 Perspective
Perspective

Affiliation:
1Department of Experimental Medicine, University of Genoa, 16132 Genoa, Italy
ORCID: https://orcid.org/0000-0002-7206-376X
Affiliation:
2Interventional Pulmonary Unit, IRCCS Ospedale Policlinico San Martino, 16132 Genoa, Italy
ORCID: https://orcid.org/0000-0001-5365-134X
Affiliation:
2Interventional Pulmonary Unit, IRCCS Ospedale Policlinico San Martino, 16132 Genoa, Italy
Affiliation:
3Molecular Pathology Unit, IRCCS Ospedale Policlinico San Martino, 16132 Genoa, Italy
Email: daniela.detotero@hsanmartino.it
ORCID: https://orcid.org/0000-0002-8481-7848
Explor Med. 2023;4:461–470 DOI: https://doi.org/10.37349/emed.2023.00156
Received: March 02, 2023 Accepted: April 19, 2023 Published: August 31, 2023
Academic Editor: Bernhard Ryffel, University of Orleans, France
The article belongs to the special issue Lung Fibrosis—Models and Mechanisms
Broncho-alveolar lavage (BAL) represents a safe tool for the differential diagnosis of various pulmonary fibrotic diseases. Idiopathic pulmonary fibrosis (IPF) belongs to a heterogeneous group of diseases, interstitial lung disease (ILD), presenting a progressive impairment of pulmonary functions. IPF is characterized by the excessive accumulation of extracellular matrix (ECM) in the alveolar parenchyma that may lead to irreversible pulmonary remodeling. Although the exact pathogenetic mechanisms leading to IPF development are still unclear it has been demonstrated that fibroblasts differentiating toward myofibroblasts are the major actors involved in this process. The possibility of obtaining and expanding fibroblasts from the BAL of ILD patients for research purposes has been recently explored. This approach is discussed here as a reliable chance, helpful to advance the scientific community knowledge and to devise two- and three-dimensional (2D/3D) pre-clinical in vitro models of these diseases, further overcoming technical and ethical concerns related to the use of fibroblasts derived from tissue biopsy.
Interstitial lung diseases (ILD) are a large and heterogeneous group of rare and chronic respiratory disorders that present progressive damage to the lung parenchyma [1]. Several ILD subtypes have pathophysiological and morphological features in common. Alveolar epithelial damage and fibroblastic proliferation leading to fibrosis are the mechanisms underlying a group of fibro-proliferative ILD; other ILD are characterized by the activation of pro-inflammatory signals in the lung parenchyma, which may under certain circumstances develop toward the fibro-proliferative pathway [2]. Idiopathic pulmonary fibrosis (IPF) is the most common ILD subtype characterized by remodeling of the pulmonary interstitium due to repetitive local micro-injuries in the context of multiple genetic and environmental risk factors. IPF, regarded as the prototype of fibrosing ILD, has a poor prognosis with a median survival of 3–5 years from diagnosis [3, 4]. Disease progression is evidenced by increasing respiratory symptoms, worsening pulmonary function, progressive fibrosis on high-resolution computed tomography (HRCT), and acute respiratory decline that finally leads to death. In addition, patients with IPF are at higher risk of developing lung cancers including squamous cell lung carcinoma (SCC). Although much of the pathogenesis of IPF remains to be elucidated, fibroblasts and epithelial cells have emerged as the major players in this disease, in particular myofibroblasts and type II alveolar epithelial cells. Myofibroblasts accumulate in IPF lungs in “fibroblastic foci” that, as the predominant sites of excess matrix production, can be thought of as the leading edge of active fibrosis. The origin of the fibroblasts remains debated being probably multiple and possibly due to the proliferation of resident fibroblasts or mesenchymal precursors, to the transition of epithelial, endothelial, or mesothelial cells toward a mesenchymal phenotype, or either to the recruitment to the lung of circulating blood mesenchymal precursors [5, 6]. Broncho-alveolar lavage (BAL) fluids collection from ILD patients is a well-tolerated and minimally invasive procedure helpful for clinical and research assessments. In this paper it is presented and discussed the perspective of using BAL from patients with IPF or other ILD to derive and expand fibroblasts, aiming to better assess their features and to improve pre-clinical models of lung fibrosis in vitro.
Already several years ago some authors tried to derive and culture fibroblasts from BAL of ILD patients to study their functional features. Fireman et al. [7–11] successfully cultured fibroblasts of BAL from patients diagnosed with IPF, sarcoidosis (SA), or hypersensitivity pneumonitis (HP), to better define their biochemical/morphological properties and to study whether particular cytokines secreted by alveolar macrophages [i.e. interleukin-1α (IL-1α), -β, prostaglandin E2 (PGE2), or endothelin-1 (ET-1)] could affect fibroblasts proliferation and increase the expression of fibrotic markers [i.e. alpha-smooth muscle actin (α-SMA), fibronectin]. Larsen et al. [12] further compared fibroblasts derived from BAL of patients with scleroderma with those of patients with asthma. The authors here highlighted, through two-dimensional electrophoresis (2DE) and sequencing dual matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-TOF), differential protein expression profiles that may reflect distinctive fibrotic processes in the two disorders [12]. They were also interested in defining if BAL-derived fibroblasts were different from those derived from bronchial biopsy in systemic sclerosis (SSc) patients with alveolitis: their successful in vitro expansion of fibroblasts from 4 out of 10 cases, evidenced that BAL-derived fibroblasts showed a more elongated and mobile phenotype with an increased proteoglycan production [13].
The origin of lung fibroblasts/myofibroblasts is however still a matter of debate deserving further studies. More or less 15 years ago, Lama et al. [14] demonstrated the presence of multipotent tissue-resident mesenchymal stem cells (MSCs), defined as fibroblast-like cells, in BAL samples obtained from 76 human lung transplant recipients. Here the authors, by plating BAL cells in liquid culture medium, derived colony forming unit fibroblasts (CFU-F) from 62% of the BAL fluids obtained. These CFU-F, expanded in culture by serial passaging, were phenotypically characterized, revealing the expression of cell-surface proteins commonly found on MSCs (i.e. CD73, CD90, and CD105). In addition, single colonies, first isolated and expanded in culture, were assessed for their capability of differentiating into the three mesenchymal cell lineages (osteocytes, chondrocytes, and adipocytes): these results suggested that resident MSCs, presented within the lung, appeared dormant in the uninjured tissue, but became activated and mobilized during the state of lung regeneration and repair. Lehtonen et al. [15] further evaluated if it could be possible to enrich and culture stromal cells also from a residual volume of BAL samples obtained for diagnostic purposes. In the presented study, fifteen milliliters of BAL fluids were collected from 98 patients with various types of lung diseases [i.e. 14 IPF, 10 non-specific interstitial pneumonia (NISP), 9 connective tissue disease-associated-ILD (CTD-ILD), and others] and the cell, successfully cultured from 62% of samples, could be further extensively characterized by immunocytochemistry, transmission electron microscopy (TEM), flow cytometry, and differentiation tests. By immunohistochemical staining the authors here observed that the cultured cells were mainly fibroblasts but included also some myofibroblasts, in accordance with TEM ultrastructure studies. In a previous study, they further showed that IPF-derived-fibroblasts were more invasive than those derived from other fibrotic diseases, each displaying its own distinctive ultrastructural characteristics [16].
We recently also tried to culture fibroblasts from residual BAL volume (10 mL) used for diagnostic purposes of patients with different types of ILD [IPF, fibrotic HP (fHP), NISP, and CTD-ILD]: fibroblasts were successfully expanded from 12 patients, but in particular we could culture fibroblasts derived from an fHP patient for a very long time (9 months, until passage 36). We therefore considered and described these cells such as an established cell line (SCI13D) [17]. It is noteworthy and unusual that uninfected and primary cells can be passaged for a long time: in our experience fibroblasts grew until passage 12–14. To our knowledge, SCI13D is the first fibroblastic cell line derived from an fHP patient and may provide a good tool to model this disease in pre-clinical studies. We indeed demonstrated, by cytofluorographic analysis and quantitative RT-PCR, that, from a phenotypical point of view, SCI13D displayed main features of myofibroblasts, showing levels of α-SMA, type I collagen, and fibronectin higher than the fetal lung fibroblastic MRC5 control cell line: interestingly type I collagen, α-SMA, and fibronectin expression appeared quite stable along the many passages of the in vitro culture. Moreover, a consistent number of SCI13D fibroblasts showed β-galactosidase (B-GAL) activity, and the mRNA levels of the two cell-cycle inhibitors P16 and P21, typical markers of senescence, were also higher when compared with the MRC5: an association between cellular senescence and progressive fibrosis in fHP could be therefore envisaged. We found also of interest the demonstration that SCI13D showed an aneuploid karyotype with a trisomy of chromosome 10 (46XY + 10). More importantly this trisomy was already observable at the early passages of in vitro expansion as well as in fibroblasts present within the patient biopsy. Since chromosome 10 harbors the fibroblast growth factor receptors 2 (FGFR2) gene we speculated that, presumably, higher levels of this receptor, due to the presence of an extra chromosome 10, may lead to the overgrowth of a proliferating clone. However further investigations are necessary to clarify this possible association.
A detailed and standardized new protocol to obtain fibroblasts from BAL fluids of ILD patients has been further recently described by Bergantini et al. [18]. By the use of this protocol here the authors reported on the establishment of a new cultured cell line (B-LSMD7) isolated from the BAL of a patient with IPF. These cells show co-expression of CD105, type I collagen, or fibronectin at similar levels observed in commercially available human lung fibroblasts (HLF) from an IPF patient. Notably, B-LSMD7 showed high reactive oxygen species (ROS) levels that, as suggested, could promote extracellular matrix (ECM) deposition in lung disease.
Lehtonen et al. [19] have further interestingly reviewed the multiple culture protocols employed in the past 3 decades to derive fibroblasts from BAL fluids, as a tool to better investigate fibrosis in ILD patients; they evidenced that only a few research groups have aimed at this object. The advantage of using BAL- versus tissue-derived fibroblasts could be still fully exploited.
Many studies, addressing the mechanisms leading to pulmonary fibrosis, took advantage of immortalized cell lines [i.e. A549 (lung adenocarcinoma alveolar basal cell line), Calu-3 (sub-mucosal adenocarcinoma cell line), and MRC5 (fetal lung fibroblast cell line)]. However, since cells immortalization may induce phenotypical/functional changes, the possibility of using patient-derived primary cells represents a better opportunity to study the cells in their physiological conditions. Indeed, analyzing primary cells, directly derived from patients, could be of help to identify particular cell subsets associated with disease pathogenesis or progression.
As a whole all the above-mentioned observations highlight that a wider use of BAL, a minimally invasive and routine procedure, will provide, in the future, the chance of deriving discrete quantities of primary fibroblasts from a greater number of IPF or other fibrotic lung disease patients, with the perspective of improving our knowledge and devising novel pre-clinical models.
The recent advent of cutting-edge technologies (i.e. gene expression profiling and single-cell RNA-sequencing) may further provide a high-resolution picture of changes in molecular phenotype and cellular heterogeneity during the disease-course and in different fibrotic lung diseases. MSCs or fibroblasts-like cells, among all the cell types that are part of the lung parenchyma (i.e. alveolar, endothelial cells), are the ones most widely studied or used in in vitro cultures, due to easier culture techniques. In addition to the demonstration of the presence of tissue-resident MSCs in BAL from IPF patients [14], Chanda et al. [20, 21] also identified, through transcriptomic analysis, a specific pattern in gene expression profile, distinguishing fibroblasts from progressive vs. stable IPF patients. In these studies the evident fibroblast growth factor 10 (FGF10) down-regulation, observed in fibroblasts from progressive patients, apparently linked to tumor growth factor (TGFβ) and sonic hedgehog (SHH) signaling hyper-activation, suggested a critical role of FGF10 in lung homeostasis maintenance. Recent studies have further indicated that “lung fibroblasts” and “lung mesenchymal cells” are quite diverse, with a variety of different functions and gene expression signatures that may vary by location [22–24]. The role of endogenous resident lung MSCs is still unclear: MSCs, physiologically involved in tissue repair mechanisms, appear instead exhausted and senescent in IPF patients, thus indicating a deficit in their regenerative potential [25]. Xia et al. [26] interestingly demonstrated that, in contrast to progenitors isolated from non-fibrotic lungs, IPF mesenchymal progenitor cells produce daughter cells manifesting the full spectrum of IPF hallmarks (high α-SMA, type I collagen, and phospho-Akt), the ability to form fibrotic lesions in zebrafish embryos and mouse lungs, and a transcriptional profile reflecting these properties. Here the authors proposed that the fibrotic ECM, typically found in the lung of IPF patients, could have a key role in stably skewing the phenotype of mesenchymal progenitors and their progeny. The importance of an altered ECM in creating a vicious feedback loop propagating the disease has been indeed confirmed by different researchers more recently [27, 28].
Although many observations have demonstrated that MSCs and fibroblasts share many common features, it is yet to be established if these cell types are subgroups of the same progenitors, if they derive from differentiation processes or if they result from aging and senescence. Indeed, whether MSCs and/or fibroblasts represent the two sides of the same coin is still a matter of debate, as reported by Soundararajan et al. [29]. A single-cell transcriptome analysis-based approach has been recently undertaken to tackle this relevant open question: the authors here examined human umbilical cord, foreskin, bone marrow MSCs (BMSCs), and adipose MSCs and demonstrated that, among 15 different cell subsets obtained through single-cell transcriptome sequencing data, fibroblast phenotype markers were shared among all subsets, whereas only 12 out of 15 displayed the MSCs-ones [30]. This finding suggested a possible hierarchical origin among these cell types and pointed out their intrinsic plasticity. Further technologically-advanced studies will probably provide more insights into the subtle differences between these two cell types.
The development of in vitro lung models, in association with transcriptomic technologies, would be therefore of help in increasing the understanding of lung cells/ECM interacting processes leading to fibrosis initiation and progression. Although 3D lung models have recently emerged, two-dimensional models still maintain their role as a primary platform of in vitro research due to easy handling and enabling to study cell response in a controlled culture setting. Sieber et al. [31], as an example, have recently investigated the dynamic molecular interactions between commercially available normal human bronchial epithelial (NHBE) cells and normal HLF (NHLF) cells, by coupling a 2D in vitro co-culture model and single-cell RNA-sequencing. By the development and thorough characterization of this in vitro lung fibrosis model, the authors determined that only direct cell-to-cell contact allowed the acquisition of phenotypic changes and genomic hallmark signatures found in cells from IPF patient lungs: NF-kappaB appeared as the critical component driving these changes. As a whole, results from this study suggested that the extensive breakdown of lung architecture in areas of active disease permits uncontrolled epithelial/mesenchymal cell contacts, leading to disease progression. Two-dimensional culture systems have been further extensively used to assay in vitro the activity of different drugs. Codullo et al. [32], aiming to test the activity of imatinib-loaded gold nanoparticles, used lung fibroblasts and macrophages derived from BAL of SSc (SSc-ILD) patients thus preliminarily demonstrating their efficacy as a potential novel therapeutic approach. Pandolfi et al. [33] further exploited the use of everolimus, encapsulated in liposomes coated with hyaluronic acid, in 2D cultures of lung fibroblasts derived from BAL of patients with CTD-ILD.
Obviously, the primary limitation of 2D cell culture is their inability to mimic the 3D nature of lung tissue: the high stiffness of the culture vessel plate might influence cell behavior, thus indicating the importance of culturing the cells in systems that take into account spatial-temporal interactions between multiple cells and ECM. Trying to bridge the gap between “in dish” and “in tissue” cell behavior, many efforts have been therefore recently applied to develop various models capable of recapitulating a more complex and physiological lung structure. In vitro 3D cell-culture models consist of multicellular compositions (spheroids and organoids), ECM supports or scaffolds (i.e. hydrogels, polymers, and decellularized lungs), bioengineered micro-devices (microfluidic devices or lung-on-a-chip), or ex vivo lung slices [34–38].
Significant advances have been made in the last years in lung organoid research. However, these studies primarily involve models of airway cell types since culturing alveolar cell types [alveolar type I cells (AEC1s) and alveolar type II cells (AEC2s)] still represents a challenge, due to the difficulty of in vitro long-term culture and maintenance of these cells [39]. Sachs et al. [40] described a method to establish long-term-expanding human airway organoids from broncho-alveolar resections or lavage material of patients with cystic fibrosis (CF): the authors here aimed to create a model that could be adapted to a variety of pulmonary diseases. Moreover, Sette et al. [41], from nasal brushing samples of CF patients, generated in vitro large amounts of airway epithelial stem cells to develop 3D airway organoids: the feasibility of these models can be envisaged as a platform to assay personalized drug-screening approaches providing each patient with his/her most appropriate therapy. Hypothetically this strategy could be also applied to IPF and related disorders trying to implement the palette of active molecules with new ones more specific than those actually in use, namely nintedanib and pirfenidone (PIRF). In the light of organoid development for IPF research, the recent work by Kathiriya et al. [42], highlighted how the co-culture of AEC2s and normal mesenchymal lung fibroblasts drove the trans-differentiation of AEC2s toward a metaplastic KRT5+ basal cell phenotype, contemporarily inducing a fibrotic IPF-like phenotype of mesenchymal cells, thus evidencing a bi-directional cross-talk. Jaeger et al. [43] further demonstrated that airway basal cells (ABC) obtained by bronchial brushing of IPF patients, showed a de-differentiated phenotype promoting fibrosis in 3D organoid- as well as in in vivo models. Organoid-based approaches have also provided suggestions on the mechanisms regulating communication and self-organization of alveolar/MSCs cells: in degenerative disorders, including fibrotic diseases, a critical role played by aged stem cells has been proposed [44].
As a whole, these studies suggested that the availability of mesenchymal/fibroblast cells directly derived from patients through scarcely invasive procedures (i.e. BAL or nasal brushing) and further expanded in vitro may provide a useful tool to clarify relationships among different cell types involved in pulmonary fibrosis.
Other experimental settings, such as decellularized ECM or basement membrane support (hydrogel, matrigel, and synthetic polymers), could further allow studying of cells’ behaviour as a consequence of the matrix/scaffold stiffness or additional mechanical properties, thus re-creating typical alterations of the pathogenetic onset [45–47].
Moreover, microfluidic and lung-on-a-chip devices, miniaturized models which reconstitute tissue-tissue and tissue-air lung interfaces, will allow for deeper investigation of the lung physiology providing an alternative to animal and clinical studies, at lower costs and higher reproducibility [48, 49].
In spite of the relevant achievements so far discussed, several open questions still remain to be thoroughly answered:
(a) In light of the extensive heterogeneity of fibroblasts within the lung, both a deeper comprehension of the origin and, possibly, of the in vivo location of BAL-derived fibroblasts may add novel insights about the role of these cells in ILD pathogenesis; in this respect, it is also possible that these cells may originate from diverse sources, depending on the different diseases among ILD.
(b) The double-edged role that MSCs may exert in fibrotic diseases adds to the complexity of their role in these tissues. Endogenous MSCs usually play a beneficial role in lung repair, while in fibrotic diseases they show signs of cellular senescence possibly leading to the loss of regenerative potential. It is however interesting to note that various pre-clinical studies have demonstrated that the infusion of allogeneic MSCs, or either their secretome or released extracellular vesicles (EVs), may regenerate damaged tissues relieving fibrotic diseases [25, 50]. It appears therefore crucial to fully understand the difference between myofibroblast-forming MSCs and anti-fibrotic MSCs, how local tissue insult might cause their pathological derangement and whether external MSCs might really represent a feasible therapeutic option.
(c) Since IPF is actually recognized as a pre-cancerous condition and pulmonary fibrosis appears relevant in cancer susceptibility, the exploitation of similarities and differences among ILD-fibrotic fibroblasts and cancer-associated fibroblasts should be better studied.
(d) Lastly, three-dimensional models need to bring together all the relevant physiological requirements of the complex lung structure before they can fully mirror the tissue organization and represent a breakthrough strategy in terms of disease-modeling, drug-screening, and personalized medicine for a proper technological translation from the bench-side to the patient’s bed-side.
In the light of the different above-cited approaches, and in spite of the yet remaining open questions, we feel confident that the utilization of patients-derived primary cells will prove to be a suitable common tool to exploit modelling and progression of IPF and related diseases; we have therefore highlighted the current and future procedures for the use of these cells in basic research and applied medicine: indeed the actual large body of literature on this topic testifies the deep interest of the scientific community (Figure 1).
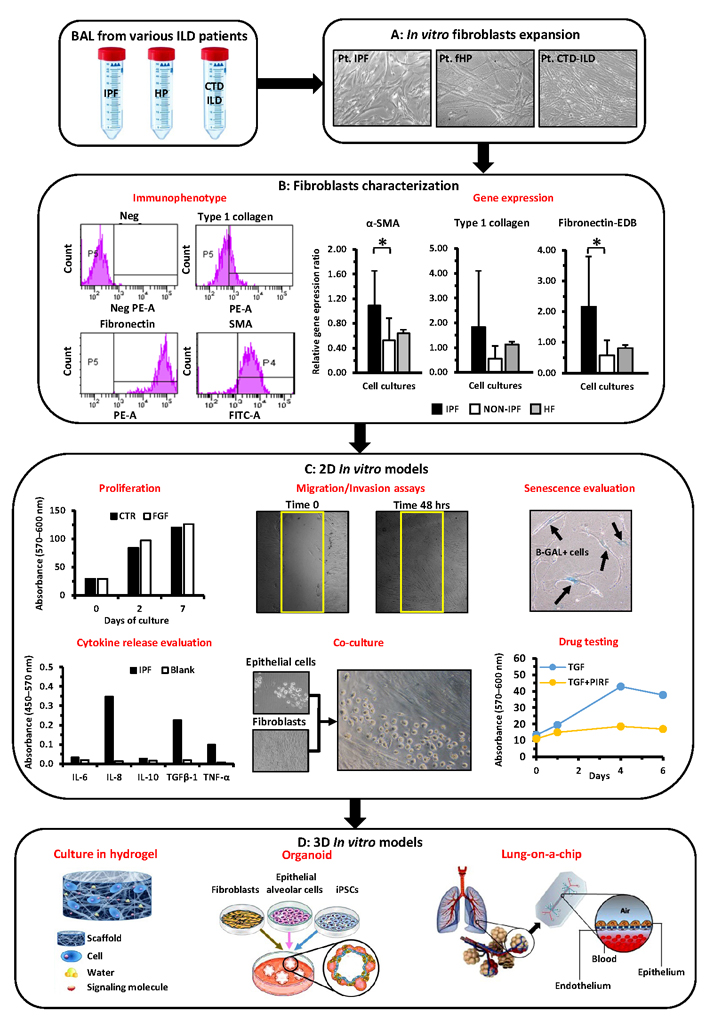
Representation of the different passages and assays potentially performed to characterize BAL-derived fibroblasts and to exploit their applications in 2D/3D pre-clinical models. (A) Mesenchymal/fibroblast cells can be derived from BAL of different ILD patients (IPF, fHP, and CTD-ILD) and further expanded in vitro; (B) these cells can be further characterized through cytofluorimetric analysis and quantitative RT-PCR to determine the expression of pro-fibrotic markers (i.e. α-SMA, type I collagen, and fibronectin-EDB); (C) these cells can be further assayed for proliferation with/without specific drugs (i.e. PIRF) or cytokines (FGF, TGFβ), for their ability to migrate, to express markers of senescence, to release specific cytokine(s), and to grow in co-culture with other cell types (i.e. epithelial cells); (D) the same cells will be of help to further devise 3D in vitro models, (i) cultures in hydrogels with tunable matrix rigidities, (ii) establishment of organoids that better recapitulate lung physiology, (iii) more complex ex vivo systems modeling pulmonary fibrosis such as “lung-on-a-chip”. Pt: plated cells; Neg: negative; PE-A: phycoerithryn; FITC-A: fluoresceine; P4 and P5: the set delimiters used to identify positive cells; fibronectin-EDB: fibronectin extra domain B; HF: human fibroblasts; CTR: controls; hrs: hours; Blank: background absorbance value; iPSC: inducible pluripotent stem cells; *: P < 0.05. Graphics or live images of cells here reported are referred to our either previously performed or ongoing studies and are intended only as examples of the different techniques that can be employed
AEC2s: alveolar type II cells
BAL: broncho-alveolar lavage
CTD-ILD: connective tissue disease-associated interstitial lung disease
ECM: extracellular matrix
fHP: fibrotic hypersensitivity pneumonitis
ILD: interstitial lung disease
IPF: idiopathic pulmonary fibrosis
MSCs: mesenchymal stem cells
α-SMA: alpha-smooth muscle actin
PG: Writing—original draft, Writing—review & editing. MG and EB: Writing—review & editing. DDT: Conceptualization, Writing—original draft, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 5089
Download: 37
Times Cited: 0
Mauro G. Silva ... Walter Manucha
Lena B. Kim, Anna N. Putyatina
