 Review
Review

Affiliation:
1Department of Physiology, College of Medicine and Health Sciences, Afe Babalola University, Ado-Ekiti 360102, Nigeria
Email: aduom@abuad.edu.ng
ORCID: https://orcid.org/0000-0002-9002-4940
Affiliation:
2Department of Human Anatomy, College of Medicine and Health Sciences, Afe Babalola University, Ado-Ekiti 360102, Nigeria
ORCID: https://orcid.org/0009-0003-1327-6217
Affiliation:
3Department of Physiology, College of Medicine, University of Medical Sciences, Ondo City 351104, Nigeria
ORCID: https://orcid.org/0000-0002-9127-7130
Affiliation:
4Department of Microbiology, Bamidele Olumilua University of Education, Science and Technology, Ikere-Ekiti 361101, Nigeria
ORCID: https://orcid.org/0009-0003-2279-7313
Affiliation:
1Department of Physiology, College of Medicine and Health Sciences, Afe Babalola University, Ado-Ekiti 360102, Nigeria
ORCID: https://orcid.org/0009-0009-7021-098X
Affiliation:
4Department of Microbiology, Bamidele Olumilua University of Education, Science and Technology, Ikere-Ekiti 361101, Nigeria
ORCID: https://orcid.org/0009-0000-5861-9138
Explor Neurosci. 2026;5:1006133 DOI: https://doi.org/10.37349/en.2026.1006133
Received: December 26, 2025 Accepted: March 30, 2026 Published: April 21, 2026
Academic Editor: Ayan Mohamud Yusuf, University of Duisburg-Essen, Germany
The article belongs to the special issue Depression: From Pathophysiology to Treatment Innovation
Major depressive disorder (MDD) is increasingly understood as a multifactorial psychiatric disorder involving interacting neural, immune, metabolic, and microbial processes. Within this framework, the microbiota–gut–brain axis and mitochondrial bioenergetics have emerged as potentially intersecting contributors to depressive symptomatology. Preclinical studies suggest that microbial metabolites—especially short-chain fatty acids (SCFAs)—can influence oxidative phosphorylation, redox balance, neuroinflammation, and synaptic plasticity, whereas inflammatory signals such as lipopolysaccharide may disrupt mitochondrial dynamics. However, the strength of evidence is uneven: mechanistic support is strongest in cell and animal models, whereas human data remain heterogeneous and largely associative. This narrative review critically synthesizes current evidence on microbiota–mitochondria crosstalk in MDD, distinguishing established findings from emerging hypotheses. It also examines recent psychobiotic trials, metabolomic and biomarker studies, and microglia–mitochondria mechanisms, and discusses the translational limitations that currently constrain clinical application. Overall, this axis represents a plausible and clinically relevant framework for hypothesis generation and adjunctive intervention development, but it should not yet be regarded as a fully validated causal pathway or stand-alone therapeutic target in MDD.
Major depressive disorder (MDD) is one of the most prevalent and disabling psychiatric conditions worldwide, affecting more than 280 million people [1]. Historically, monoamine theories, focusing on dysfunctions in serotonin, norepinephrine, and dopamine, have dominated neurochemical investigations of MDD. However, existing pharmacotherapies based on these theories often exhibit minimal efficacy, slow onset of action, and high relapse rates, leaving approximately 30 percent of patients unresponsive to treatment [2, 3]. These limitations have encouraged broader models of depression that incorporate neuroimmune dysfunction, altered metabolism, chronic low-grade inflammation, and bidirectional gut–brain signaling [4, 5]. A central component of this expanded framework is the gut–brain axis (GBA), a communication network linking the gastrointestinal tract and the central nervous system (CNS) through neural, endocrine, immune, and metabolic pathways [6, 7]. Within this system, the gut microbiota produces neurotransmitter precursors, short-chain fatty acids (SCFAs), bile-acid derivatives, and other signaling molecules that may influence stress responsivity, affective behavior, and cognitive function [8–10]. At the same time, converging evidence suggests that mitochondrial dysfunction contributes to MDD through impaired oxidative phosphorylation, excessive reactive oxygen species (ROS) production, altered synaptic energetics, and inflammatory amplification [11–14].
These converging literatures suggest that microbiota–mitochondria crosstalk may represent an important bioenergetic interface within the GBA. SCFAs such as butyrate, propionate, and acetate have been proposed to influence mitochondrial biogenesis, redox homeostasis, and inflammatory tone, whereas microbial endotoxins such as lipopolysaccharide (LPS) may impair mitochondrial dynamics and promote neuroinflammatory signaling [14–18]. Yet several aspects of this model remain unresolved. Much of the mechanistic literature is derived from rodent, germ-free (GF), or in vitro systems, and human studies are often cross-sectional, small, and confounded by diet, antidepressant exposure, and clinical heterogeneity. This review, therefore, aims to provide an updated and critical overview of microbiota–mitochondria crosstalk within the GBA; to distinguish well-supported observations from emerging hypotheses; to evaluate preclinical and clinical evidence relevant to MDD; and to discuss psychobiotics, prebiotics, metabolomics-guided biomarkers, and mitochondrial-targeted strategies while highlighting current translational barriers.
The GBA represents a multifaceted, bidirectional communication network that integrates signals from the gastrointestinal tract with the CNS. This intricate system encompasses neural, endocrine, metabolic, and immune pathways, all of which profoundly impact mental health by regulating mood, stress responses, and cognition [19, 20]. Communication within this axis occurs through both direct and indirect means. Direct pathways involve microbiota-derived metabolites acting on neural cells, while indirect pathways modulate host immune responses and inflammatory signaling [21].
The vagus nerve, the tenth and longest cranial nerve, is an essential component of the neural pathway, originating in the medulla oblongata and innervating various visceral organs, including the heart, lungs, spleen, and gut. As the principal parasympathetic fiber, it forms a critical anatomical and functional link between the gut and the brain [22]. Approximately 80% of vagal fibers are afferent, transmitting sensory information from the periphery to the brain, whereas the remaining fibers are predominantly efferent and convey motor output [23, 24]. Vagal signaling is implicated in gastrointestinal, metabolic, and neuropsychiatric processes, including those relevant to depression [23, 24]. Intestinal vagal afferents project to the nucleus tractus solitarius (NTS), relaying visceral information to higher autonomic centers in the brain [24]. These fibers monitor microbial metabolites and transmit signals to the brain, influencing mood and stress levels via neural pathways (Figure 1) [24]. Beyond neural interconnections, a complex interplay exists between the gut microbiota, the immune system, and the CNS. Gut microbes regulate immune homeostasis through their metabolic products [25]. The oral and gastrointestinal mucosa, particularly the mucosa-associated lymphoid tissue (MALT), plays a significant role in microbe–immune interactions. Commensal microorganisms constantly interact with this network to maintain immune stability [25]. Microbial peptides and polysaccharides promote the growth of regulatory T cells (Tregs) and the production of anti-inflammatory cytokines like IL-10, influencing innate lymphoid cell activity [26, 27]. Certain microbes, such as Anaeroplasma species, modulate T follicular helper cells, which subsequently adjust IgA production—the most common mucosal antibody. This bidirectional interaction ensures that microbial composition is influenced by the immune system, and microbial metabolites impact immune cell activities [25]. Peripheral immune cells express receptors for neurotransmitters (dopamine and serotonin), allowing gut-derived neuromodulators to regulate cytokine secretion, migration, and differentiation [25]. Immune activation leads to the production of pro-inflammatory cytokines (e.g., IL-6, TNF-α, IL-1β), which can cross or disrupt the blood-brain barrier (BBB), interfering with neurotransmission, altering neuroplasticity, and thereby contributing to MDD pathophysiology [28]. Furthermore, increased gut permeability allows endotoxins, such as LPS, to enter the circulation, fostering neuroinflammation and depressive symptoms [29–31]. The gut also functions as the largest endocrine organ in the human body, with its endocrine arm of the GBA mediated by enteroendocrine cells (EECs) [32]. EECs release hormones that control intestinal motility, secretion, and neurotransmission. Notably, approximately 90% of the body’s serotonin is produced in the gut by enterochromaffin cells [32]. Gut-derived serotonin stimulates vagal afferent fibers, which relay impulses to the NTS in the brainstem. Serotonergic and noradrenergic projections then extend to higher brain centers, including the hypothalamus, amygdala, and cortex, where they contribute to emotion and cognition [32]. This EEC-vagal loop interacts with the hypothalamic–pituitary–adrenal (HPA) axis, forming a neuroendocrine feedback loop sensitive to environmental stressors [15]. An imbalance in this circuit is a significant contributor to the mood and physiological changes associated with stress in depressive disorders.
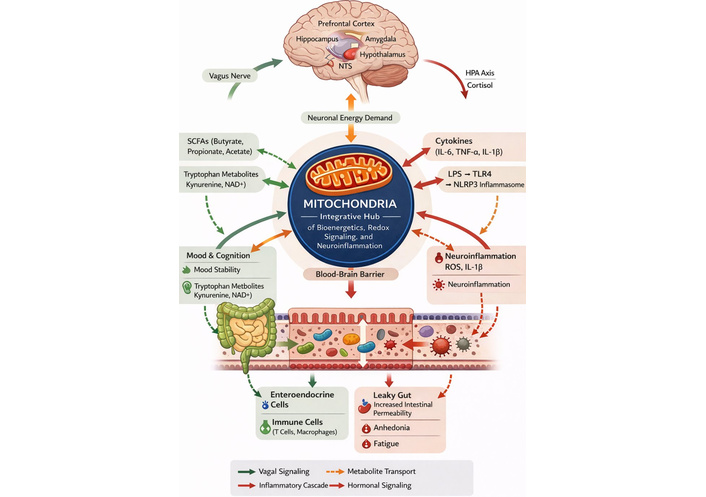
Gut–brain axis pathways in MDD, highlighting mitochondria as an integrative hub linking microbial metabolites, immune signaling, and neuroinflammation. BBB: blood-brain barrier; HPA: hypothalamic–pituitary–adrenal; IL: interleukin; LPS: lipopolysaccharide; MDD: major depressive disorder; NAD+: nicotinamide adenine dinucleotide; NLRP3: nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3; NTS: nucleus tractus solitarius; ROS: reactive oxygen species; SCFAs: short-chain fatty acids; TLR4: toll-like receptor 4; TNF-α: tumor necrosis factor alpha.
Gastrointestinal microbial metabolites are crucial mediators of gut-brain communication. Among these, SCFAs—acetate, propionate, and butyrate—are the most extensively researched, produced through the fermentation of dietary fibers [23]. SCFAs exert beneficial effects on mental health by enhancing tryptophan hydroxylase 1 (TPH1), a rate-limiting enzyme in serotonin production, and by strengthening both gut and BBB integrity [15, 32]. They serve as energy sources for colonocytes, stimulate the maturation of microglia, and function as communication molecules within the CNS [23]. Specifically, butyrate possesses anti-inflammatory properties, promoting IL-10 production by Tregs to maintain immune tolerance and reduce systemic inflammation [33]. Conversely, low concentrations of butyrate are associated with depressive-like behavior and increased neuroinflammation [34–36]. SCFAs have also been proposed to influence central neuroimmune signaling and barrier integrity, thereby offering plausible neuroprotective effects, although the exact transport and receptor mechanisms remain context-dependent [15, 37].
Tryptophan derivatives and bile acids also regulate systemic inflammation and mitochondrial function. Dysfunction of bile acid signaling can disturb tight-junction homeostasis and compromise barrier function [38]. Furthermore, the tryptophan metabolite system, particularly through the kynurenine pathway, impacts neurotoxicity and oxidative stress homeostasis [18]. Collectively, these processes position the microbiota as a major controller of innate and adaptive immune responses, directly applicable to the pathophysiology of MDD.
Intestinal permeability, often referred to as “leaky gut”, can be exacerbated by stress, microbial imbalance, dietary factors, and dysbiosis. When this barrier is compromised, microbial antigens and toxins can translocate into the systemic circulation, triggering inflammatory processes and the activation of pro-inflammatory cytokines [24]. This disturbance has been implicated in the etiology of various neurological and psychiatric disorders, including Alzheimer’s, Parkinson’s, anxiety, and depression [21]. Clinical research has linked MDD to increased gut permeability, as demonstrated by lactulose/mannitol challenge tests showing dysfunctional barriers in adolescents with depressive symptoms [39]. Similarly, bacterial translocation and the passage of microbial by-products through the gut barrier have been associated with systemic inflammatory processes and altered neurobehavior [40]. Analogous to the intestinal barrier, the BBB maintains CNS homeostasis. The BBB is composed of endothelial cells, astrocyte end-feet, and tight-junction proteins like occludin and claudin [41, 42]. Claudin-5, a major regulator of BBB permeability, has been linked to dysfunction in depression, obsessive-compulsive disorder, bipolar disorder, and schizophrenia [43]. The combined disruption of both the gut barrier and the BBB reinforces a self-sustaining loop of inflammatory processes that underlie MDD pathogenesis.
Mitochondrial dysfunction has emerged as a key pathophysiological mechanism in MDD. These organelles are fundamental for producing adenosine triphosphate (ATP), which powers critical cellular functions [11]. The brain, with its exceptionally high metabolic rate and limited glucose storage capacity, relies heavily on continuous mitochondrial ATP production through oxidative phosphorylation to sustain neuronal function [44]. In MDD, mitochondrial oxidative phosphorylation is frequently impaired, leading to reduced ATP levels, synaptic dysfunction, and increased neuronal vulnerability [45–47]. Consistent findings in postmortem human brain tissue and MDD animal models reveal structural abnormalities of mitochondria and ATP depletion, which contribute to impaired neurotransmitter release and loss of synaptic plasticity [14, 48, 49]. Furthermore, impaired mitochondrial trafficking and localization at synapses may exacerbate energy imbalance and compromise the stability of neural circuits [45]. A primary driver of mitochondrial pathology in MDD is oxidative stress and inflammation. Excessive production of ROS can damage mitochondrial DNA (mtDNA), which lacks the protective histones of nuclear DNA, leading to mutagenesis, defective transcription, and a vicious cycle of oxidative stress [47]. Damaged mtDNA can also be released as damage-associated molecular patterns (DAMPs), activating inflammatory pathways, including NF-κB-related signaling [47, 50]. Chronic neuroinflammation, in turn, impairs energy metabolism, exacerbates purine imbalance, and worsens depressive pathology [51].
Beyond energy impairment, mitochondrial dysfunction plays a crucial role in apoptotic signaling and neuronal death. Overproduction of ROS and depolarization of the mitochondrial membrane can open the mitochondrial permeability transition pore, leading to the release of cytochrome c and the initiation of apoptosis [52, 53]. N-methyl-D-aspartate receptor (NMDAR) overactivation can promote calcium overload, aggravate mitochondrial dysfunction, disrupt mitochondrial membrane potential, and facilitate cytochrome c release into the cytosol [54, 55]. Subsequent activation of caspases triggers neuronal cell death, contributing to synaptic loss and the manifestation of depressive symptoms [56].
Recent literature also indicates that dysregulation of mitochondrial dynamics—fusion, fission, and mitophagy—may contribute to depressive phenotypes [14, 48]. Impaired mitochondrial turnover can promote the accumulation of damaged organelles, reduced ATP generation, and inflammatory signaling, but most mechanistic data derive from preclinical models rather than direct human brain measurements. Emerging peripheral biomarker studies provide more concrete, although still preliminary, human evidence. In a relatively large peripheral blood mononuclear cell analysis, patients with MDD showed increased expression of ER-stress genes (XBP1u, XBP1s, and ATF4) and the mitochondrial biogenesis-related gene MFN2 after adjustment for age, sex, BMI, and medication status, while ASC and MFN2 expression correlated positively with C-reactive protein [57–59]. These findings are important because they move the field beyond generic oxidative-stress narratives toward assayable signatures of organelle stress, inflammasome activation, and mitochondrial remodeling. However, these biomarkers remain peripheral rather than brain-specific, are largely cross-sectional, and have not yet been standardized for diagnosis, prognosis, or treatment stratification. Collectively, the available data support mitochondrial dysfunction as a plausible contributor to MDD pathophysiology, but not as a singular or universally dominant mechanism (Table 1).
Summary of mechanistic pathways.
| Mitochondrial dysfunction | Pathophysiological mechanism | Neurobiological consequences of MDD |
|---|---|---|
| Impaired oxidative phosphorylation | Reduced ATP and synaptic energy | Cognitive and mood deficits |
| Oxidative stress and mtDNA damage | Excessive ROS and apoptosis | Neurodegeneration, neuronal loss |
| Altered fission/fusion dynamics | Fragmented mitochondria | Impaired plasticity, stress vulnerability |
| Defective mitophagy | Accumulation of damaged mitochondria | Chronic inflammation |
| Epigenetic dysregulation | Silencing of mitochondrial biogenesis genes | Reduced neurogenesis, metabolic failure |
MDD: major depressive disorder; ATP: adenosine triphosphate; mtDNA: mitochondrial DNA; ROS: reactive oxygen species.
Building upon the understanding of mitochondrial dysfunction as a core mechanism in MDD, this section explores the profound influence of the gut microbiota on mitochondrial activity. The gut microbiota exerts a tremendous impact on brain physiology and mitochondrial function via the GBA, controlling neural energy metabolism, oxidative homeostasis, and inflammation [60, 61]. This regulation is mediated by microbial metabolites, signaling molecules, and host-microbe interactions that collectively modulate mitochondrial gene expression and bioenergetic capacity.
SCFAs—acetate, propionate, and butyrate—are key microbial metabolites resulting from the fermentation of dietary fibers and are among the most plausible mediators of microbiota–mitochondria crosstalk [37, 62]. Butyrate can function as a histone deacetylase inhibitor and, in experimental systems, has been linked to AMPK activation, PGC-1α signaling, and increased expression of mitochondrial biogenesis genes [17, 63, 64]. These mechanisms may improve oxidative phosphorylation efficiency, reduce oxidative stress, and support neuronal resilience [65, 66]. SCFAs may also influence neurotrophic and neuroimmune signaling, although evidence for effects on brain-derived neurotrophic factor (BDNF), enterochromaffin serotonin synthesis, and glial immunometabolism in MDD remains indirect and largely preclinical [37, 67]. Nevertheless, direct demonstration that these pathways operate in the same way in humans with MDD remains limited. Most data come from cell culture, intestinal tissues, or rodent studies, and SCFA effects are likely context-dependent, varying with dose, host diet, microbiome composition, and BBB transport [67].
Certain gut microbes, including Bifidobacterium and Lactobacillus species, have been proposed to influence host NAD+ metabolism and downstream sirtuin signaling [68, 69]. This hypothesis is biologically attractive because SIRT1/3 activity can promote PGC-1α deacetylation, mitochondrial biogenesis, and antioxidant defense [70, 71]. However, evidence linking specific gut taxa to clinically meaningful changes in host NAD+ availability in MDD is still indirect. At present, the microbiome–NAD+–sirtuin axis should be considered an emerging mechanism rather than an established human pathway, and future work will need integrated metabolomic and flux-based studies to determine its magnitude and disease relevance [72].
Beyond beneficial metabolites, pro-inflammatory microbial products such as LPS may impair mitochondrial function through TLR4 signaling, ROS generation, and inflammasome activation [73–76]. In experimental systems, these processes can alter mitochondrial dynamics, enhance mitochondrial ROS generation, and amplify inflammasome-associated cytokine release [75–78]. Recent work has added mechanistic depth by implicating microglial organelle crosstalk in this process. In a chronic social defeat stress model, augmented endoplasmic reticulum-mitochondria contacts in hippocampal microglia promoted formation of the IP3R3–GRP75–VDAC1 complex, increased mitochondrial calcium transfer, intensified mitochondrial damage and superoxide generation, and facilitated NLRP3 inflammasome activation, together contributing to depressive-like behavior [79]. Complementary evidence indicates that inflammatory stimulation can also drive mitochondrial fragmentation and energetic reprogramming in reactive microglia, supporting the idea that microglial mitochondrial state is not merely a bystander but part of the inflammatory machinery itself [77, 78]. These studies strengthen the case for microglia-mitochondria interactions as a relevant intermediate mechanism linking stress, neuroinflammation, and depressive phenotypes, although the evidence remains predominantly preclinical and model-specific.
Overall, gut-derived inflammatory signals likely interact with mitochondrial dynamics in ways that are biologically credible, yet the field is still moving from mechanistic plausibility to human validation. Claims that the gut microbiota uniformly controls mitochondrial fission, fusion, or mitophagy in clinical depression should therefore be interpreted cautiously until supported by longitudinal human studies with cell-specific and metabolite-level resolution.
Representative microbial metabolites, their principal mitochondrial targets, and downstream neurobiological correlates are summarized in Table 2.
Microbial metabolites and their mitochondrial targets in GBA.
| Microbial metabolite | Primary mitochondrial target or pathway | Mechanism of action | Neurobiological/Behavioral outcome | Key references |
|---|---|---|---|---|
| Butyrate | HDAC inhibition; AMPK-related mitochondrial signaling | Supports mitochondrial biogenesis and redox balance in experimental systems | Reduced neuroinflammation and improved behavioral resilience in preclinical models | [17, 66, 67] |
| Propionate | SCFA receptor and metabolic signaling | Can influence lipid oxidation and cellular energy metabolism | Potential support for energy homeostasis and stress adaptation | [37, 68] |
| Acetate | Energy substrate and immune-metabolic signaling | Supports cellular respiration and may modulate neuroimmune responses | Potential reduction of neuroinflammation in preclinical settings | [37, 68] |
| Tryptophan metabolites (e.g., kynurenine, serotonin) | Kynurenine/NAD+ related signaling | Links microbial metabolism with redox state and serotonergic pathways | Possible effects on oxidative stress and depressive phenotypes | [18, 69] |
| Bile acids (secondary) | FXR/TGR5-related signaling | Can influence barrier integrity and metabolic signaling relevant to mitochondrial function | Potential support for neuronal homeostasis; evidence remains indirect | [38] |
| Lipopolysaccharide (LPS) | Toll-like receptor 4 (TLR4); NLRP3 inflammasome | Promotes mitochondrial stress, mtROS, and inflammatory cytokine release | Neuroinflammation and depressive-like behavior in experimental models | [73, 77, 78] |
The control of mitochondrial biogenesis and energy homeostasis involves AMPK (AMP-activated protein kinase) and PGC-1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha). Activation of SIRT1 (sirtuin 1) supports mitochondrial integrity and neuroprotection. LPS-induced TLR4 activity is associated with inflammation-related mitochondrial injury in MDD. SCFAs such as butyrate, propionate, and acetate act as metabolic and epigenetic modulators that may support neural plasticity. SCFA: short-chain fatty acid; NLRP3: nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3; MDD: major depressive disorder.
GF animals remain important experimental tools because they demonstrate that the absence of commensal microbiota alters stress responsivity, barrier integrity, immune maturation, and neurochemical signaling [20, 21]. However, GF models also have major limitations for depression research. These animals exhibit profound developmental and immunological differences that do not resemble the clinical trajectory of most human MDD, making it difficult to separate depression-relevant effects from consequences of life-long microbial deprivation. Thus, GF studies are most useful for mechanistic inference, not for direct clinical extrapolation.
FMT studies provide stronger causal leverage than observational human microbiome comparisons because they test whether donor communities can transfer behavioral phenotypes to recipient animals. Several reports suggest that microbiota from patients with MDD can induce anhedonia- or immobility-related behaviors in rodents, whereas microbiota from healthy donors may reverse some stress-induced abnormalities [80, 81]. Even so, these effects are model-dependent. Outcomes vary according to donor selection, recipient strain and sex, antibiotic pretreatment, colonization efficiency, housing conditions, and the behavioral assays used. Accordingly, FMT evidence supports causal plausibility in animals, but it does not establish that the human depressive phenotype is reducible to microbiota composition alone.
Human psychobiotic trials present a more nuanced and clinically mixed picture than preclinical work. The current literature consists largely of small adjunctive randomized controlled trials, strain-specific interventions, and heterogeneous patient populations, making direct comparison difficult. In a randomized double-blind trial involving 120 adults with MDD, a combination of probiotics, magnesium orotate, and coenzyme Q10 improved some depressive outcomes during the 8-week treatment phase, although benefits were less clear at follow-up, and the multi-ingredient design limits attribution of effect to the probiotic component alone [82]. By contrast, an 8-week placebo-controlled trial of Lactobacillus plantarum PS128 in 32 antidepressant-treated patients found no significant between-group improvement in depressive symptoms, inflammatory markers, gut permeability markers, or overall microbiota composition [83]. Another randomized study in women receiving SSRIs reported improvement in depressive severity and sexual outcomes with adjunctive probiotics, but its single-population design limits generalizability [84]. Viewed together with recent evidence syntheses, the emerging clinical signal is best interpreted as modest, context-dependent, and probably strain-specific rather than class-wide [85]. Accordingly, psychobiotics should currently be framed as investigational or adjunctive tools with short-term potential in selected subgroups, not as established stand-alone antidepressant therapies.
Metabolomic and biomarker studies help bridge the gap between taxonomic observations and host physiology, but they are also mostly associative. Importantly, metabolomics has begun to show that the microbiome may be relevant not only through compositional shifts but also through altered biochemical output. Multi-omics analysis in unmedicated patients with MDD has demonstrated concurrent disturbances in gut microbiota, serum, and urine metabolomes, with abnormalities involving glycerophospholipid metabolism, primary bile acid biosynthesis, linoleic acid metabolism, amino acid pathways, purine metabolism, and other energy-relevant networks [86]. These pathways are biologically relevant because they can influence mitochondrial membrane composition, substrate handling, redox balance, and neurotransmitter precursor availability. In a separate study of 126 patients, gut microbial composition and fecal metabolomic profiles differed between SSRI responders and non-responders; taxa such as Ruminococcus, Bifidobacterium, and Faecalibacterium were enriched in responders, and functional analyses pointed to acetate degradation and neurotransmitter-related pathways as potential treatment-response correlates [87]. Metabolite-centered experimental studies are also beginning to add mechanism: a 2024 Cell Metabolism report identified gut bacteria-driven homovanillic acid as a modulator of synaptic integrity and depression-related behavior in mice, illustrating how specific microbiome-derived small molecules may influence central phenotypes [88]. Peripheral mitochondrial biomarkers are likewise promising but immature; altered ER-stress, inflammasome, and mitochondrial biogenesis-related gene expression have been reported in peripheral blood mononuclear cells, yet these findings still await replication, standardization, and prospective validation [57]. Collectively, the human literature supports association, biological plausibility, and potential stratification value, but not a singular causal model of MDD.
The microbiota–mitochondria axis offers a useful framework for exploring new treatments for MDD, but current therapeutic evidence should be interpreted cautiously. Most supportive data come from preclinical models, small adjunctive trials, or multi-component interventions. At present, microbiota- or mitochondria-targeted strategies are better regarded as experimental or adjunctive approaches than as replacements for evidence-based antidepressant, psychotherapeutic, or lifestyle care.
Probiotics, especially strains within Lactobacillus and Bifidobacterium, are the most clinically advanced microbiota-targeted interventions. Proposed mechanisms include modulation of gut permeability, inflammatory tone, neurotransmitter precursors, and redox balance [8]. However, clinical results are inconsistent. Positive findings have been reported in some adjunctive trials, whereas other placebo-controlled studies have failed to show significant between-group effects [82–84]. These discrepancies likely reflect heterogeneity in strain composition, baseline diet, concomitant antidepressant use, illness severity, treatment duration, and outcome measures. Therefore, psychobiotics should currently be framed as promising but not yet established adjuncts for MDD. A conceptual overview of probiotic-mediated effects on mitochondrial biogenesis, oxidative stress, inflammatory signaling, and depressive phenotypes is shown in Figure 2.
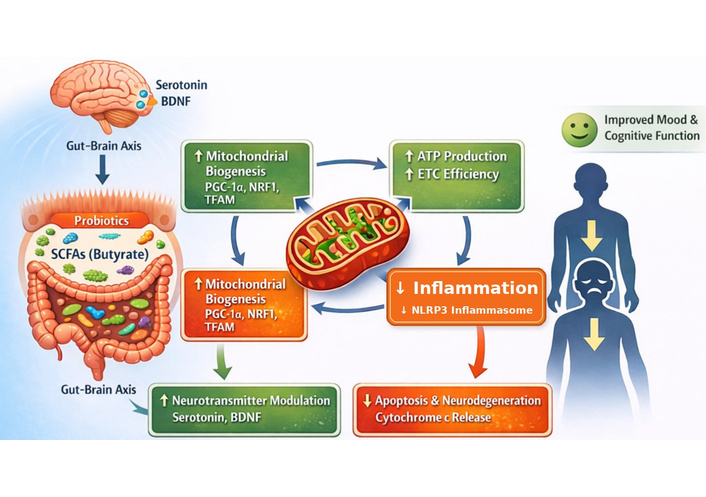
Proposed mechanisms through which probiotics modulate mitochondrial function and depressive phenotypes. ATP: adenosine triphosphate; BDNF: brain-derived neurotrophic factor; ETC: electron transport chain; NLRP3: nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3; NRF1: nuclear respiratory factor 1; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; SCFAs: short-chain fatty acids; TFAM: mitochondrial transcription factor A.
Prebiotics and postbiotic/metabiotic approaches are mechanistically attractive because they may enhance SCFA production or deliver defined microbial metabolites without the variability of live-organism colonization [8]. Experimental work suggests that compounds such as butyrate can influence histone acetylation and mitochondrial signaling in preclinical systems [17, 66]. Yet human evidence remains sparse, and dose-response relationships, safety with chronic use, and the durability of benefits are not well characterized. For now, prebiotics and postbiotics should be considered emerging supportive strategies rather than validated antidepressant treatments.
FMT remains one of the most direct methods for manipulating the gut microbial ecosystem, but its role in depression is still investigational. Although preclinical studies suggest that donor microbiota can influence depressive-like behavior [81], human psychiatric data remain limited, and issues of donor selection, engraftment durability, adverse-event monitoring, and regulatory oversight remain unresolved [89]. At present, FMT should be discussed as an experimental research tool rather than a ready-to-implement treatment for MDD.
Mitochondrial peptides and gene-based approaches are conceptually intriguing because they target oxidative stress, apoptosis, and organellar quality control at a deeper mechanistic level. However, the evidence base for depression remains very early. Most data come from preclinical neurological models or broader mitochondrial medicine rather than controlled MDD trials [90]. Consequently, these interventions should be presented as exploratory rather than near-term clinical options.
Lifestyle interventions are currently the most pragmatic component of this framework because exercise, diet quality, sleep regularity, and circadian alignment all influence both microbial ecology and mitochondrial function. Even here, however, the microbiota–mitochondria pathway should not be oversold as the sole mechanism of benefit. These interventions likely act through multiple overlapping biological and psychosocial routes, and their antidepressant effects are best understood within multimodal care rather than a single-axis model [91].
Taken together, the therapeutic literature supports a cautious conceptual framework rather than a clinically mature treatment paradigm. Related preclinical probiotic work also suggests that prolonged administration of selected strains may help preserve neurometabolic and behavioral stability under chronic stress, but these findings remain indirect for MDD treatment design [92]. Future progress will depend on adequately powered randomized trials, standardized microbiome and metabolomic readouts, clearer patient stratification, and mechanistic linkage between peripheral biomarkers and central symptom change. Until such evidence is available, microbiota–mitochondria interventions should be viewed as investigational or adjunctive, not as stand-alone remission-promoting therapies.
Despite growing interest in microbiota-mitochondria crosstalk in MDD, the field remains constrained by conceptual and methodological limitations (Table 3). Much of the mechanistic evidence comes from GF, stress-exposed, or cell-based models, whereas human studies are often cross-sectional, small, and vulnerable to confounding by diet, obesity, sex, medications, and sampling protocols. Longitudinal, interventional, and multi-omic studies are therefore required before this axis can be translated into clinically reliable biomarkers or targeted therapies.
Summary of key challenge areas.
| Challenge area | Research priority | Potential outcome |
|---|---|---|
| Microbiota variability | Stratify patients based on microbiota–host profiles | Precision psychobiotic therapy |
| Biomarker identification | Develop composite microbial–mitochondrial panels | Early diagnosis and response monitoring |
| Multi-omics integration | Combine metagenomic, proteomic, and metabolomic data | Network-level understanding of MDD |
| AI in psychiatry | Predict individualized treatment response | Digital microbiome–mitochondria modeling |
| Ethics and translation | Establish global safety and equity standards | Ethical and accessible implementation |
MDD: major depressive disorder.
One of the most significant challenges in microbiome research is the inherent variability in microbial composition and metabolic output across individuals. Genetics, diet, age, sex, geographical location, and medication use are all known to significantly alter microbial diversity and functionality [4, 21]. In clinical cohorts, antidepressant exposure may also confound microbiome analyses, complicating the interpretation of gut-brain interactions in treated individuals [20, 21]. Furthermore, individual host-microbe interactions profoundly impact the production of microbial metabolites, such as SCFAs and bile acids, which play a major role in mitochondrial activity and neuroprotection. This high degree of inter-individual variability necessitates a shift towards precision psychiatry, where therapeutic interventions are personalized based on an individual’s genetic history, microbiota composition, and lifestyle. Future research must focus on identifying robust microbial signatures that transcend individual differences or developing stratification strategies for patient cohorts.
The discovery of valid microbiome-mitochondrial biomarkers is a breakthrough point for clinical utility. Current MDD diagnostic systems predominantly rely on subjective symptomatology rather than objective molecular markers [4]. Developing composite biomarkers that incorporate microbial metabolites (e.g., SCFAs, tryptophan derivatives), mitochondrial respiration patterns, and inflammatory cytokines would be invaluable for determining disease severity, predicting treatment response, and monitoring adherence [89]. However, reproducibility remains a significant issue in biomarker research. Heterogeneous sampling approaches, sequencing platforms, and analysis pipelines contribute to discrepancies across studies. To achieve clinical reliability and widespread adoption, the standardization of data acquisition, normalization, and interpretation protocols will be critical.
The inherent complexity of MDD pathophysiology necessitates the use of multi-omics integration, including genomics, transcriptomics, proteomics, metabolomics, and metagenomics, to more comprehensively uncover interactions between the gut microbiota and host bioenergetic biology [86, 87]. Single-layer analyses often fail to capture the network-level biomolecular changes in bioenergetic and proinflammatory pathways that become visible in integrated systems approaches [86, 87]. For example, joint analysis of metagenomic, metabolomic, and host transcriptomic data can help identify microbial taxa and metabolites that are associated with mitochondrial signaling, oxidative phosphorylation, and treatment response in MDD [86, 87]. Computational integration of these datasets may help generate testable networks linking microbial metabolites, mitochondrial signaling, and treatment response in depression research [86, 87]. Such integrative methods are important for biomarker discovery, patient stratification, and response prediction in depression research [86, 87].
The rapid growth of computational biology and machine-learning approaches may eventually support more individualized MDD stratification, particularly when integrated with microbiome and metabolomic datasets. At present, however, these applications remain largely prospective rather than clinically validated in microbiota-mitochondria research [86, 87].
Despite the immense research potential and possibilities of microbiota-mitochondria modulation in psychiatry, significant challenges remain regarding translation and ethics. The long-term safety profiles of probiotics, prebiotics, and FMT are not yet fully characterized, particularly with respect to prolonged use, donor screening, engraftment durability, and adverse-event monitoring in psychiatric populations [85]. Ethical principles must also guide both microbiome manipulation and emerging mtDNA-editing strategies [93]. Moreover, ensuring equitable access to personalized psychobiotic interventions is crucial to avoid exacerbating existing mental health disparities globally. Future studies must prioritize cross-cultural follow-ups, research in low-resource settings, and the seamless incorporation of microbiota-derived therapies into general healthcare systems. Addressing these translational and ethical dimensions is paramount for the responsible and impactful advancement of this promising field.
MDD is a multifactorial condition involving complex neural, immune, metabolic, and microbial interactions. Current evidence supports microbiota–mitochondria crosstalk as a plausible contributor to MDD pathophysiology, particularly through pathways related to bioenergetics, oxidative stress, neuroinflammation, and stress responsivity. However, the evidentiary landscape is uneven: mechanistic depth is greatest in preclinical models, whereas human data remain heterogeneous and largely associative. Findings on SCFAs, psychobiotics, peripheral mitochondrial biomarkers, and metabolomic signatures are promising, but they do not yet justify treating this axis as a single unifying cause of MDD or as a fully validated therapeutic target. The most defensible conclusion at present is that microbiota-mitochondria interactions provide a useful integrative framework for future precision psychiatry research and for the development of adjunctive interventions. Progress will depend on rigorous randomized trials, longitudinal multi-omics, biomarker standardization, and careful attention to translational and ethical issues.
ATP: adenosine triphosphate
BBB: blood-brain barrier
CNS: central nervous system
EECs: enteroendocrine cells
FMT: fecal microbiota transplantation
GBA: gut–brain axis
GF: germ-free
LPS: lipopolysaccharide
MDD: major depressive disorder
mtDNA: mitochondrial DNA
NTS: nucleus tractus solitarius
ROS: reactive oxygen species
SCFAs: short-chain fatty acids
Tregs: regulatory T cells
OMA: Conceptualization. Data curation, Investigation, Writing—original draft, Writing—review & editing, Supervision. CAC, OEO, OCA, and ODA: Data curation, Investigation, Writing—review & editing. SJA: Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

