 Review
Review

Affiliation:
1Department of Physiology, Faculty of Medicine, Istanbul Medeniyet University, 34700 Istanbul, Türkiye
2Science and Advanced Technologies Research Center (BILTAM), Istanbul Medeniyet University, 34700 Istanbul, Türkiye
Email: Mustafa.beker@medeniyet.edu.tr; m.caglarbeker@gmail.com
ORCID: https://orcid.org/0000-0002-9476-8488
Affiliation:
3Department of Neurology, University Hospital Essen, University of Duisburg-Essen, 45147 Essen, Germany
ORCID: https://orcid.org/0000-0003-0198-3152
Affiliation:
1Department of Physiology, Faculty of Medicine, Istanbul Medeniyet University, 34700 Istanbul, Türkiye
2Science and Advanced Technologies Research Center (BILTAM), Istanbul Medeniyet University, 34700 Istanbul, Türkiye
ORCID: https://orcid.org/0000-0001-6494-8923
Explor Neurosci. 2026;5:1006127 DOI: https://doi.org/10.37349/en.2026.1006127
Received: December 08, 2025 Accepted: January 27, 2026 Published: March 17, 2026
Academic Editor: Ludmilla A. Morozova-Roche, Umea University, Sweden
The circadian clock orchestrates cellular physiology by synchronizing transcriptional, metabolic, and signaling networks with the environmental light-dark cycle. Basic helix-loop-helix ARNT-like protein 1 (BMAL1), a core transcriptional regulator of circadian timing, contributes to rhythmic gene expression and is implicated in cellular responses to stress and energy demand. Emerging evidence suggests an interplay between BMAL1 and the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, a central hub controlling cell survival, metabolism, and angiogenesis. In ischemic conditions, BMAL1 is associated with increased PI3K/AKT activity and downstream mTOR signaling, which may help preserve mitochondrial integrity, limit oxidative stress, and support neuronal and vascular recovery. Conversely, BMAL1 deficiency is linked to impaired AKT phosphorylation and redox imbalance, exacerbating ischemic injury. Proteomic and functional studies further suggest that BMAL1 may contribute to metabolic reprogramming through PI3K/AKT-dependent regulation of oxidative phosphorylation and antioxidant defenses. This review is based on a focused narrative evaluation of experimental and translational studies retrieved from PubMed, emphasizing circadian regulation of PI3K/AKT signaling in ischemic and vascular contexts. Collectively, these findings support the concept that BMAL1 functions as a temporal modulator of PI3K/AKT signaling, integrating circadian and metabolic cues to promote cellular resilience. Understanding this regulatory axis may offer novel therapeutic perspectives for ischemic and neurovascular disorders associated with circadian misalignment.
Circadian rhythms are integral biological processes that regulate the timing of various physiological functions within all living organisms, ensuring that internal systems remain synchronized with the external light-dark cycle [1]. Circadian regulation is widely implicated in homeostasis and influences processes such as the sleep-wake cycle, metabolic regulation, immune function, and cellular growth [2–4]. Central to the circadian system is the suprachiasmatic nucleus (SCN), the master pacemaker that coordinates the timing of peripheral clocks located in various tissues, including the brain, liver, and heart [5–7]. These peripheral oscillators, although capable of operating autonomously, are synchronized by signals from the SCN to ensure coordinated physiological functions across the body.
A key player in the molecular regulation of circadian rhythms is basic helix-loop-helix ARNT-like protein 1 (BMAL1), a core clock protein that, together with circadian locomotor output cycles kaput (CLOCK), regulates the expression of downstream clock genes through transcriptional-translational feedback loops. Beyond its canonical role in circadian timing, BMAL1 has been implicated in the modulation of multiple cellular processes, including metabolism, cellular stress responses, and tissue repair [8–10]. Recent studies have suggested an interaction between BMAL1 and the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway, a central regulator of cell survival, growth, and metabolic adaptation. Although PI3K/AKT signaling has been investigated across diverse biological and pathological contexts, the present review specifically focuses on its circadian regulation by BMAL1 in the context of ischemic injury and vascular repair, where precise temporal control of survival and metabolic pathways is critical for neurovascular resilience [11–14].
This review focuses on the relationship between BMAL1 and the PI3K/AKT signaling pathway, exploring how circadian regulation by BMAL1 modulates PI3K/AKT activity in different physiological and pathological conditions. In ischemic injury and vascular repair settings, BMAL1 has been associated with changes in PI3K/AKT activation and downstream signaling that may support cellular resilience and tissue recovery, in a context- and timing-dependent manner. Understanding this complex interaction may offer novel insights into therapeutic strategies targeting circadian rhythms and signaling pathways, particularly in the context of diseases linked to circadian disruption.
This review was conducted as a focused narrative synthesis of the literature. Relevant experimental and translational studies were identified through searches of PubMed using combinations of keywords including “BMAL1”, “circadian rhythm”, “PI3K/AKT”, “ischemia”, “stroke”, and/or “vascular repair”. Priority was given to peer-reviewed studies providing mechanistic, temporal, or cell-type-specific insights into BMAL1-PI3K/AKT crosstalk in ischemic and neurovascular contexts.
Circadian rhythms are essential biological phenomena that regulate the timing of various physiological processes in virtually all living organisms. These 24-h cycles help synchronize internal processes with the Earth’s light-dark cycle, optimizing physiological and behavioral responses. Circadian regulation influences key biological functions such as the sleep-wake cycle, metabolic processes, hormone secretion, immune responses, and body temperature regulation [2]. This timing mechanism is fundamental to homeostasis and helps keep physiological systems aligned with external environmental cues [3].
In mammals, the circadian system is structured as a hierarchical network, with central and peripheral oscillators working together to regulate the timing of various physiological processes [15]. At the core of this system lies the SCN in the anterior hypothalamus, which functions as the master pacemaker of circadian rhythms (Figure 1). It synchronizes internal biological clocks across the body and coordinates various functions, including sleep, neuroendocrine activity, and body temperature regulation [16]. The SCN is essential for aligning internal biological clocks with external environmental cues, making it the primary driver of circadian timing [17]. Its synchronization with the environment is largely mediated by light, the most powerful external cue, or zeitgeber. Light is detected by intrinsically photosensitive retinal ganglion cells containing melanopsin, which relay signals via the retinohypothalamic tract to the SCN. This initiates a cascade of molecular and cellular events that reset the SCN’s internal clock, ensuring that the organism’s circadian rhythms remain synchronized with the day-night cycle [18, 19].
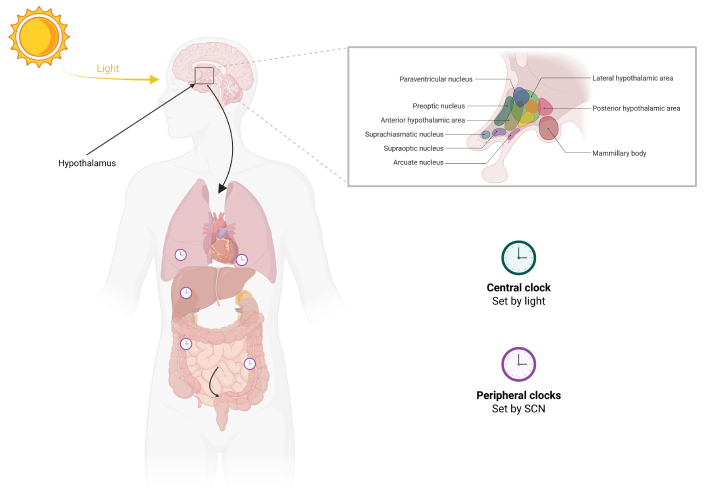
Schematic representation of the central and peripheral circadian clocks in mammals. Light signals are detected by the retina and transmitted to the SCN of the hypothalamus, which functions as the central clock. The SCN synchronizes circadian rhythms throughout the body via neural and hormonal outputs. SCN: suprachiasmatic nucleus. Created in BioRender. Beker, M. Ç. (2026) https://BioRender.com/lgdta09.
The SCN not only serves as the central pacemaker but also synchronizes peripheral oscillators in tissues like the liver, heart, lungs, and specific brain regions, such as the hippocampus [20]. Although these peripheral clocks operate autonomously, they rely on the SCN’s signals to maintain temporal synchronization (Figure 1). This dynamic interplay between the SCN and peripheral oscillators is essential for the coordinated regulation of physiological processes across multiple organs, ensuring systemic harmony and optimal function. For instance, the SCN regulates the release of melatonin, a hormone produced by the pineal gland that facilitates sleep, as well as cortisol, which plays a key role in the body’s response to stress [21]. By modulating the timing of these hormones, the SCN contributes to the regulation of sleep-wake cycles and metabolic function, while also supporting adaptation to environmental stressors such as oxidative stress [21, 22].
Beyond neural signaling, the SCN also communicates with peripheral tissues through humoral signals [21]. These include transforming growth factor-α and prokineticin receptor 2, which help synchronize peripheral clocks in various tissues. This humoral communication is crucial for maintaining the temporal alignment of biological processes throughout the body [23]. The coordination between the central SCN and peripheral clocks establishes a finely tuned system known as interval synchrony. This synchronization ensures that the body’s physiological rhythms are tightly coordinated, promoting efficient metabolic regulation, immune responses, and overall health.
The molecular regulation of the circadian rhythm is regulated through a series of intricate transcription-translation feedback loops (TTFLs) involving key transcription factors. Central to this process is the heterodimerization of CLOCK and BMAL1, also known as MOP3 or Arnt3, which drives the transcription of various clock-regulated genes. These genes include the negative arm components, cryptochrome (CRY) and period (PER), which play essential roles in feedback inhibition [24–27].
In the canonical model, the CLOCK-BMAL1 complex activates the transcription of CRY and PER genes, which then form a complex in the cytoplasm (Figure 2). After a delay, the CRY-PER complex translocates to the nucleus, where it inhibits or attenuates the transcriptional activity of CLOCK-BMAL1 by binding to the E-box elements in the promoters of target genes, thus completing the feedback loop [2, 24, 26, 28]. However, more recent findings reveal a nuanced regulation. CRY proteins (CRY1 and CRY2) directly interact with the CLOCK-BMAL1 complex, blocking its transcriptional activity even in the absence of PER proteins, revealing an additional mode of repression. In the presence of PER, the CRY-PER complex can further enhance repression by displacing CLOCK-BMAL1 from the E-box, resulting in transcriptional silencing [27, 29, 30].
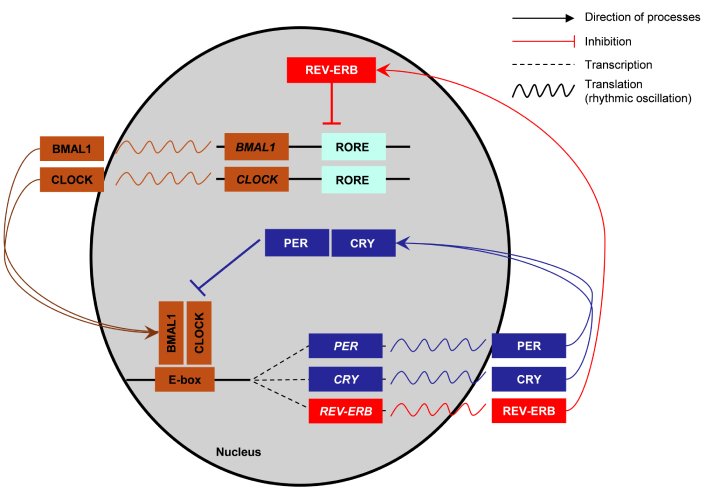
Molecular architecture of the mammalian circadian transcription-translation feedback loop. The core clock machinery consists of transcriptional activators BMAL1 and CLOCK, which form a heterodimer that binds to E-box elements in target gene promoters, initiating the transcription of period (PER) and cryptochrome (CRY) genes. The translated PER and CRY proteins accumulate in the cytoplasm, form complexes, and translocate back into the nucleus to inhibit BMAL1-CLOCK activity, thereby generating a negative feedback loop. In a secondary regulatory loop, BMAL1-CLOCK also drives the expression of REV-ERB genes via E-box elements. REV-ERB proteins subsequently repress BMAL1 transcription by binding to RORE sites, creating a stabilizing feedback mechanism that maintains rhythmic oscillations in gene expression and cellular physiology. BMAL1: basic helix-loop-helix ARNT-like protein 1; CLOCK: circadian locomotor output cycles kaput; REV-ERB: nuclear receptor subfamily 1 group D member (NR1D1/2); RORE: retinoic acid receptor-related orphan receptor response element. Adapted from [31]. © 2015 Chen and Yang. Distributed under CC BY 4.0.
At the molecular level, the stability of PER and CRY proteins is tightly regulated by E3 ubiquitin ligase complexes, specifically the Skp1-Cullin-F-box complex, which involves beta-transducin repeat containing protein (β-TrCP) and F-box and leucine-rich repeat protein 3 (FBXL3) [25, 27]. This regulatory mechanism helps regulate the degradation of PER and CRY proteins by the 26S proteasome after phosphorylation by kinases like casein kinase 1 epsilon and delta (CK1ε/δ) and AMP-activated protein kinase (AMPK), thus controlling the duration of their repression [2, 27]. When levels of PER and CRY decline, the inhibition on CLOCK-BMAL1 is increased, allowing the cycle to restart, thus ensuring the continuation of the circadian rhythm. Additionally, nuclear receptor subfamily 1 group D member 1 (NR1D1; REV-ERBα) and nuclear receptor subfamily 1 group D member 2 (NR1D2; REV-ERBβ), as well as the retinoic acid receptor-related orphan receptors (RORs), play important roles in modulating BMAL1 expression. REV-ERBs act as transcriptional repressors of BMAL1, while RORs positively regulate its expression by binding to ROR response elements (ROREs) in the BMAL1 promoter [32].
Beyond these core mechanisms, the circadian clock is influenced by various additional factors. These include intracellular calcium flux, membrane depolarization, and the activation of cyclic AMP (cAMP) signaling, all of which contribute to the rhythmic activity of the SCN neurons. Specifically, calcium influx and the periodic activation of cAMP play crucial roles in modulating the rhythmic expression of circadian regulators. The activation of calcium/cAMP response element-binding protein (CREB) through phosphorylation leads to the binding of CREB to regulatory elements in the promoters of key clock genes such as PER1, further influencing circadian regulation [33].
The PI3K/AKT signaling pathway is a critical regulator of cellular functions such as growth, survival, metabolism, and differentiation, impacting a wide variety of processes, including cell proliferation, apoptosis, autophagy, and angiogenesis. The activation of this pathway is often triggered by extracellular stimuli such as growth factors, cytokines, and hormones. These signaling molecules bind to receptor tyrosine kinases (RTKs) or G-protein coupled receptors, which initiate a cascade of intracellular signaling events. For example, epidermal growth factor, platelet-derived growth factor (PDGF), and insulin-like growth factor can activate PI3K through the autophosphorylation of tyrosine residues in the cytoplasmic domains of RTKs [34].
PI3Ks are a family of lipid kinases essential for regulating various cellular functions, including vesicular trafficking, cellular metabolism, and cell growth [35, 36]. These kinases catalyze the phosphorylation of phosphatidylinositol lipids, producing important second messengers that help orchestrate cellular responses. PI3Ks are classified into three distinct classes—Class I, Class II, and Class III—based on their structural properties, substrates, and cellular functions (Table 1) [37].
Classification and functional overview of PI3Ks.
| Class | Catalytic isoforms | Regulatory subunits | Primary lipid product | Predominant expression | Key functions |
|---|---|---|---|---|---|
| Class I(IA & IB) | p110α (PIK3CA), p110β (PIK3CB), p110δ (PIK3CD), p110γ (PIK3CG) | IA: p85α/β, p55γ; IB: p101, p84 | PIP3 | α/β: ubiquitous; δ/γ: immune cells | Growth factor signaling, metabolism, survival, motility |
| Class II | PI3KC2α (PIK3C2A), PI3KC2β (PI3KC2B), PI3KC2γ (PI3KC2G) | None (monomeric) | PI(3)P, PI(3,4)P2 | C2α/β: broad; C2γ: liver-enriched | Endocytosis, exocytosis, vesicular trafficking, ciliogenesis |
| Class III | VPS34 (PIK3C3) | VPS15 (PIK3R4) + partners | PI(3)P | Ubiquitous | Autophagy initiation, endo/lysosomal trafficking, phagocytosis |
PI3Ks: phosphatidylinositol 3-kinases; PIP3: phosphatidylinositol (3,4,5)-trisphosphate; PI(3)P: phosphatidylinositol 3-phosphate; PI(3,4)P2: phosphatidylinositol 3,4-bisphosphate.
Class I PI3Ks consist of four distinct isoforms: p110α, p110β, p110γ, and p110δ, encoded by the genes PIK3CA, PIK3CB, PIK3CG, and PIK3CD, respectively [38]. These isoforms contribute to various cellular functions. Specifically, p110α is a key regulator of growth factor-mediated signaling, playing a crucial role in cellular proliferation, metabolism, and oncogenesis [39]. p110β, in addition to its involvement in RTK signaling, has been implicated in G protein-coupled receptor signaling and is essential for sustaining tumorigenic processes in certain cancers [40]. p110γ and p110δ, predominantly expressed in hematopoietic cells, are integral to immune cell activation, differentiation, and migration [41, 42]. Specifically, p110γ is vital for chemokine signaling in leukocytes, whereas p110δ is critical for B and T cell receptor signaling [43, 44]. The p85 subunit of Class I PI3Ks serves not only as a regulatory scaffold but also helps integrate cellular signals, further enhancing the specificity of downstream signaling activation [45]. Additionally, Class I PI3Ks are known to activate small monomeric GTPases and tyrosine kinases, amplifying the signaling pathways initiated at the membrane.
Class II PI3Ks differ significantly from Class I in that they lack regulatory subunits and can function as monomers [46]. These isoforms—PI3KC2α, PI3KC2β, and PI3KC2γ—play key roles in cellular processes such as membrane trafficking and endosomal functions [47, 48]. PI3KC2α and PI3KC2β are widely expressed across tissues, whereas PI3KC2γ is more specific to the liver [36, 49]. Although Class II PI3Ks share some structural similarities with Class I enzymes, their functions are less understood. They are primarily involved in the regulation of vesicular trafficking and act as critical modulators of endocytosis and exocytosis, which are essential for maintaining cellular homeostasis and responding to external stimuli. These enzymes have been implicated in diverse physiological processes, including neurotransmitter release and insulin signaling, but their full biological roles remain an active area of research [50].
Class III PI3K is unique among the three classes due to its distinct structure and function [36, 51]. The prototypical enzyme in this class is VPS34 (also known as PIK3C3), which is involved in regulating autophagy and macrophage phagocytosis [52, 53]. Class III PI3Ks are heterodimers composed of a catalytic subunit and a regulatory subunit, and their primary function is the regulation of autophagic vesicle formation [52, 53]. Through this activity, VPS34 helps modulate the degradation of cellular components, playing a crucial role in cellular quality control [36, 54]. Additionally, Class III PI3Ks are essential for regulating lysosomal and endosomal trafficking, processes that are crucial for maintaining cellular integrity and responding to stress [37]. Recent studies have also highlighted the role of Class III PI3Ks in immune cell activation, where they regulate the phagocytosis of pathogens, an essential function in the innate immune response [36, 43, 55].
AKT is a serine/threonine protein kinase that plays a crucial role in regulating cellular responses to stress, growth factors, and metabolic signals [56]. In mammals, three isoforms of AKT exist: AKT1, AKT2, and AKT3. These isoforms are tissue-specific; AKT3 is primarily expressed in the brain, where it serves as the predominant AKT isoform in neurons. It accounts for approximately 50% of the total AKT protein in the brain and contributes to about 30% of the AKT presence in the spinal cord [57, 58]. AKT1 and AKT2 are enriched in tissues such as the liver, pancreas, and adipose tissue [59].
The activation of AKT begins at the plasma membrane with the conversion of phosphatidylinositol (4,5)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3) by the catalytic subunit of PI3K. This conversion is a key step that facilitates the recruitment of AKT to the membrane via its pleckstrin homology (PH) domain, which binds to PIP3 [38].
Once at the membrane, AKT is partially activated through phosphorylation at threonine 308 (T308) in the activation loop by 3-phosphoinositide-dependent kinase-1 (PDK1) [60]. While this phosphorylation marks the initial activation of AKT, it is insufficient for full enzymatic activity. Full activation occurs when AKT is further phosphorylated at serine 473 (S473) in the hydrophobic C-terminal motif. This secondary phosphorylation is catalyzed either by the mechanistic target of rapamycin complex 2 (mTORC2) or DNA-dependent protein kinase (DNA-PK), both of which are important for the completion of AKT activation [61, 62]. Once fully activated, AKT is capable of phosphorylating a wide range of downstream targets that are involved in cellular survival, metabolism, protein synthesis, and cell cycle progression [36].
The activation of AKT leads to the modulation of several critical substrates. For example, AKT phosphorylates the forkhead box O (FOXO) transcription factors, which are important regulators of apoptotic pathways [63]. Phosphorylation of FOXO proteins results in their sequestration in the cytoplasm, preventing their nuclear localization and subsequent transcription of pro-apoptotic genes. This mechanism is a key aspect of AKT’s role in promoting cell survival and inhibiting apoptosis. In addition to FOXO, AKT phosphorylates and inactivates glycogen synthase kinase 3 (GSK3), which further promotes glycogen synthesis and glucose metabolism, supporting cellular energy homeostasis [64].
AKT also plays a pivotal role in regulating mTORC1, a master regulator of cell growth and protein synthesis. AKT activates mTORC1 by phosphorylating two key inhibitors of the pathway: proline-rich AKT substrate of 40 kDa (PRAS40) and tuberous sclerosis protein 2 (TSC2). Phosphorylation of PRAS40 and TSC2 relieves the inhibitory effect of the TSC complex on mTORC1, thereby promoting its activation [65, 66]. Activated mTORC1, in turn, phosphorylates downstream targets such as eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) and ribosomal protein S6 kinase 1 (S6K1), which are crucial for mRNA translation and cell growth [66]. These downstream effects of AKT-driven mTORC1 activation lead to increased protein synthesis, ribosome biogenesis, and cellular proliferation.
Beyond its effects on cell survival and growth, AKT also modulates various other cellular functions. For example, AKT regulates angiogenesis by phosphorylating endothelial nitric oxide synthase (eNOS), which promotes the production of NO and induces the formation of new blood vessels [67]. Moreover, AKT plays a key role in the regulation of autophagy under stress conditions. During nutrient deprivation or metabolic stress, AMPK is activated and inhibits AKT signaling through the phosphorylation of TSC2, enhancing its GTPase-activating protein (GAP) activity, which inhibits mTORC1 and promotes autophagy as an energy-conserving mechanism [68].
AKT signaling is tightly regulated by both positive and negative feedback mechanisms to maintain cellular homeostasis. Negative regulation is particularly important to prevent unchecked activation of AKT, which can contribute to oncogenesis. One of the key negative regulators of AKT signaling is phosphatase and tensin homolog (PTEN), a tumor suppressor that dephosphorylates PIP3 back to PIP2, thereby preventing further AKT activation [69]. PTEN’s action ensures that AKT signaling is appropriately dampened, preventing excessive cellular proliferation and survival.
Furthermore, AKT signaling is modulated by various phosphatases that dephosphorylate AKT and its upstream regulators. Protein phosphatase 2A (PP2A) dephosphorylates T308, reducing AKT’s activation [70]. Similarly, PH-domain leucine-rich-repeat-containing protein phosphatases (PHLPPs; PHLPP1/2) dephosphorylate S473, leading to AKT inactivation and attenuating downstream signaling [71]. Additionally, the TSC1/TSC2 complex, when phosphorylated by AKT, acts as a negative regulator of mTORC1. By inhibiting the GAP activity of TSC2, AKT promotes mTORC1 activation, which is essential for cellular growth and metabolism [72].
The PI3K/AKT signaling pathway also interacts with other important signaling networks, such as the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway. This pathway, consisting of key components like ERK1/2, c-Jun N-terminal kinase (c-JNK), and p38, regulates various cellular responses, including proliferation, differentiation, and stress responses. The activation of these pathways is implicated in neurological diseases such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, Huntington’s disease, and stroke [13, 73–78].
In summary, the AKT signaling cascade is initiated by the binding of PIP3 at the membrane, followed by phosphorylation at T308 by PDK1 and full activation through phosphorylation at S473 by mTORC2 or DNA-PK. Fully activated AKT orchestrates a variety of cellular processes by phosphorylating key substrates such as FOXO, GSK3, and TSC2, which influence apoptosis, metabolism, and protein synthesis. AKT signaling is carefully controlled by negative feedback mechanisms, including PTEN and PP2A, to prevent aberrant activation that could lead to uncontrolled cell growth or tumorigenesis. This finely tuned regulation is crucial for maintaining cellular homeostasis and ensuring appropriate cellular responses to internal and external signals.
Stroke remains a major global health challenge, ranking among the leading causes of mortality and long-term disability worldwide. Each year, more than 12 million individuals experience a stroke, resulting in approximately 6.5 million deaths and millions of survivors requiring sustained medical and social support [79]. Despite advances in acute care, recombinant tissue plasminogen activator (rt-PA) remains the only FDA-approved thrombolytic therapy, and its narrow therapeutic window restricts its use to a small fraction of patients, underscoring the need for alternative and adjunct therapeutic strategies [80].
Importantly, acute ischemic stroke is not a uniform disease entity, but a heterogeneous clinical syndrome composed of distinct etiological subtypes, including cardioembolic, lacunar, atherothrombotic, and cryptogenic infarctions. These subtypes differ substantially in vascular pathology, risk factor profiles, stroke severity, and clinical outcome, and are increasingly recognized to exhibit distinct molecular and biomarker signatures. Accordingly, etiologically stratified analyses are essential for accurate clinical interpretation and for improving the translational relevance of mechanistic stroke research [81]. In addition to etiological diversity, biological sex contributes to variability in stroke presentation and outcome. Clinical cohort studies indicate that female sex is associated with differences in vascular risk factors, a higher prevalence of cardioembolic stroke, greater stroke severity at admission, and less favorable in-hospital outcomes compared with male patients [82]. These observations highlight that stroke subtype and sex interact to shape disease severity and prognosis, emphasizing the importance of demographic and etiological stratification when evaluating molecular pathways implicated in ischemic injury.
Disruption of BMAL1 expression in ischemic conditions impairs circadian regulation and is associated with diminished PI3K/AKT pathway activity. In myocardial ischemia, for instance, decreased BMAL1 levels correlate with reduced phosphorylation of AKT and ERK, accompanied by increased oxidative stress and apoptosis [83]. Key neurological and neurovascular evidence linking BMAL1 to PI3K/AKT-mTOR signaling in ischemic and vascular contexts is summarized in Table 2. Treatment with hydrogen sulfide (H2S) restores BMAL1 mRNA levels and rhythmicity, reactivating PI3K, AKT, and 14-3-3γ, which together mitigate oxidative damage and cell death, highlighting the association between BMAL1 and PI3K/AKT signaling to protect cells during ischemic events [83].
Summary of key neurological and neurovascular studies linking BMAL1/circadian regulation to PI3K/AKT-mTOR signaling under ischemic and vascular stress.
| Ref. | Model/Context | CLOCK/BMAL1 axis | PI3K/AKT node(s) emphasized | Principal finding |
|---|---|---|---|---|
| [84] | Mouse focal cerebral ischemia | Time-of-day (ZT) + BMAL1 rhythmicity | pAKT T308, ERK1/2 phosphorylation, survival signaling mediators (mTOR/S6; Bad/PRAS40 axis) | Ischemic damage is time-of-day dependent; higher BMAL1 phase associates with enhanced AKT activation and improved survival signaling. |
| [85] | OGD (± reoxygenation) | BMAL1-melatonin interaction | PDK1, mTOR, PTEN, GSK3α/β, p70S6K phosphorylation panel | BMAL1/melatonin context amplifies phosphorylation across PI3K/AKT cascade, consistent with stress resilience. |
| [86] | Mouse focal cerebral ischemia + proteomics | BMAL1 overexpression vs. knockdown/deletion | mTOR (Ser2448) + mitochondrial/metabolic modules (OXPHOS/TCA/ATP synthase proteins) | BMAL1 gain-of-function reduces infarct and neuronal injury and reprograms metabolic pathways consistent with pro-survival PI3K/AKT-mTOR tone. |
| [87] | PC12 OGD/R | BMAL1 overexpression | Indirect support of PI3K/AKT via redox: Nrf2/HO-1, Bcl-2/Bax | BMAL1 enhances antioxidant defense and anti-apoptotic balance under ischemic stress. |
| [88] | Endothelial/hematopoietic BMAL1 deletion (vascular injury) | Cell-type-specific BMAL1 loss | pAKT S473, mTORC2/RICTOR implication; eNOS/NO signaling | BMAL1 deletion reduces pAKT (S473) and worsens microvascular outcomes (nitrative stress, degeneration). |
| [89] | Disturbed flow/vascular regions | BMAL1/CLOCK regulation by hemodynamic context | eNOS and AKT phosphorylation dynamics | Disturbed flow is associated with BMAL1/CLOCK changes and endothelial signaling remodeling. |
| [90] | VSMC proliferation under growth factor | BMAL1 induction via ROS–ERK–EGR1 | ERK modulation (PI3K/AKT adjacent crosstalk) | BMAL1 participates in growth-factor-driven vascular proliferation via MAPK/ERK coupling. |
BMAL1: basic helix-loop-helix ARNT-like protein 1; PI3K: phosphatidylinositol 3-kinase; CLOCK: circadian locomotor output cycles kaput; ZT: zeitgeber time; pAKT: phospho AKT; T308: threonine 308; Bad: Bcl-2-associated agonist of cell death; PRAS40: proline-rich AKT substrate of 40 kDa; OGD: oxygen-glucose deprivation; PDK1: 3-phosphoinositide-dependent kinase-1; PTEN: phosphatase and tensin homolog; GSK3: glycogen synthase kinase 3; p70S6K: p70 ribosomal protein S6 kinase; OXPHOS: oxidative phosphorylation; TCA: tricarboxylic acid; Nrf2: nuclear factor erythroid 2-related factor 2; HO-1: heme oxygenase-1; S473: serine 473; mTORC2: mechanistic target of rapamycin complex 2; RICTOR: rapamycin-insensitive companion of mTOR; eNOS: endothelial nitric oxide synthase; VSMC: vascular smooth muscle cell; ROS: reactive oxygen species; ERK: extracellular signal-regulated kinase; EGR1: early growth response protein 1; MAPK: mitogen-activated protein kinase.
In focal cerebral ischemia, temporal fluctuations in BMAL1 expression are associated with changes in AKT activation. At zeitgeber time (ZT) 18 (ZT18), when BMAL1 levels peak, phosphorylation of AKT (T308) together with increased phosphorylation of ERK1/2, mTOR, and ribosomal protein S6—is significantly enhanced, promoting neuronal survival through the regulation of apoptotic factors such as Bcl-2-associated agonist of cell death (Bad) and PRAS40 [84]. Furthermore, under oxygen-glucose deprivation, both BMAL1 and melatonin intensify the phosphorylation of key components in the PI3K/AKT cascade—including PDK1, mTOR, PTEN, GSK3α/β, ribosomal S6 kinase 1 (RSK1), ribosomal protein S6 kinase (rpS6K), and p70 rpS6K (p70S6K)—thus promoting cellular resilience [85]. Notably, inhibition of BMAL1, however, not only reduces these phosphorylation events but also disrupts PI3K/AKT signaling, indicating a bidirectional regulatory relationship between BMAL1 and this pathway (Figure 3).
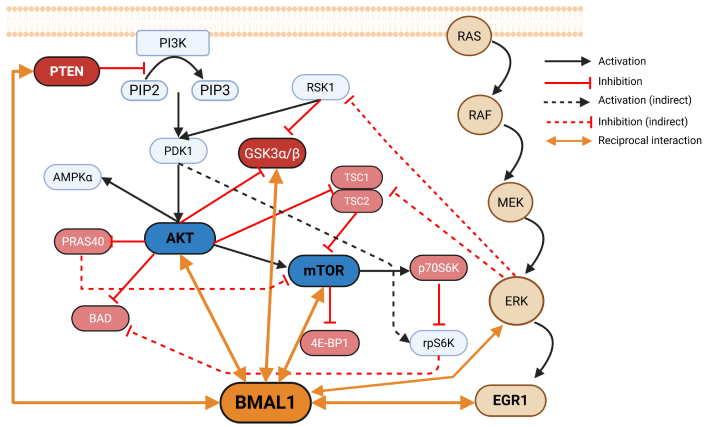
Crosstalk between BMAL1 and the PI3K/AKT/mTOR signaling network. Upon activation of PI3K, the conversion of PIP2 to PIP3 facilitates the recruitment and phosphorylation of AKT by PDK1, leading to activation of mTOR signaling through phosphorylation-dependent inhibition of TSC2 and PRAS40. AKT phosphorylates GSK3α/β, resulting in reduced kinase activity, and phosphorylates PRAS40 to relieve its inhibitory constraint on mTOR. Activated mTOR modulates protein synthesis via 4E-BP1 and p70S6K, while AKT suppresses pro-apoptotic mediators such as BAD and TSC1/2. BMAL1 receives input from both AKT-mTOR and ERK cascades, integrating metabolic and proliferative signals to fine-tune circadian transcriptional outputs. In turn, BMAL1 influences the activity of AKT, GSK3β, and ERK, thereby establishing a feedback loop that synchronizes cellular growth, metabolism, and circadian timing. Arrows indicate activation, blunt lines represent inhibition, dashed lines denote indirect effects, and bidirectional arrows indicate reciprocal interactions. PI3K: phosphatidylinositol 3-kinase; PTEN: phosphatase and tensin homolog; PIP2: phosphatidylinositol (4,5)-bisphosphate; PIP3: phosphatidylinositol (3,4,5)-trisphosphate; PDK1: 3-phosphoinositide-dependent kinase-1; AMPKα: AMP-activated protein kinase α; PRAS40: proline-rich AKT substrate of 40 kDa; BAD: Bcl-2-associated agonist of cell death; RSK1: ribosomal S6 kinase 1; GSK3: glycogen synthase kinase 3; BMAL1: basic helix-loop-helix ARNT-like protein 1; TSC1: tuberous sclerosis protein 1; 4E-BP1: eukaryotic translation initiation factor 4E-binding protein 1; p70S6K: p70 ribosomal protein S6 kinase; rpS6K: ribosomal protein S6 kinase; RAS: rat sarcoma (small GTPase); RAF: rapidly accelerated fibrosarcoma; MEK: mitogen-activated protein kinase/extracellular signal-regulated kinase kinase; ERK: extracellular signal-regulated kinase; EGR1: early growth response protein 1. Created in BioRender. Beker, M. Ç. (2026) https://BioRender.com/suc1om4.
Recent findings have demonstrated that BMAL1 acts as a master integrator of circadian and metabolic resilience during cerebral ischemia. Lentiviral BMAL1 overexpression in mice exposed to transient middle cerebral artery occlusion markedly reduced infarct size and neuronal DNA fragmentation, whereas BMAL1 knockdown or genetic deletion aggravated ischemic damage [86]. Elevated BMAL1 levels increase phosphorylation of mTOR (Ser2448) in ischemic cortex, linking circadian control to metabolic adaptation. Quantitative proteomics has revealed broad BMAL1-dependent modulation of mitochondrial and metabolic pathways, including oxidative phosphorylation, the tricarboxylic acid (TCA) cycle, and subunits of ATP synthase and cytochrome c oxidase (ATP5ME, ATP6V1H, ATP5F1C, ATP5PB, ATP6V1A, COX4I1, COX6B1, COX7A2). This metabolic reprogramming preserves ATP synthesis, stabilizes calcium dynamics, and limits excessive reactive oxygen species (ROS) formation, maintaining neuronal energy balance during ischemia [86].
Complementary in vitro evidence suggests that BMAL1 may indirectly sustain PI3K/AKT-mTOR signaling by stabilizing the cellular redox environment and energy metabolism. In PC12 cells exposed to oxygen-glucose deprivation/reoxygenation, BMAL1 overexpression activated the nuclear factor erythroid 2-related factor 2/heme oxygenase-1 (Nrf2/HO-1) antioxidant axis, reduced ROS accumulation, and restored the Bcl-2/Bax equilibrium, thereby suppressing apoptosis and inflammatory cytokine release [87]. Given that oxidative stress and mitochondrial dysfunction can inhibit PI3K/AKT signaling, these findings support the possibility that BMAL1-mediated redox regulation contributes to the maintenance of AKT phosphorylation and downstream mTOR activity under ischemic stress, potentially reinforcing a time-dependent interplay between circadian control and metabolic survival pathways.
Endothelial- and hematopoietic-specific deletion of BMAL1 further underscores its critical role in vascular PI3K/AKT regulation [88]. In Bmal1flox/flox; Tek-Cre mice, total and phosphorylated AKT (Ser473) levels were markedly decreased in retinal vessels despite preserved eNOS expression, leading to impaired NO signaling, increased nitrative stress, and extensive microvascular degeneration. The resulting threefold rise in acellular capillaries and enhanced neointimal hyperplasia were accompanied by reduced progenitor cell proliferation, highlighting the dependence of vascular repair on BMAL1-AKT signaling. Loss of BMAL1 likely disrupts mTORC2 [rapamycin-insensitive companion of mTOR (RICTOR)]-mediated phosphorylation of AKT, thereby destabilizing vascular PI3K/AKT-mTOR activity and compromising endothelial survival (Figure 3).
Beyond ischemic injury, BMAL1 also contributes to vascular remodeling under hemodynamic stress. In regions exposed to disturbed flow, such as the aortic arch or ligated carotid arteries, expression of BMAL1 and CLOCK increases in parallel with changes in eNOS and AKT phosphorylation [89]. BMAL1 directly modulates eNOS Ser1177 phosphorylation, thereby influencing endothelial tone and NO signaling. It is known that PDGF-BB activates RTKs and engages the canonical rat sarcoma (RAS; small GTPase)-rapidly accelerated fibrosarcoma (RAF) kinase MAPK/ERK kinase (MEK) signaling cascade, culminating in ERK phosphorylation. Activated ERK induces transcriptional programs via immediate early genes such as early growth response protein 1 (EGR1), thereby supporting proliferative responses. In this setting, PDGF-BB-induced ERK activity upregulates BMAL1 through a ROS–ERK–EGR1 axis, while BMAL1, in turn, modulates ERK signaling to fine-tune PDGF-BB-driven vascular smooth muscle cell proliferation [90]. Collectively, these findings indicate that BMAL1 integrates circadian and growth factor signaling to preserve vascular function and structural integrity under ischemic and mechanical stress conditions (Figure 3).
Evidence across ischemic brain and heart models, vascular endothelium, and metabolic contexts collectively suggests a coherent mechanism: BMAL1 appears to align cellular energy state and stress defenses with environmental time by tuning PI3K/AKT activity. When BMAL1 levels or phase are preserved, upstream PI3K signaling, AKT phosphorylation (Thr308/Ser473), and downstream nodes (mTORC1/2, PRAS40, TSC2, GSK3β, FOXO) shift toward pro-survival, pro-angiogenic, and pro-metabolic phenotypes. This configuration sustains mitochondrial function, limits ROS, maintains NO bioavailability, and stabilizes blood-brain barrier integrity, thereby improving ischemic resilience and vascular repair. Conversely, BMAL1 loss or circadian misalignment dampens PI3K/AKT flux, amplifies oxidative and nitrative stress, and worsens ischemic and metabolic injury. The synthesis of mechanistic, proteomic, and functional data supports a model in which BMAL1 acts both upstream of and responsive to AKT-mTOR signaling, forming a time-coded feedback circuit that coordinates transcriptional timing with kinase-driven survival programs.
Importantly, circadian robustness is not governed solely by photic entrainment. Accumulating evidence indicates that systemic non-photic stressors, through their effects on redox balance, mitochondrial function, and metabolic homeostasis, can disrupt SCN-peripheral clock coupling [91–93]. Experimental and observational studies conducted under hypomagnetic or extreme environmental conditions highlight the vulnerability of circadian organization under such stressors [93–95] and provide a conceptual framework by which SCN-peripheral desynchrony may weaken BMAL1-dependent temporal gating of PI3K/AKT signaling. Although direct experimental evidence linking non-photic environmental perturbations to PI3K/AKT dysregulation in ischemic disease remains limited, these observations reinforce the central premise of this review that intact SCN-to-periphery coupling is required to preserve time-of-day-dependent coordination between circadian transcriptional programs and kinase-driven survival pathways, particularly under systemic stress.
Beyond these mechanistic insights, emerging evidence from vascular and cerebrovascular biology indicates that circadian clocks modulate endothelial function, inflammatory signaling, and treatment responsiveness, suggesting that the efficacy of neurovascular interventions may vary according to circadian phase [96, 97]. In this context, chronotherapy—defined as aligning therapeutic interventions with endogenous circadian timing—represents a promising but still insufficiently explored direction for future research. Systematic preclinical and translational studies incorporating circadian phase, stroke subtype, and sex-specific effects, as well as cell-type-specific responses across neurons, endothelial cells, and immune cells, will be essential to determine whether temporal optimization of BMAL1-PI3K/AKT-targeted strategies can enhance therapeutic efficacy while minimizing oncogenic or unintended pro-proliferative signaling in off-target tissues.
A limitation of the present review is that the available clinical and experimental literature does not uniformly stratify findings by circadian phase, stroke subtype, or biological sex, which may contribute to heterogeneity across reported outcomes.
AMPK: AMP-activated protein kinase
BMAL1: basic helix-loop-helix ARNT-like protein 1
cAMP: cyclic AMP
CLOCK: circadian locomotor output cycles kaput
CREB: cyclic AMP response element-binding protein
CRY: cryptochrome
DNA-PK: DNA-dependent protein kinase
EGR1: early growth response protein 1
eNOS: endothelial nitric oxide synthase
ERK: extracellular signal-regulated kinase
FOXO: forkhead box O
GAP: GTPase-activating protein
GSK3: glycogen synthase kinase 3
MAPK: mitogen-activated protein kinase
mTORC2: mechanistic target of rapamycin complex 2
NO: nitric oxide
PDGF: platelet-derived growth factor
PDK1: 3-phosphoinositide-dependent kinase-1
PER: period
PH: pleckstrin homology
PHLPPs: pleckstrin homology-domain leucine-rich-repeat-containing protein phosphatases
PI3K: phosphatidylinositol 3-kinase
PIP2: phosphatidylinositol (4,5)-bisphosphate
PIP3: phosphatidylinositol (3,4,5)-trisphosphate
PP2A: protein phosphatase 2A
PRAS40: proline-rich AKT substrate of 40 kDa
PTEN: phosphatase and tensin homolog
REV-ERBα: nuclear receptor subfamily 1 group D member 1
RORs: retinoic acid receptor-related orphan receptors
ROS: reactive oxygen species
rpS6K: ribosomal protein S6 kinase
RTKs: receptor tyrosine kinases
S473: serine 473
SCN: suprachiasmatic nucleus
T308: threonine 308
TSC2: tuberous sclerosis protein 2
MCB: Conceptualization, Writing—original draft, Writing—review & editing. DMH: Conceptualization, Writing—review & editing. EK: Conceptualization, Writing—review & editing, Funding acquisition. All authors read and approved the final version of the manuscript.
Dirk M. Hermann, who is the Editor-in-Chief and Guest Editor of Exploration of Neuroscience; Ertugrul Kilic, who is the Associate Editor of Exploration of Neuroscience, had no involvement in the decision-making or the review process of this manuscript. The other author declares no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by Turkish Academy of Sciences (TUBA; to EK). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 1719
Download: 28
Times Cited: 0
