 Systematic Review
Systematic Review

Affiliation:
1Department of Oncology, Alfred Health, Melbourne 3004, Australia
ORCID: https://orcid.org/0000-0002-0110-2813
Affiliation:
1Department of Oncology, Alfred Health, Melbourne 3004, Australia
2School of Translational Medicine, Monash University, Melbourne 3004, Australia
Email: malaka.ameratunga@monash.edu
ORCID: https://orcid.org/0000-0002-7171-6781
Explor Neurosci. 2025;4:1006116 DOI: https://doi.org/10.37349/en.2025.1006116
Received: August 12, 2025 Accepted: September 17, 2025 Published: November 28, 2025
Academic Editor: Katrin Sak, NGO Praeventio, Estonia
The article belongs to the special issue Current Approaches to Malignant Tumors of the Nervous System
Background: Glioblastoma multiforme (GBM) is the most common primary malignant brain tumor in adults, with a poor prognosis despite advances in treatment options. T-cell-engager therapies, which have an antibody-based structure connecting immune cells to target cancer cells with high affinity, offer a promising strategy but face four key barriers: antigen heterogeneity, immune escape, the blood-brain barrier (BBB), and the immunosuppressive tumor microenvironment (TME). This systematic review synthesizes preclinical developments in bispecific T-cell engager (BiTE), tri-specific T-cell engager (TriTE), and multi-specific T-cell engagers for GBM over the last 10 years, evaluating their capacity to overcome these barriers.
Methods: A systematic search was conducted in OVID Medline, Embase, and ClinicalTrials.gov for pre-clinical and clinical studies. A descriptive analysis without meta-analysis was formulated in which data were grouped thematically by the ability of treatments to overcome GBM-specific barriers.
Results: Among the 14 studies meeting inclusion criteria, all studies were preclinical, with 12/14 (85.7%) utilizing an in vivo mouse model. BiTEs were used in 12/14 (85.7%) studies, while 4/14 (28.6%) studies targeted multiple antigens through either TriTEs or multivalent BiTEs. There was a range of antigen targets with the most common being interleukin 13 receptor alpha 2 (IL13Rα2) as well as epidermal growth factor receptor (EGFR) or EGFR variant III (EGFRvIII) in 7/14 (50.0%) studies. Most studies (85.7%) addressed two or more barriers, with 13/14 (92.9%) showing evidence of affecting the TME.
Discussion: In the last decade, T-cell engager therapies have evolved in both antigenic targets and delivery vehicles used to overcome the key barriers. An emerging area within T-cell engager therapies is targeting multiple antigens through multi-specific T-cell engager therapies, such as the TriTEs. Studies have explored chimeric antigen receptor T-cells (CAR-Ts) as a potential delivery vehicle for BiTEs. A future clinical trial using multi-specific T-cell engager therapies or a CAR-T-secreting BiTE in adult patients is required to determine the potential clinical utility of T-cell engagers.
Glioblastoma multiforme (GBM) is the most aggressive form of glioma and the most common primary malignant brain tumor in adults [1]. Despite advances in medical, surgical, and radiotherapy treatment options, the prognosis has historically remained poor. The current standard of treatment for GBM frequently involves surgery followed by radiotherapy and concurrent temozolomide (chemo-radiotherapy) as part of the Stupp Protocol, which demonstrated a median survival of 14.6 months [1, 2]. Unlike other cancer types, there is no approved immunotherapy for glioblastoma, with pivotal negative studies such as Checkmate 143 [3]. Historically, the brain has been considered an immune-privileged site; however, there is emerging evidence supporting innate and adaptive immunity in the central nervous system (CNS) [4]. Consequently, there has been increased interest in immunotherapy strategies to target glioblastoma [5]. Strategies have included checkpoint inhibitors, which have largely been unsuccessful [3]; oncolytic viruses, which reported exciting initial results but have not become clinically applicable a decade since their initial reporting [6]; and, more recently, chimeric antigen receptor T-cell (CAR-T) therapy.
Excitement has grown surrounding CAR-T therapy since a 2024 study using intraventricularly infused CAR-T cells in three participants with recurrent glioblastoma targeting epidermal growth factor receptor variant III (EGFRvIII) and wild-type EGFR through secretion of a T-cell-engaging antibody molecule (TEAM) demonstrated transient radiographic tumor regression in two out of the three participants [7]. This contrasts with the antibody-drug conjugate (ADC) depatuxizumab mafodotin, targeting the same antigen, which did not prolong survival in this condition [8]. CAR-T therapy is complicated by high costs and risks of on-target toxicity [9] and significant infrastructure requirements for administration. Moreover, to date, CAR-T therapy has had limited success in solid tumors.
An emerging immunotherapy modality with increasing data in solid tumors is T-cell engager therapies [10, 11]. A subset within antibody-based immunotherapies, T-cell engagers are antibody-based structures that are designed to connect immune cells to the target cancer cell, with high-affinity binding [10]. Bispecific T-cell engagers (BiTEs) were the first T-cell engagers developed, and blinatumomab was initially approved by the Food and Drug Administration (FDA) for treatment of B-cell acute lymphoblastic leukemia (B-ALL) [10]. In the context of solid tumors, the two FDA-approved BiTEs are Tarlatamab and Tebentafusp, which target delta-like ligand 3 in patients with previously treated small-cell lung cancer and glycoprotein 100 in metastatic uveal melanoma, respectively [12, 13]. Notably, an extended follow-up after a median of 12.1 months, Tarlatamab demonstrated metastatic small lung cancer CNS metastases shrinkage in 62.5% of patients with a baseline CNS lesion ≥ 10 mm, highlighting the capacity of BiTEs to have efficacy within the CNS [12–14]. However, no previous studies of approved FDA BiTEs have been conducted in GBM patients.
Structurally, BiTEs consist of two single-chain variable fragments (scFvs), derived from monoclonal antibodies: one targeting CD3 on the T-cell receptor (TCR) complex (anti-CD3), and the other targeting a tumor-associated antigen (such as CD19 in B-ALL), connected by a short peptide linker [10]. Activation of both arms triggers cytotoxic T-cell activation with the release of perforin and granzyme, causing cytolytic tumor cell destruction as well as activation of the innate immune system [15–17]. As BiTEs are not limited by TCR specificity, BiTEs can engage tumor-infiltrating lymphocytes and cytotoxic CD8+ T-effector memory cells independently of the major histocompatibility complex (MHC) and can bypass MHC downregulation in GBM [17, 18]. Consequently, by adding additional scFvs targeting additional tumor antigens, tri-specific T-cell engager (TriTE) and multi-specific T-cell engager therapies have been developed to expand the potential therapeutic utility of engager therapies [10].
Although manufacturing T-cell engager therapies is simpler than CAR-T, as they can be created without requiring patient individualization, key limiting factors are their short serum half-life and suboptimal tissue penetration, often necessitating frequent infusions [18–20]. Despite advances in immunotherapy, treating GBM remains a challenge due to a combination of factors, including limited penetration through the blood-brain barrier (BBB), the antigenic heterogeneity of GBM, immune escape mechanisms, and insufficient understanding of the impact of the tumor microenvironment (TME) [5].
While previous reviews have discussed the key challenges facing T-cell engager therapies, there is currently no framework to systematically evaluate whether emerging therapies address these barriers in a clinically meaningful manner. This review will synthesize the developments of BiTE, TriTEs, and multi-specific T-cell engager therapies within the last 10 years for GBM, focusing on advances in identifying new antigenic targets, novel delivery strategies, and providing a framework for assessing mechanisms to overcome immune resistance. This framework aims to highlight novel solutions and guide future developments in T-cell engager therapies for GBM.
A systematic search was conducted on OVID Medline and Embase on 13th September 2025, using the terms grouped into either T-cell engager therapies or brain tumors. The search terms relating to T-cell engager therapies included “bispecific T-cell engager*”, “bi-specific T-cell engager*”, “trispecific T-cell engager*”, “tri-specific T-cell engager*”, and “multispecific engager*”. While the search terms related to brain cancer included “glioblastoma*”, “GBM”, “Brain tumour*”, “Brain tumor*”, “Brain cancer”, “Brain malignanc*”, “astrocytoma grade 4”, “grade 4 astrocytoma”, “high-grade glioma”. Only studies published within the last 10 years (from 2015 onwards) were included, to reflect the recent advances in the rapidly evolving field of immunotherapy. The same search was performed on ClinicalTrials.gov to identify any studies in progress.
After removing duplicates, studies were subjected to inclusion and exclusion criteria in a two-stage selection process. Inclusion criteria were: (1) publication in a peer-reviewed journal, (2) GBM patients or tissue samples were tested, (3) full-text availability in English, and (4) use of BiTE, TriTEs, or multi-specific T-cell engager therapies. Due to the rapidly evolving nature of the therapy, preprint articles published on bioRxiv were also considered. Review articles and studies that don’t specifically test T-cell engager therapies for GBM were excluded. Initially, titles and abstracts of all records were screened, and potentially eligible full-text articles were obtained and subjected to a second round of screening.
Data were extracted into an Excel spreadsheet, which included information on study design, target antigens, and key findings.
Due to significant heterogeneity in study design, with different animal models used, and a mixture of in vivo and in vitro results, a meta-analysis was considered unsuitable, and instead a descriptive summary of treatment data was formulated. Data were grouped thematically by the ability of treatments to overcome GBM-specific barriers, including antigen heterogeneity [21], immune escape [22], BBB [16], and the TME [23]. To assess the extent to which each study addressed the core challenges of GBM immunotherapy, a framework was developed to evaluate each study. Other barriers, such as pharmacokinetics and systemic toxicity, were considered but not included as all included studies were preclinical and therefore would not be as relevant at this stage of T-cell engager therapy development. Studies were considered to have overcome antigen heterogeneity through the usage of treatment modalities targeting multiple different antigens, while studies were regarded as mitigating immune escape if there was evidence of durable tumor control through mechanisms such as bystander T-cell recruitment, sustained T-cell activity, and memory T-cell formation. Studies were considered to have bypassed the BBB if there was evidence of T-cell engager therapy reaching the CNS and exerting antitumor activity in GBM models. This included either systemic delivery with confirmed CNS penetration and efficacy or local delivery methods (intracranial or intraventricular injection). Studies were further classified based on delivery route (systemic or local delivery). Evaluating the effect of the therapy on the TME is a complex challenge, which was inferred by therapies that enhanced intratumoral T-cell infiltration or activity, downregulated immunosuppressive cells, and sustained local immune activity.
The literature search identified 79 unique records after 26 duplicates were removed (Figure 1). On title and abstract screening, 54 records were excluded, and the remaining 25 articles were retrieved for full-text screening. Of these, 14 papers were included in the final analysis and summarized in Table 1. Three of the articles were preprints published on bioRxiv, which had not yet been peer reviewed but were included due to their relevance [16, 24, 25]. There were two phase 1/2 clinical trials identified (Table 2), NCT04903795 and NCT06814496, but they were not included as they were estimated to be completed in 2026 and 2030, respectively. Another phase 1 clinical trial (NCT03296696) of a BiTE therapy targeting EGFRvIII-positive tumor antigens had an interim analysis completed in 2019 [26], but was prematurely terminated with only an abstract written [27]. As there was no full-text article written about the trial, it was not included in the analysis.
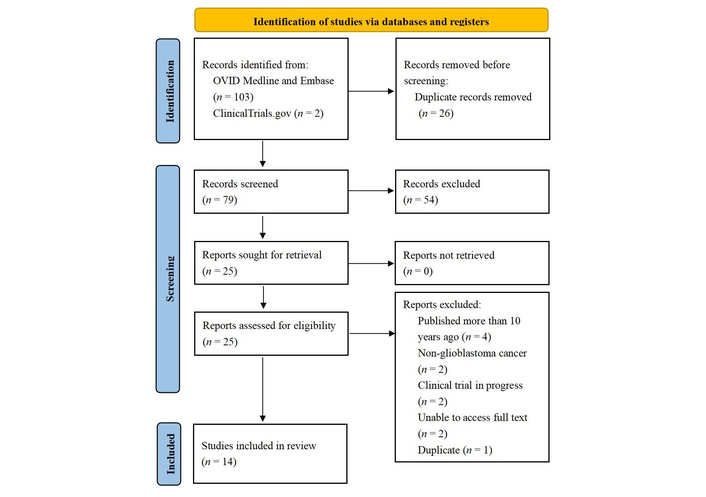
PRISMA flow diagram summary of study selection. Adapted from “The PRISMA 2020 statement: an updated guideline for reporting systematic reviews” by Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. BMJ. 2021;372:n71 (https://www.bmj.com/content/372/bmj.n71). CC BY 4.0.
Summary of bi-, tri-, and multi-specific T-cell engager therapies in preclinical GBM models and their effects against key immunological barriers.
| Study, year | Construct, model type (in vivo, in vitro), delivery method | Antigen target | Antigen heterogeneity | Immune escape | Blood-brain barrier | Tumor microenvironment |
|---|---|---|---|---|---|---|
| Choi et al. [25], 2025 | Construct: BiTE (encoded by oncolytic adenoviruses) + CAR-T cellsModel (in vitro + in vivo): in vitro (U87, U251 cell lines, 2D/3D, BBB spheroid); in vivo (subcutaneous GBM xenograft, NSG mice)Delivery: intratumoral injection of BiTE followed by systemic infusion of CAR-T | IL13Rα2 (BiTE), EGFR, and EGFRvIII (CAR-T) | √Multimodal BiTE & CAR-T targeting multiple antigens | ×Significant reductions in tumor luminescence intensity; however, no evidence of durable control, memory, or bystander T-cell recruitment | ×The subcutaneous tumor model is not able to accurately assess the BBB | √Increased intratumoral CD3+ T-cell recruitment and infiltration |
| Zannikou et al. [24], 2025 | Construct: BiTE Model (in vivo + in vitro): immunocompetent mice (genetically engineered mouse model, orthotopic GL261-IL13Rα2) + murine glioma cell lines (GL261, SMA560, and CT2A) Delivery: systemic (IV) | IL13Rα2 | ×Single target | √Enhanced memory T-cell formation, sustained T-cell activity | √BiTE detected in the brain following systemic IV administration | √Increased intracranial CD8+ T-cells, memory T-cells, and regulatory T-cells (Tregs). Reduced glioma volume and viability, reduced intracranial immunosuppressive myeloid cells |
| Brosius et al. [16], 2024 | Construct: BiTE Model (in vivo + in vitro): orthotopic high-grade glioma xenografts in nude mice + in vitro human/mouse co-culturesDelivery: injected intracranially | EGFR | ×Single target | ×Not specifically addressed | √Bypassed via local delivery of migratory cortical inhibitory interneuron precursors (MCIPs), which migrated intracranially to tumors and secreted BiTEs within the CNS | √In vitro, killing of GBM cells was induced by CD8+ T-cells, but no clear evidence of TME modulation in vivo |
| Park et al. [21], 2024 | Construct: TriTE Model (in vivo + in vitro): immunocompromised mice, as well as a patient in vitro assay using patient peripheral blood mononuclear cells (PBMCs)Delivery: intramuscular injection | EGFRvIII and IL13Rα2 | √Targets both EGFRvIII and IL13Rα2 in a single construct | √Durable tumor clearance, sustained expression (up to 105 days), and effective tumor control in a heterogeneous GBM model, including post-radiotherapy and post-chemotherapy PBMC | ×Unclear if crosses BBB or acts via peripheral immune activation | √Induces activation of CD4+, CD8+, and natural killer T-cells; promotes antitumor cytokine release (IFN-γ, TNF-α, IL-2) |
| Baugh et al. [1], 2024 | Construct: BiTE (delivered via oncolytic HSV-1 G207)Model (in vitro only): GBM cell lines, patient-derived mesenchymal glioma stem cellsDelivery: no in vivo models | NKG2DL | ×Single target | ×Not addressed—no demonstration of durable tumor control, memory T-cell formation, or bystander activation | ×Entirely in vitro | √CD4+ and CD8+ T-cell activation with increased CD25, CD69, IFN-γ, granzyme B, perforin, and CD107a in the presence of GBM cells; activity synergized with sublethal radiation and temozolomide, enhancing antigen expression and T-cell activation |
| Park et al. [22], 2023 | Construct: multivalent BiTEs Model (in vivo + in vitro): mice Delivery: intramuscular injection plus electroporation | EGFRvIII and HER2 | √Targeting both EGFRvIII and HER2 resulted in enhanced cytotoxicity and 80% tumor clearance | √Potent and durable CD4+ and CD8+ T-cell activation; mitigated immune escape in 80% of the challenged mice | ×Not directly addressed | √Activated CD4+ and CD8+ T-cells with increased secretion of IFN-γ, TNF-α, IL-2, and activation of CD107a (marker for degranulation). Tumor regression in orthotopic models |
| Bhojnagarwala et al. [15], 2022 | Construct: BiTE Model (in vivo + in vitro):immunodeficient NSG mice; In vitro: U87, U251, U373 GBM lines Delivery: systemic (IV) via DNA electroporation | IL13Rα2 | ×Single target | ×Not specifically addressed | √Peripherally delivered DNA-based BiTE crossed the BBB and controlled orthotopic GBM growth | √CD4+ and CD8+ T-cell activation, cytokine release (IFN-γ, IL-2, TNF-α), granule secretion (perforin, granzyme A and B), and tumor cytolysis |
| Huynh et at. [28], 2022 | Construct: dual antigen T-cell engager Model (in vitro only): 3D GBM spheroid models (GBM08, BT935) Delivery: no in vivo models; in vitro used hydrogel-based local release system | CD133 | ×Single target | √Small increase in CD45RO+ effector memory T-cells | ×Not addressed (no in vivo or BBB-relevant model) | ×TME effects are not specifically addressed, as no in vivo or stromal component |
| Yin et al. [11], 2022 | Construct: BiTE Model (in vivo + in vivo): orthotopic miceDelivery: infusion (route not specified) | EGFR and IL13Rα2 | √Dual antigen targeting (EGFR and IL13Rα2) using bivalent BiTE constructs | ×No evidence demonstrated | ×Unclear if systemic delivery crossed the BBB | √Enhanced T-cell activation (CD69), cytokine production (IFN-γ, TNF-α, IL-2), tumor infiltration, and tumor suppression |
| Li et al. [29], 2021 | Construct: BiTE Model (in vivo + in vitro): mice; U87/U251/A172/T98G cell linesDelivery: local intratumoral injection | Fn14 | ×Single target | ×No evidence demonstrated | √Injected directly into the lesion and suppressed tumor growth | √Increased CD3+ T-cell infiltration and tumor suppression |
| Pituch et al. [18], 2021 | Construct: BiTE (secreted by neural stem cells)Model (in vivo + in vitro): in vivo (mice), in vitro (human PBMCs, GBM6, GBM12, GBM39)Delivery: local intratumoral injection | IL13Rα2 | ×Single target | ×Partially addressed as it engages local CD3+ T-cells and produces granzyme B, but no durable control, memory T-cells, bystander cell recruitment | √Local intracranial NSC delivery enables CNS access, where it persists in the tumor | √Increased CD3+ infiltration, IFN-γ/TNF-α/IL-2 cytokine production, activation of exhausted tumor-infiltrating T-cells |
| Arnone et al. [30], 2021 | Construct: BiTE (encoded by oncolytic adenoviruses) Model (in vitro + in vivo): in vitro human GBM cancer cell lines (U373, U87) and an in vivo xenograft mouse modelDelivery: local intratumoral injection | EphA2 | ×Single target | √Increase memory T cells, increase activation of CD4 and CD8 T cells | √Injected directly into the lesion with detection of infiltrating T-cells in the tumor | √Enhanced intratumoral T-cell infiltration, activation of T-cell effector function, including Th1 cytokines (IFN-γ), and increased chemokine production |
| Gardell et al. [19], 2020 | Construct: BiTEs (secreted by genetically engineered human macrophages)Model (in vivo + in vitro): mice (subcutaneous and intracranial GBM U87 EGFRvIII xenografts); in vitro (human T-cells + GEMs + EGFRvIII+ GBM cells)Delivery: local intratumoral injection | EGFRvIII | ×Single target | ×Tumor rebound observed in both models; no survival benefit from BiTE GEMs alone; modest effect enhanced by IL-12 co-secretion. In vitro upregulation of memory-associated gene (PRDM1) and cytokines associated with T-cell survival (IL-2, IL-7, IL-15), but not formally assessed in vivo | √Bypasses BBB via direct intracranial injection; GEMs enable local BiTE secretion and CNS-targeted immune activation | √Increased CD3+, CD8+ T-cell infiltration (↑ CD25, CD69, CD107a, IFN-γ, granzyme B), increased cytokines (IFN-γ, TNF-α, IL-2/7/15), chemokines (CXCL9/13), cytotoxic markers (GZMB, LAMP3); downregulation of immunosuppressant TGFB1 |
| Choi et al. [31], 2019 | Construct: BiTEs (secreted by CAR-T cells)Model (in vivo + in vitro): mice (orthotopic and heterogenous GBM xenografts), in vivo (primary human T-cells and GBM cells)Delivery: local intraventricular injection | EGFRvIII | √Able to mitigate antigen escape by redirecting bystander T-cells against EGFR-positive, EGFRvIII-negative tumor cells | √Redirect non-specific bystander T-cells and Tregs to exert cytotoxicity; reverses exhaustion when CAR + BiTE co-stimulation used (reduced PD-1, TIM-3 and LAG-3) | √Intraventricular delivery enables local BiTE production within the CNS. BiTE was not detected systemically | √Increased T-cell infiltration and cytokine secretion (IFN-γ, TNF-α) |
×: Does not fulfill criteria; √: fulfills criteria. BBB: blood-brain barrier; BiTE: bispecific T-cell engager; CAR-T: chimeric antigen receptor T-cell; CNS: central nervous system; EGFR: epidermal growth factor receptor; EGFRvIII: epidermal growth factor receptor variant III; EphA2: erythropoietin-producing human hepatocellular carcinoma A2 receptor; Fn14: fibroblast growth factor-inducible 14; GBM: glioblastoma multiforme; GEMs: genetically engineered macrophages; IL13Rα2: interleukin 13 receptor alpha 2; NKG2DL: natural killer group 2 member D ligands; TME: tumor microenvironment.
Ongoing clinical trials of T cell engager therapies in glioblastoma.
| NCT number | Intervention | T-cell engager antigen target | Indications | Trial phase | Primary endpoint | Delivery route |
|---|---|---|---|---|---|---|
| NCT04903795 | Bispecific T cell engager therapy post radiation plus temozolomide | EGFRvIII | Grade 4 glioma with EGFRvIII mutation | Phase 1 | Dose-limiting toxicity | Bolus injection |
| NCT06814496 | Combination bispecific T cell engager therapy (Tarlatamab) with radiation | DLL3 | Glioblastoma, melanoma, Merkel cell carcinoma, medullary thyroid cancer, sinonasal undifferentiated cancer, esthesioneuroblastoma, bladder cancer, testicular cancer, cervical cancer, large cell neuroendocrine lung cancer, and non-small cell lung cancer | Phases 1 and 2 | Dose-limiting toxicity | Infusion |
EGFRvIII: epidermal growth factor receptor variant III; DLL3: delta-like ligand 3.
All studies were preclinical, with no studies performed on living humans. Overall, 12/14 (85.7%) studies utilized an in vivo mouse model, while 2/14 (14.3%) only performed in vitro experiments using patient-derived mesenchymal glioma stem cells or 3D GBM spheroid models. Although 12/14 (85.7%) studies utilized BiTEs, there were variances in the delivery methods and antigenic targets. The most common T-cell engager targets were EGFR or EGFRvIII in 6/14 (42.9%) studies, and interleukin 13 receptor alpha 2 (IL13Rα2) in 6/14 (42.9%) of studies. Five studies had unique antigen targets of HER2, natural killer group 2 member D ligand (NKG2DL), CD133, erythropoietin-producing human hepatocellular carcinoma A2 receptor (EphA2), and fibroblast growth factor-inducible 14 (Fn14). Beyond BiTEs, 4/14 (28.6%) studies targeted multiple antigens through either TriTEs, multivalent BiTEs, or a combination of BiTE and CAR-T. In terms of delivery methods, 3/14 (21.4%) were systemic infusions, 7/14 (50.0%) were locally administered intratumorally, 2/14 (14.3%) were intramuscular injections, and 2/14 (14.3%) were in vitro only. In terms of studies addressing the four criteria limiting T-cell engager therapy efficacy in GBM, 5/14 (35.7%) of studies addressed antigen heterogeneity, 6/14 (42.9%) demonstrated evidence of overcoming immune escape, 8/14 (57.1%) were able to bypass the BBB successfully, and 13/14 (92.9%) showed evidence of affecting the TME. Overall, as demonstrated in Table 3, 2/14 (14.3%) studies addressed one barrier, 7/14 (50.0%) two barriers, 4/14 (28.6%) three barriers, and 1/14 (7.1%) all four barriers.
Matrix heatmap summarizing whether included studies addressed the four key barriers to T-cell engager therapy in GBM.
| Study | Antigen heterogeneity | Immune escape | BBB | TME |
|---|---|---|---|---|
| Choi et al. [25], 2025 | √ | × | × | √ |
| Zannikou et al. [24], 2025 | × | √ | √ | √ |
| Brosius et al. [16], 2024 | × | × | √ | √ |
| Park et al. [21], 2024 | √ | √ | × | √ |
| Baugh et al. [1], (2024 | × | × | × | √ |
| Park et al. [22], 2023 | √ | √ | × | √ |
| Bhojnagarwala et al. [15], 2022 | × | × | √ | √ |
| Huynh et al. [28], 2022 | × | √ | × | × |
| Yin et al. [11], 2022 | √ | × | × | √ |
| Li et al. [29], 2021 | × | × | √ | √ |
| Pituch et al. [18], 2021 | × | × | √ | √ |
| Arnone et al. [30], 2021 | × | √ | √ | √ |
| Gardell et al. [19], 2020 | × | × | √ | √ |
| Choi et al. [31], 2019 | √ | √ | √ | √ |
Green and √ = barrier addressed; red and × = barrier not addressed. GBM: glioblastoma multiforme; BBB: blood-brain barrier; TME: tumor microenvironment.
Although our understanding of the CNS as an immune-privileged site has evolved to recognize the role of the CNS in the cancer-immunity cycle, the immunosuppressive TME of GBM has limited treatment options. Due to the complexity of treating GBM, a framework was developed to focus on four of the key barriers preventing optimal treatment of GBM: antigen heterogeneity, immune escape mechanisms, the BBB, and the TME. Emerging immunotherapies such as T-cell engager therapies have been developed to overcome specific aspects of these barriers. This systematic review synthesizes the preclinical developments within the last decade to provide a structure when translating this therapy into clinical trials.
Antigen heterogeneity in the context of GBM refers to the intra-tumor phenotypic and genetic diversity that occurs during the course of tumor progression [32]. Studies using fluorescent in situ hybridisation of individual tumor samples have demonstrated antigenic mosaicism, with varying patterns of tyrosine kinase receptor expression [32]. A study assessing potential ADC targets in the CNS identified HER3/ERBB3, CD276/B7-B3, and NECTIN4 expression in adult GBM, while HER2 expression was absent [33]. Conversely, a different study of 43 patients with high-grade glioma (WHO grades III–IV) identified interpatient heterogeneity of antigen expression such that 11.6% were double positive for EGFR and IL13Rα2, 11.6% for IL13Rα2 and HER2, 25.6% for EGFR and HER2, 23.3% triple positive, and only 7.0% triple negative [34]. Consequently, 93.0% of patients expressed at least one of EGFR, IL13Rα2, or HER2, highlighting their utility as antigen targets. Within the context of T-cell engager therapies, a range of different antigens have been investigated, including IL13Rα2, EGFR, Fn14, NKG2DL, EphA2, and HER2. Although the GD2 tumor antigen is highly expressed in GBM tissue, it has not yet been investigated as a target for T-cell engager therapies and could be a novel target in future studies [35]. When designing potential T-cell engager therapies, the ideal approach would require choosing a universally expressed antigen on a population level that is highly specific for GBM and not expressed in other tissues in the body [17]. Among the antigens explored in the included studies, IL13Rα2 is an established glioma-specific antigen associated with a more aggressive disease and a poor prognosis [15, 24]. Expressed in approximately 75% of individuals with GBM as well as non-CNS tumors and brain metastases, IL13Rα2 is not present in normal tissues, highlighting its high specificity as a tumor target [15, 24]. Amplification and overexpression of EGFR is the most common mutation in GBM, occurring in 34–63% cases, while EGFRvIII, the most frequent mutant form of EGFR, is expressed in 25–64% of diagnosed GBM and is undetectable in normal tissue [34]. Conversely, Fn14, which was an antigen target by Li et al. [29], is expressed in normal tissues, and consequently, despite intralesional delivery, there was cross-reactivity between the Fn14× CD3 BiTE and normal tissue. Despite the need for novel targets such as Fn14, the risk of off-tumor side effects may limit potential clinical applications. EphA2 was selected by Arnone et al. [30], as it is more accessible during malignant cell division and not on normal cells. Among the four different barriers used to assess the included studies, antigenic escape was the least addressed, with only 5/14 (35.7%) successfully overcoming it. An emerging area within T-cell engager therapies is targeting multiple antigens through multi-specific T-cell engager therapies, such as the TriTEs utilized by Park et al. [21], which targeted EGFRvIII and IL13Rα2. Notably, Choi et al. [25] used a combination of an intratumorally delivered BiTE targeting IL13 Rα2, which was followed by a systemic infusion of CAR-T targeting either EGFR and EGFRvIII. Although the study was performed on a subcutaneous GBM mouse model, there was evidence of T-cell recruitment and reduced tumor mass, highlighting the potential of combination therapy approaches [25].
Whereas antigenic heterogeneity illustrates the importance of therapy design and the rationale for multi-specific agents, immune escape highlights dynamic mechanisms of recurrence and resistance that can arise despite targeted antigen approaches. Immune escape mechanisms include downregulation and selective survival of antigen-deficient tumor populations, limiting the potential utility of single antigen-targeted treatments [22]. Notably, antigen loss has been previously demonstrated in recurrent GBM following treatment with CAR-T cells targeting EGFRvIII and IL13Rα2 [36, 37]. Consequently, combination therapies targeting multiple tumor antigens, such as tri-specific antibodies or co-delivery of multiple BiTEs, are an emerging approach [11, 22]. Therapies can be considered to address immune escape mechanisms if they are able to achieve durable tumor control despite tumor evolution and adaptation, such as by recruiting bystander T-cells or persistent T-cell activity. For example, the TriTEs utilized by Park et al. [21] not only were able to induce natural killer (NK) T-cells but also induced CD8+ T-cell-mediated cytotoxicity in cell lines with variable antigen expression, including difficult-to-treat MGMT-unmethylated cell lines. A previous study by Park et al. [22] used a multivalent approach of co-delivery of BiTEs targeting EGFRvIII and HER2, which demonstrated enhanced tumor clearance and survival. Mice that received combined treatment had an 80% survival rate compared with murine models that received single antigen therapy, which only had a 20% (EGFRvIII) and 10% (HER2) survival rate, highlighting the significant improvement in mitigating immune escape [22]. Choi et al. [31] developed a CAR-T BiTE that was able to target multiple antigens, activate bystander T-cells, and enhance local BiTE production, reducing the potential need for continuous infusions. Choi et al. [31] support the complementary use of CAR-T and BiTEs, as CAR costimulatory domains can protect T-cells from BiTE-driven exhaustion. There was an overlap between studies that addressed both antigenic heterogeneity and immune escape, with 3/8 (37.5%) addressing both, 2/8 (25.0%) addressing antigenic heterogeneity only, and 3/8 (37.5%) addressing immune escape only. Future studies should continue to explore multi-specific T-cell engagers due to their demonstrated capacity to simultaneously overcome antigenic heterogeneity and immune escape mechanisms.
A consistent challenge of treating GBM is identifying the optimal delivery mechanism for treatments to bypass the BBB, to reach the CNS, an immune-privileged space with restricted immune infiltration [16]. Despite the small size of BiTEs providing a theoretical advantage compared to larger monoclonal antibodies, the movement of effector cytotoxic T-cells to GBM is still limited by the BBB [17]. There are limitations to directly injecting BiTEs into the brain, such as its short half-life of two to four hours, which necessitates continuous infusions using two-, four-, or seven-day bags, complicating delivery and increasing infection risk [16, 38]. Additional inherent limitations of BiTEs include the lack of biodistribution and inability to self-amplify once infused [15, 39]. To address these issues, several innovative delivery systems have been explored in the context of T-cell engager therapies, including using migratory cortical inhibitory interneuron precursors (MCIPs), using a DNA-launched platform to deliver BiTEs, using neural stem cells as a cellular carrier, human monocyte-derived macrophages secreting BiTEs, and CAR-T secreting BiTEs. Among the studies that were able to overcome the BBB, 2/7 (28.6%) were given via a systemic infusion, while the remaining 5/7 (71.4%) were all given locally intracranially. The first study, which was given systemically, was by Bhojnagarwala et al. [15], who utilized a DNA-launched BiTE targeting IL13Rα2, and that BiTE was able to bypass the BBB and control the GBM models. The second systemically administered study was by Zannikou et al. [24], who utilized a BiTE construct also targeting IL13Rα2, which was subsequently detected in the brain post-delivery. Notably, Bhojnagarwala et al. [15] were limited by the short half-life, requiring continuous infusions. Among the therapies given locally, Brosius et al. [16] injected intracranially a BiTE targeting EGFR that utilized a cellular delivery system harnessing the innate ability of MCIPs to overcome the BBB. As MCIP cells are native to the brain and can functionally integrate, MCIPs are theoretically capable of continuously secreting BiTEs locally to the tumor, limiting off-tumor effects, and potentially facilitating a wider array of antigenic targets [16]. Using direct intracranial administration of modified neural stem cells to secrete IL13Rα2-targeting BiTEs, Pituch et al. [18] demonstrated that BiTEs were able to persist 7 days post administration, which was eventually limited by antigenic escape. Another study by Gardell et al. [19] used a human macrophage lentivirus vector engineered to secrete BiTEs targeting EGFRvIII that was able to overcome the BBB and induce T-cell granulation and cytokine release, causing reduced tumor growth. Although the BiTE was delivered intratumorally via injection rather than systemic delivery, the use of genetically engineered macrophages (GEMs) provided several advantages, including facilitating antigenic presentation, supporting T-cell effector functions, and proinflammatory responses in the TME [19]. Both Choi et al. [25] and Arnone et al. [30] delivered BiTEs intratumorally, which were encoded by oncolytic adenoviruses, which is an emerging strategy that aims to augment immune activation and responsiveness to immunotherapies. While Arnone et al. [30] showed tumor-infiltrating T-cells, due to the subcutaneous model used by Choi et al. [25], they could not accurately assess the BBB. These different approaches highlight that cellular carriers may be an effective strategy to enhance BBB penetration, prolong intratumoral BiTE activity, and reduce off-target toxicity. Notably, among the two clinical trials in progress that are using BiTEs in GBM patients, both are being given via infusions. While neither study specifically addressed the BBB in their design descriptions, both studies are using BiTEs in combination with cranial radiotherapy, which has some evidence that it can influence BBB permeability [40]. A 2022 systematic review identified a high risk of bias, including publication and heterogeneity among studies assessing radiotherapy’s effect on the BBB, and therefore, the results of the two clinical trials will help inform future approaches in BiTE design [40].
The GBM TME is a highly complex, dynamic system comprising cellular (endothelial cells, neuronal cells, astrocytes, oligodendrocytes, immune cells), and non-cellular (signaling molecules, extracellular matrix) components [23]. Not only is the TME immunosuppressive but most gliomas are intrinsically poorly immunogenic, with a lower availability of immunogenic neoantigens [23]. Although microglia and glioma-associated macrophages (GAMs) are among the most prevalent immune cells within the TME, other immune cells such as myeloid-derived suppressive cells (MDSCs) are present [23]. Not only do GBMs secrete immunosuppressive cytokines such as TGF-β, IL-6, IL-10 which can reduce MHC class 2 expression on microglia, but they also can induce M1 to M2 phenotype switching of macrophages, which has been associated with tumor progression [4]. NK cells are relatively sparse in GBM, and T-cells poorly infiltrate with CD8+ and CD4+ T-cells typically dysfunctional in GBM due to a combination of senescence, tolerance, anergy, exhaustion, and ignorance [41]. Although immune checkpoint inhibitors have had clinically meaningful responses in melanoma brain metastases [42], they have conversely demonstrated poor responses in primary gliomas, which is possibly related to the low mutational load of GBM [4, 23]. A phase 3 study comparing nivolumab versus bevacizumab in recurrent GBM did not demonstrate a survival advantage using immunotherapy, while a study assessing pembrolizumab in recurrent GBM only had a response rate of 8% and a median OS of 13.1 months [3, 43]. Conversely, a study of combination durvalumab plus radiotherapy for new diagnosis unmethylated MGMT GBM had an OS of 15.1 months [44]. In the context of GBM, T-cell engager therapies can be considered to address the TME if they enhance intratumoral T-cell infiltration or activity, promote formation of memory T-cells, downregulate immunosuppressive cells and sustain local immune activity over time. Among the studies included, 13/14 (92.9%) demonstrated a meaningful impact on the TME. Attempting to specifically address the TME, Baugh et al. [1] targeted a unique antigen of NKG2DL using an engineered oncolytic herpes simplex virus to secrete BiTEs from infected cells. Although the study demonstrated an increase in T-cell activation, the authors acknowledged that cancer cells have previously evolved tumor evasion methods by dysregulating the NKG2D response, limiting the potential use of this antigen target [1]. An important limitation of the existing studies is their reliance on immunodeficient models that may not accurately capture the adult human GBM microenvironment [18]. Consequently, Zannikou et al. [24], specifically assessed BiTEs on immunocompetent mouse models to evaluate the effect of the TME. The BiTEs used by Zannikou et al. [24] addressed the TME by triggering T-cell activation, increasing memory T-cell formation, and reducing immunosuppressive intracranial myeloid cells with resultant reduction in glioma volume and viability.
Due to the emerging nature of T-cell engager therapies, there is a paucity of studies performed in humans, with only one clinical trial performed using BiTEs, which was prematurely stopped. An interim analysis on the phase 1 trial was performed on AMG 596, a BiTE targeting EGFRvIII which showed adverse events in all patients analysed (14/14), with serious adverse events in 7/14 (50%) [26]. Adverse events were considered tolerable with none causing discontinuation, such that headache and reduced consciousness in 2/14 (14.3%) were the most common grade 3 or above adverse events [26]. In the 8/14 patients that had sufficient follow-up, 1/8 (12.5%) achieved partial response, 2/8 (25.0%) had stable disease, and 4/8 (50.0%) had progressive disease [26]. Among clinical trials in progress, as shown in Table 2, there is a phase 1 clinical trial (NCT04903795) using BiTEs targeting EGFRvIII and another phase 1/2 study (NCT06814496), which is planning to assess palliative and consolidative radiotherapy in combination with Tarlatamab in multiple malignancies, including GBM. Unfortunately, there are no other active clinical trials assessing T-cell engager therapies in GBM. A challenge that was not explored due to the preclinical nature of the included studies is the practicalities of scaling T-cell engager therapies, including costs, regulatory hurdles, and manufacturing feasibility. As a platform, there have been several important evolutions which newer generations of BiTE molecules, including improving the tolerability through a fully human anti-CD3-binding domain, enhancing the stability through disulfide bonds, and extending the half-life through BiTE core to a single-chain Fc (scFc) [45]. T-cell engager therapy in GBM is an emerging strategy, and further research will be required to optimize translation into clinical use.
Aside from the studies by Baugh et al. [1] and Huynh et al. [28], all the included studies were performed in vivo on mouse models. It is important to highlight the differences between murine and human T-cells, as specific modifications to human BiTEs to improve their pharmacokinetics and plasma half-life will likely be required to ensure therapeutic levels in humans [24]. Due to the heterogeneity in experimental design and models both in vivo and in vitro, a meta-analysis could not be accurately performed. Once the safety of T-cell engager therapies is established in GBM, future studies potentially could explore combination approaches with other classes of emerging immune adjuvant drugs, such as IL-2 or Pi3K delta inhibition. For example, there is currently a clinical trial (NCT07063875) exploring Tebentafusp in combination with IL-2 for metastatic uveal melanoma.
Ultimately, T-cell engager therapies are an emerging therapeutic option for GBM, with preclinical studies demonstrating promising strategies to overcome key treatment barriers. In the last decade of research, there has been an evolution in both the antigenic targets and delivery vehicles to which the therapy has been administered to improve efficacy. Notable advancements include multi-specific approaches to address antigen heterogeneity and immune escape, novel delivery methods to bypass the BBB, and a combination of strategies to optimize the TME for tumor killing. Further pharmacokinetic research is required to extend the half-life of T-cell engager therapies, enabling systemic delivery and minimizing off-targeting toxicity. All 14 of the included studies are preclinical, and as 85.7% utilized an in vivo mouse model, there remains uncertainty regarding their potential translation into human studies. Given that the only clinical trial performed was prematurely terminated, hopefully, the two planned clinical trials will be followed through to completion. Further preclinical research using adult human GBM tissue models rather than murine models would be beneficial, as the murine TME does not entirely reflect the adult human TME. As most of the research has been primarily pre-clinical in murine models, the eventual goal is another clinical trial using multi-specific T-cell engager therapies targeting EGFRvIII and IL13Rα2 or a CAR-T secreting BiTE in adult patients to advance the translation as a potential treatment for GBM.
ADC: antibody-drug conjugate
B-ALL: B-cell acute lymphoblastic leukemia
BBB: blood-brain barrier
BiTE: bispecific T-cell engager
CAR-Ts: chimeric antigen receptor T-cells
CNS: central nervous system
EGFR: epidermal growth factor receptor
EGFRvIII: epidermal growth factor receptor variant III
EphA2: erythropoietin-producing human hepatocellular carcinoma A2 receptor
FDA: Food and Drug Administration
Fn14: fibroblast growth factor-inducible 14
GBM: glioblastoma multiforme
IL13Rα2: interleukin 13 receptor alpha 2
MCIPs: migratory cortical inhibitory interneuron precursors
MHC: major histocompatibility complex
NK: natural killer
NKG2DL: natural killer group 2 member D ligand
scFvs: single-chain variable fragments
TCR: T-cell receptor
TME: tumor microenvironment
TriTE: tri-specific T-cell engager
AHL: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. MA: Conceptualization. Both authors read and approved the submitted version.
Both authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
All datasets analyzed for this study are included in the main text and the references.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Zohreh Khosravi Dehaghi
Maria Ciscar-Fabuel ... Andreu Gabarros-Canals
Maria Ciscar-Fabuel ... Andreu Gabarros-Canals
Julius Mulumba ... Yong Yang
