | 0.904 | 0.871–0.939 | < 0.001 |
| SOD | 1.593 | 1.347–1.886 | < 0.001 |
| CAT | 1.190 | 1.124–1.259 | < 0.001 |
| GSH | 0.553 | 0.439–0.696 | < 0.001 |
| Zn | 0.016 | 0.001–0.189 | 0.001 |
| Se | 0.369 | 0.166–0.822 | 0.015 |
| Vitamin C | 0.861 | 0.781–0.949 | 0.003 |
| Vitamin E | 0.880 | 0.661–1.771 | 0.380 |
MDA: malondialdehyde; TAC: total antioxidant capacity; SOD: superoxide dismutase; CAT: catalase; GSH: glutathione.
Pearson correlation of MDA with antioxidant parameters.
| Variables | MDA |
|---|---|
| SOD | r = 0.281, p = 0.001 |
| CAT | r = 0.179, p = 0.034 |
| GSH | r = –0.247, p = 0.003 |
| TAC | r = –0.221, p = 0.009 |
| Zn | r = –0.101, p = 0.235 |
| Se | r = –0.139, p = 0.156 |
| Vitamin C | r = –0.060, p = 0.483 |
| Vitamin E | r = –0.045, p = 0.594 |
r = Pearson correlation. SOD: superoxide dismutase; CAT: catalase; GSH: glutathione; TAC: total antioxidant capacity.
Pearson correlation of biochemical parameters with positive, negative, and general symptoms in the schizophrenia population.
| Biochemical variable | Positive symptoms | Negative symptoms | General symptoms |
|---|---|---|---|
| MDA | r = –0.095, p = 0.417 | r = 0.054, p = 0.648 | r = 0.033, p = 0.77 |
| Zinc | r = –0.071, p = 0.544 | r = 0.152, p = 0.193 | r = 0.075, p = 0.521 |
| Selenium | r = –0.086, p = 0.923 | r = 0.021, p = 0.906 | r = –0.017, p = 0.902 |
| Vitamin C | r = 0.156, p = 0.183 | r = 0.156, p = 0.182 | r = 0.037, p = 0.753 |
| Vitamin E | r = –0.018, p = 0.874 | r = –0.003, p = 0.982 | r = –0.100, p = 0.396 |
| CAT | r = –0.187, p = 0.473 | r = –0.008, p = 0.947 | r = –0.115, p = 0.325 |
| SOD | r = –0.030, p = 0.790 | r = –0.067, p = 0.569 | r = –0.177, p = 0.128 |
| GSH | r = –0.285, p = 0.013 | r = 0.101, p = 0.390 | r = –0.029, p = 0.804 |
| TAC | r = –0.083, p = 0.458 | r = 0.059, p = 0.616 | r = 0.151, p = 0.197 |
r = Pearson correlation. MDA: malondialdehyde; CAT: catalase; SOD: superoxide dismutase; CAT: catalase; GSH: glutathione; TAC: total antioxidant capacity.
Schizophrenia is a disorder of multiple etiological pathways, which converge into a common pathway and account for the array of clinical symptoms [54]. Free radicals mediated abnormalities have been implicated in the development of such important consequences in schizophrenia [16, 55]. The elevated lipid peroxidation observed in this study may reflect oxidative imbalance in schizophrenia, rather than serving as a determining factor in the disorder. The increased serum MDA activity in schizophrenia patients observed is consistent with previous findings [14, 56]. This MDA activity serves as a marker of peroxidative damage to the phospholipid membrane, a process that has been suggested to relate to neuronal membrane instability, which can result in disruption of the cell cycle and neurotransmitter release and uptake [9]. Oxidative stress has been suggested to contribute to the pathophysiology of schizophrenia; in particular, the oxidative damage in lipids, proteins, and DNA seen in schizophrenia may impair cell viability and function, and may be associated with increased disease risk [55, 57]. The increased activity of SOD and CAT observed in this study is consistent with previous findings [58, 59], which may represent an adaptive or compensatory response to elevated oxidative stress resulting from excessive free radical production [60].
Furthermore, evidence points towards an alteration in the activities of the enzymatic and non-enzymatic antioxidant system, which is clearly shown in the low level of non-enzymatic antioxidant enzymes, which agrees with a previous study [29, 61]. Plasma level of vitamin C decreases in schizophrenia related to that of normal controls. The antioxidant function of ascorbic acid may protect against dopamine dysregulation-induced neurodegenerative processes [62]. Although vitamin E did not show a statistically significant difference in the present study, its role in antioxidant defense through potential synergistic interactions with vitamin C in redox regulation remains biologically plausible [44, 63]. Previous studies have reported that vitamin C supplementation may reduce oxidative stress and contribute to clinical improvement in schizophrenia, possibly reflecting increased antioxidant requirements under conditions of elevated redox activity [64, 65].
Lower zinc levels were also demonstrated in this study, and a recent review suggests possible links between micronutrient status and psychosis [66]. The observed differences may reflect nutritional, metabolic, or lifestyle factors rather than direct biological effects of schizophrenia. Selenium and zinc are essential micronutrients with antioxidant properties, and while differences were detected between groups, most micronutrients and antioxidant indices did not correlate significantly with symptom subscales, indicating that lower circulating levels do not necessarily correspond to more severe clinical symptoms [25, 67, 68]. Although Selenium and Zinc are essential micronutrients that have important roles in reducing oxidative stress and protecting DNA from attack by ROS, interestingly, our study showed a difference in selenium levels relative to the control. Antioxidant micronutrients such as Zn, Se, vitamin E, vitamin C, and TAC show no relationship with positive, negative, and general symptoms of schizophrenia in this study [69, 70]. This may be due to the population presenting with mild to moderate symptoms and being drug naïve; the situation might be different if they are chronic cases [71].
A significant inverse correlation was found between GSH levels and positive symptoms of schizophrenia. GSH regulates glutamate metabolism and maintains redox balance in the brain [72]. Dysregulation of the glutamate neurotransmitter system, particularly involving the NMDA receptor, has been linked to schizophrenia [26, 28]. However, the strength of this association was modest (r ≈ −0.285), indicating only a weak relationship. GSH depletion may disrupt glutamate neurotransmission, which may contribute to the development of positive symptoms in schizophrenia [73]. This is supported by reduced GSH levels in schizophrenia patients in this study. Additionally, genetic variations in the glutamate-cysteine ligase catalytic subunit (GCLC), a key enzyme involved in GSH synthesis, have been associated with an increased risk of schizophrenia [74]. Inflammation and immune dysregulation have also been implicated in schizophrenia [75]. GSH helps modulate inflammation and protect against neuroinflammation. Disturbances in GSH levels may therefore contribute to neuroinflammatory processes, further influencing positive symptoms [76]. Other antioxidant markers, including CAT, SOD, vitamin C, vitamin E, selenium, and zinc, showed no significant relationship with symptom severity, which may result from variations in redox regulation across different illness phases [17, 55].
Abnormality in glutamatergic neurotransmission mediated by NMDA receptor hypofunction has been linked with the pathophysiology of schizophrenia [77]. The link between the glutamatergic system and GSH may suggest why positive symptoms correlated negatively with GSH concentration in this study [78, 79]. Previous studies have demonstrated that GSH is significantly associated with positive symptoms of PANSS [80]. However, other studies have found a link between GSH and negative symptoms [81]. Other antioxidants did not correlate with all the symptoms of the PANSS. SOD and CAT, important antioxidant enzymes, correlated positively with MDA concentration, while negatively with GSH and TAC levels. These patterns likely reflect compensatory biochemical responses to oxidative stress rather than direct clinical phenotype effects [82]. It is important to note that the other antioxidant parameters did not exhibit significant positive correlations with MDA; in fact, some showed negative correlations. For example, reduced GSH and TAC were negatively correlated with MDA, indicating that higher levels of MDA are associated with lower levels of these antioxidants [31, 83]. Overall, the correlations observed suggest complex interactions between oxidative stress markers and antioxidant parameters in individuals with schizophrenia, but they do not demonstrate that these biomarkers determine clinical manifestations [31, 56].
This study has several limitations that should be considered when interpreting the findings. First, dietary intake was not formally assessed using validated tools such as food frequency questionnaires or 24-hour dietary recalls. Consequently, reduced serum micronutrient levels may reflect low dietary intake, altered absorption or metabolism, increased oxidative utilization, or a combination of these factors. The relative contribution of these mechanisms could not be disentangled within the current design. Second, smoking status was not systematically recorded or matched between groups. Given the higher prevalence of cigarette smoking among individuals with schizophrenia and its established role in increasing oxidative stress while reducing circulating antioxidants (e.g., vitamin C), smoking represents a potential confounder that may have influenced the observed associations. More broadly, several potential confounding variables, including dietary patterns, physical activity, socioeconomic status, body mass index, environmental exposures, and other lifestyle factors, were not comprehensively measured or statistically controlled. The absence of adjustment for these variables may have introduced residual confounding [84–86]. Third, although efforts were made to recruit controls from both hospital staff and community volunteers to improve representativeness, the inclusion of hospital staff may have introduced some degree of selection bias, as healthcare workers may differ in health awareness, nutritional habits, or access to care compared to the general population. However, this potential bias is likely attenuated by the mixed recruitment strategy incorporating community participants [87]. Fourth, the cross-sectional case–control design precludes causal inference. The findings therefore reflect associations rather than directionality or temporality, and it cannot be determined whether redox imbalance and micronutrient alterations are causes, consequences, or correlates of schizophrenia. Fifth, certain statistical considerations should be acknowledged. Tests of normality were not formally conducted prior to applying parametric analyses, and non-parametric alternatives were not systematically explored. In addition, multicollinearity diagnostics among predictor variables were not formally assessed, which may affect the stability and interpretability of regression coefficients. Furthermore, predictors were analyzed in their original measurement scales; as a result, some odds ratios may appear disproportionately large or small per unit change depending on the scale of measurement [88]. These effect estimates should therefore be interpreted with caution, particularly when comparing variables with different units or ranges. While the findings provide important insights into the interplay between redox-oxidative stress and micronutrients in schizophrenia, they should be interpreted within the context of these methodological and analytical constraints [89].
This study demonstrates that drug-naïve Nigerian individuals with schizophrenia exhibit a distinct pattern of redox imbalance characterized by increased lipid peroxidation, altered enzymatic antioxidant activity, and reduced circulating levels of selected non-enzymatic antioxidants and micronutrients when compared with healthy controls. These biochemical alterations may be associated with clinical symptom dimensions, with a modest inverse correlation observed between GSH levels and positive symptom severity. The findings support the growing body of evidence implicating redox dysregulation in the pathophysiology of schizophrenia, particularly in early or untreated stages of the illness. Future longitudinal and interventional studies are warranted to clarify temporal relationships, explore underlying mechanisms, and determine whether modulation of oxidative stress pathways may have therapeutic relevance in schizophrenia.
AAS: atomic absorption spectrophotometry
CAT: catalase
GSH: glutathione
MDA: malondialdehyde
PANSS: Positive and Negative Syndrome Scale
ROS: reactive oxygen species
SOD: superoxide dismutase
TAC: total antioxidant capacity
TBARS: thiobarbituric acid reactive substances
We acknowledge the invaluable support received from the staff of the Neuropsychiatric Hospital, Aro, and the University of Lagos throughout the course of this project. Special appreciation is extended to the Centre for Genomics of Non-Communicable Diseases and Personalized Healthcare, University of Lagos, Akoka, Lagos, Nigeria, for their technical guidance, resources, and collaborative efforts that significantly contributed to the success of this work.
TAO: Methodology, Investigation, Resources, Data curation, Writing—original draft, Project administration, Visualization. SJA: Methodology, Formal analysis, Resources. BMS: Methodology, Formal analysis, Resources. BWG: Methodology, Formal analysis, Resources. OAO: Methodology, Formal analysis, Investigation, Resources. OJO: Formal analysis, Investigation. AO: Resources, Data curation, Visualization. JDA: Conceptualization, Data curation, Writing—review & editing, Supervision, Project administration. OA: Conceptualization, Writing—original draft, Writing—review & editing, Supervision, Project administration, Visualization. All authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
This study was approved by the Health Research Ethics Committee of Neuropsychiatric Hospital, Aro, Abeokuta (PR003/19). The study adhered to the principles of the Declaration of Helsinki (2013 revision).
Written informed consent was obtained from each participant.
Not applicable.
All data generated and gathered during this study are available to the corresponding author on reasonable request.
This research received no external funding.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

