 Review
Review

Affiliation:
1Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy
ORCID: https://orcid.org/0000-0002-5751-0277
Affiliation:
2Corporate Preclinical R&D, Chiesi Farmaceutici S.p.A., 43122 Parma, Italy
ORCID: https://orcid.org/0000-0003-1655-609X
Affiliation:
1Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy
ORCID: https://orcid.org/0000-0003-2719-7272
Affiliation:
2Corporate Preclinical R&D, Chiesi Farmaceutici S.p.A., 43122 Parma, Italy
ORCID: https://orcid.org/0009-0005-4384-6545
Affiliation:
1Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy
ORCID: https://orcid.org/0000-0003-0552-9205
Affiliation:
2Corporate Preclinical R&D, Chiesi Farmaceutici S.p.A., 43122 Parma, Italy
Email: s.pontis@chiesi.com
ORCID: https://orcid.org/0000-0002-9873-4998
Explor Med. 2023;4:1116–1134 DOI: https://doi.org/10.37349/emed.2023.00199
Received: July 14, 2023 Accepted: September 18, 2023 Published: December 29, 2023
Academic Editor: Bernhard Ryffel, University of Orleans, France
The article belongs to the special issue Lung Fibrosis—Models and Mechanisms
Chronic obstructive pulmonary disease (COPD) is an inflammatory lung pathology characterized by persistent airflow limitation and is the third leading cause of death globally. COPD pathophysiology includes both environmental and host risk factors and the presence of comorbidities contributes to its harmful outcome. Cardiovascular disease (CVD) is closely related to COPD and their coexistence is associated with worse outcomes than either condition alone. COPD impairs the cardiovascular system favoring mostly endothelial dysfunction that is a significant COPD prognostic factor at different stages of the disease. The mechanisms promoting endothelial dysfunction in the systemic and/or pulmonary circulation of COPD patients are different and include systemic inflammation, alteration of adhesion and pro-inflammatory molecules, oxidative stress, cellular senescence, and apoptosis. Nevertheless, the role of endothelium in the onset and progression of COPD and CVD is not yet fully understood. Hence, the purpose of this narrative review is to analyze the literature and provide evidence supporting the importance of endothelial dysfunction in COPD.
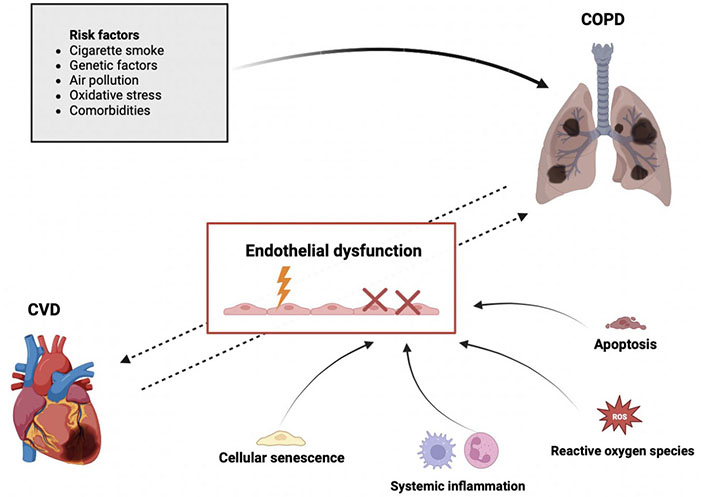
The role of endothelium and its potential relationship in COPD and CVD. The dotted arrows in the figure indicate that the factors linking COPD and CVD are not yet fully understood and are likely to be interconnected through endothelial dysfunction. While the solid arrows represent the well-known risk factors for COPD and the mechanisms associated with endothelial dysfunction. Created with BioRender.com
Chronic obstructive pulmonary disease (COPD) is an incurable inflammatory pathology [1] characterized by persistent limitation of the airflow and lung tissue destruction [2]. Approved pharmacological treatments (e.g., long-acting muscarinic antagonists, long-acting beta-agonists, corticosteroids) display the limited ability to halt disease progression mainly because they do not significantly tackle the underlying inflammation. Considerable efforts have been made to develop efficient anti-inflammatory compounds however drugs that can reverse the patient’s pathological state are still lacking [3]. For all these reasons, COPD remains a major worldwide health problem and global burden of disease studies have estimated COPD as the third leading cause of death worldwide [4].
The pathophysiology of COPD is complex and it includes both environmental and host risk factors. Long-term exposure to irritant gases or particles and more frequently, to cigarette smoke (CS) are among the first [5]. Whereas host factors include polymorphisms of genes like those encoding for metalloproteinase matrix 12 and glutathione S-transferase both associated with decreased lung function [6, 7] or increased risk of COPD onset [8]. Moreover, a small percentage of individuals (about 1–5%) develop COPD secondary to a genetic deficiency in alpha-1 antitrypsin (AAT). Beyond genetics, lung cell hyperresponsiveness to stimuli and poor lung growth during childhood play crucial roles in the development and severity of COPD [9, 10].
Patients experience cough, mucus hypersecretion, wheezing, chest tightness, and breathing difficulty, especially during physical activities [11]. In addition to the daily symptom burden, COPD may be punctuated by periods of acute worsening of respiratory symptoms requiring additional therapeutic interventions [12] and resulting in a significant increase in both morbidity and mortality [13]. These events are called exacerbations and are mainly triggered by viral and bacterial respiratory infections. This complex and faceted clinical scenario is classified according to Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines that rank the severity of airflow limitation according to specific spirometric cut-offs as it follows: GOLD 1 (mild), forced expiratory volume in 1 s (FEV1) ≥ 80% predicted; GOLD 2 (moderate), 50% ≤ FEV1 < 80% predicted; GOLD 3 (severe), 30% ≤ FEV1 < 50% predicted; GOLD 4 (very severe), FEV1 < 30% predicted [14]. Moreover, a post-bronchodilator FEV1/forced vital capacity (FVC) < 0.70 confirms the presence of persistent obstruction. Spirometry is the most reproducible and objective measurement of airflow limitation, representing the fundamental test used to diagnose COPD nowadays.
Even if COPD is heterogenous in its manifestations, it usually includes two major pathologies: small airway disease with peribronchiolar fibrosis and emphysema with alveolar wall destruction [15]. In chronic bronchitis, the principal alteration is airflow obstruction caused mainly by airways remodeling. On one side, bronchial tube become inflamed and narrowed while on the other, lung produces more mucus because of submucosal glands hypertrophy and hyperplasia that can further affect the narrowed tubes [16]. In emphysema, the pre-eminent physio-pathological alteration is the loss of elastic return of the lung by parenchymal destruction. These conditions are not mutually exclusive but can occur together [17].
A relevant aspect of the mechanism of both airway disease and emphysema is inflammation. Indeed, COPD is mainly an inflammatory disease and different inflammatory pathways can be activated [18]. The inflammatory response involves both innate and adaptive immunity and is most pronounced in the bronchial walls with neutrophilic infiltration, the commonest inflammatory phenotype of COPD. Exposure to CS, pollutants, and oxidants [19] causes the release of chemotactic factors such as interleukin (IL)-4, IL-5, IL-6, IL-8, IL-13, interferon-γ, monokine, and leukotriene B4 by the airway epithelium and macrophages, which are the key inflammatory cells coating the airway mucosa [20]. Excessive release of IL causes accumulation in the airways of neutrophils, CD8 T cells, and CD4 helper T cells [21]. This causes an extended immune response that causes elastin degradation mainly in alveoli, the consequence of which is a lack of elastic pulmonary recoil and emphysema. In addition, the accumulation and the prolonged activation of these factors result in hypertropia and hyperplasia of goblet cells, with an increased production of mucus. Along with chemotactic factors, macrophages, and epithelial cells secrete transforming growth factor-β (TGF-β), which causes fibroblast activation [22]. Fibroblasts produce a connective tissue growth factor, a chemokine that promotes the senescence of lung epithelial cells, decreasing cell rebuilding and leading to emphysema [23].
It is known that the inflammatory phenotype most associated with COPD is the neutrophil phenotype, but the most recent searches show a 10–40% increase of eosinophilic inflammation in the sputum and blood of COPD patients [24]. Eosinophilic inflammation is related to a type 2 inflammatory profile that leads to the activation of IL-5, the main inflammatory cytokine responsible for the maturation, survival, and activation of eosinophils. Although increased IL-5 has been detected in patients’ sputum, anti-IL-5 therapies are not effective in preventing exacerbations, highlighting that the mechanism by which eosinophils contribute to COPD is not yet to be understood [25]. However, eosinophilic inflammation and an increase in the eosinophilic count can be found in lung tissue, sputum, and blood both during stable disease and during exacerbations, so for this reason, eosinophils could be considered as biomarker of disease [26].
Another level of complexity that describes the heterogeneity of the COPD patient population is given by comorbidities which influences the prognosis of COPD. Comorbidities not primarily involving the vascular system such as diabetes, metabolic syndrome, and lung cancer can be distinguished from those including cardiovascular disease (CVD) [27]. CVD is often linked to COPD and their coexistence is associated with worse outcomes than either condition alone [28]. COPD impairs the cardiovascular system, especially the endothelium, favoring the development of atherosclerotic plaques. As a result, CVD mortality accounts for 25% of all causes of death in COPD patients [29]. Not by chance, COPD and CVD share common risk factors, pathophysiological processes, clinical signs, and symptoms [30]. Among them, chronic low-grade systemic inflammation emerged as a possible link between the two disorders [31].
Based on these contentions, several questions remain open: Are COPD and CVD two different pathologies? How is one reflected in the other? And above all, is COPD a risk factor for CVD or the opposite?
In the attempt to answer these questions, is relevant to highlight the importance of endothelium and its dysfunctions in both COPD and CVD as shown in Figure 1. Several studies suggest that endothelial dysfunction contributes to the progression of CVD and at the same time, it is linked to COPD severity [32]. In COPD, endothelial dysfunction occurs in the early stages of the disease and worsens with pulmonary obstruction severity and during acute exacerbations [33]. In this scenario, it becomes very attractive to study and explore the endothelium, through which inflammatory cells must transmigrate [transendothelial migration (TEM)] to reach the lung parenchyma and cause damage ultimately leading to COPD onset and progression. Understanding the mechanisms underlying the trafficking of leukocytes, neutrophils, cytokine, chemokine, and inflammatory molecules across the endothelium to the lung parenchyma, new treatments may be developed and the risk of COPD onset may be reduced.
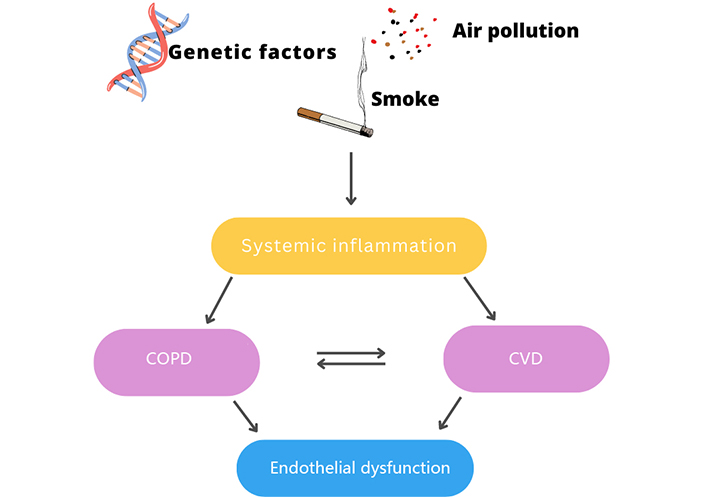
Exposure to harmful substances leads to the development of inflammatory mechanisms causing COPD and CVD. The two diseases are closely related and can lead to endothelial dysfunction. Figure was created with Canva
Vascular endothelial cells (ECs) cover the internal surface of the vessels forming a continuous monolayer [34]. ECs rest on a basal membrane consisting of collagen and glycoproteins, while on the luminal part, there are glycocalyces, a structure of proteoglycans and glycoproteins that have different functions [35]. The basal membrane is also constituted by a non-continuous layer of pericytes that are mediators of different microvascular processes, such as angiogenesis and ECs proliferation [36]. Endothelium is involved in multiple and different mechanisms such as vascular permeability, angiogenesis, leukocyte trafficking, inflammation, and coagulation [37, 38]. In the respiratory system, it has another important role: it detoxifies xenobiotics incoming via the airways, CS, and air pollution [39]. The participation of pulmonary capillary ECs in the blood-air barrier makes the integrity of pulmonary endothelium critical for the maintenance of the lung physiological performance [40].
The first description of vascular injury, including capillary loss, in emphysematous lungs of COPD patients comes from Liebow [41]. Thanks to histological studies, he concluded that, in centrilobular emphysema, a lower inflow of blood in the pre-capillaries contributes to alveolar septa damage [41]. What Liebow [41] discovered for years remained obscure, but later, significant advances were made in the study of endothelial dysfunction in lung diseases. Several years later, Dinh-Xuan et al. [42] described that endothelial dysfunction occurs in the pulmonary arteries of COPD patients with end-stage disease as evaluated by in vitro experiments measuring relaxation of pulmonary artery rings using increasing doses of exogenous acetylcholine and adenosine diphosphate (ADP). They suggested that endothelial dysfunction in end stage COPD might contribute to the development of pulmonary arterial hypertension in COPD. However, a subsequent study conducted by Peinado et al. [43] reported that endothelial dysfunction appears not only in the end stage but also as an early feature of COPD. This relevant clinical study was conducted on 41 patients, divided into non-smokers, smokers with normal lung function, and patients with COPD to check whether patients with mild COPD showed endothelial dysfunction of the pulmonary artery. Endothelial-dependent relaxation, mediated by nitric oxide (NO), was evaluated in vitro in pulmonary artery rings exposed to different concentrations of acetylcholine and ADP. Authors suggested that the intima of the small pulmonary arteries was thickened and level of relaxation reduced in both patients with mild COPD and smokers with regular lung activity suggesting that smoking may cause morpho-functional changes in COPD [43]. They also detected endothelial apoptosis and endothelial dysfunction in the lungs of COPD patients thanks to computed tomography that revealed reduced and reshaped peripheral vascularization [44].
The mechanisms that promote endothelial dysfunction in the systemic and/or pulmonary circulation of COPD patients are different and include systemic inflammation, alteration of adhesion and pro-inflammatory molecules, oxidative stress, cellular senescence, and apoptosis as shown in Figure 2.
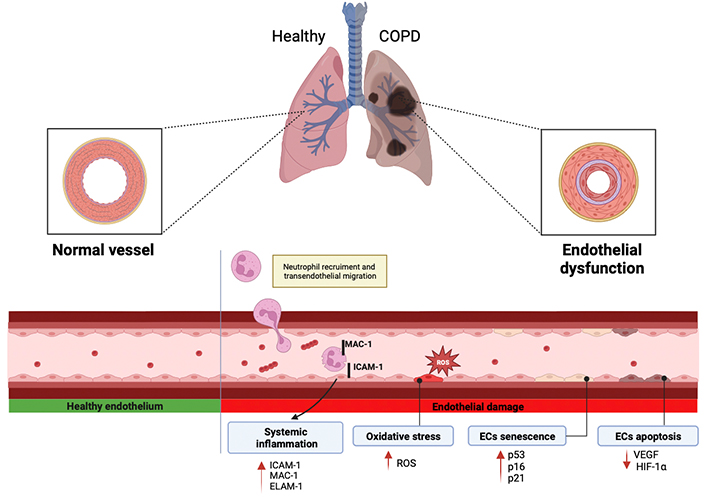
Different mechanisms by which endothelial dysfunction is promoted in COPD, including systemic inflammation, oxidative stress, ECs senescence, and apoptosis. ROS: reactive oxygen species; ICAM-1: intracellular adhesion molecule-1; MAC-1: macrophage-1 antigen; ELAM-1: endothelial-leukocyte adhesion molecule; HIF-1α: hypoxia-inducible factor 1α; VEGF: vascular endothelial growth factors. Red arrows indicate an increase or decrease in the corresponding factors involved in the mechanisms. Created with BioRender.com
Systemic inflammation occurring in COPD patients may contribute to the development of both pulmonary and systemic endothelial dysfunction. Activation and alteration of adhesion molecules in ECs promote the inflammatory reactions occurring in pulmonary vessels. The surface activation of cellular adhesion molecules represents a common endothelial response to a variety of toxic and inflammatory stimuli and their shedding into the systemic circulation provides evidence of endothelial dysfunction.
As described above, the endothelium produces and activates molecules and inflammatory factors, which exit the bloodstream and reach the target tissue through the endothelium. Trafficking of leukocytes out of the circulation into tissues is a highly regulated, complex, multistep process. Physiologically, inflammation is initiated by a relatively loose adhesion of leukocytes to ECs followed by a firmer adhesion and transmigration of leukocytes across pulmonary ECs [45]. Specifically, neutrophils are provided by two different mechanisms: The first one is called paracellular transmigration and consists of a connection with the apical domain of ECs via cell surface proteins (e.g., pseudopods) over one or between two ECs. The second mechanism is called TEM by which neutrophils can extravasate at junction regions between two ECs. This process consists of multiple steps of interaction between neutrophils and ECs and has a relevant role in the inflammatory response [46]. TEM is a mechanism by which endothelium may play a role in COPD and therefore may be important in the development of inflammation and in the pathogenesis of COPD. Patients with COPD have high levels of TEM and during this process, the MAC-1, an important human cell surface receptor found on B and T lymphocytes, is upregulated [47]. MAC-1 binds to ICAM-1 on the surface of ECs resulting in increased ICAM-1. This mechanism could be clinically relevant, as when ICAM-1 serum levels are high, lung function is reduced. Conversely, when ICAM-1 is turned off, pulmonary inflammation is reduced, supporting the hypothesis that ICAM-1 upregulation could relate to the increased inflammation seen in COPD patients [48]. Furthermore, another study by Johnston et al. [49] suggested that the upregulation of ICAM-1 could be reduced by an anti-ICAM-1 molecule, administered at the initial stage of the disease, thus preventing COPD exacerbations and its adverse outcomes [49, 50]. Another relevant adhesion molecule involved in TEM is ELAM-1. This has also been found upregulated in COPD patients’ serum and its levels are particularly high in patients with chronic bronchitis. Altogether, this evidence further supports the involvement of adhesion molecules in lung inflammation and COPD pathogenesis [51].
In the last years, scientists improved the research about a variety of COPD inflammatory biomarkers [52]. This research field is interesting for the study of disease progression, since inflammatory biomarkers are diagnostic and prognostic factors of COPD patients with acute exacerbation of COPD (AECOPD). For example, are studied as markers for degradation and repair processes factors like neutrophil elastase (NE) or matrix metalloproteases (MMP), which are detectable in bronchoalveolar lavage fluid (BALF) or sputum [53]. Among the most significative systemic markers of COPD there are fibrinogen and acute C-reactive protein (CRP), which is one of the predictive factors. This project, called “Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE)”, is a three-year longitudinal study whose main objective was to identify factors that predict disease progression in individuals with different endotype of COPD [54]. Literature studies show that serum CRP is associated with mortality, morbidity, number of exacerbations and is inversely related to lung function. Increased serum concentrations of IL-6, tumor necrosis factor α (TNFα), and monocyte chemoattractant protein-1 (MCP-1) are also related to systemic inflammation in COPD patients [55].
As described in the sections above, the inflammatory endotype in COPD can be mediated by neutrophils [T helper 1 (Th1)] and by eosinophils (Th2) [56]. Eosinophil recruitment to inflammation sites is mediated by eotaxins [eotaxin-1 (CCL11), eotaxin-2 (CCL24), eotaxin-3 (CCL26)]. These chemokines are considered as candidate biomarkers to monitor COPD development and progression since they are upregulated in COPD patients [57]. The main source of eotaxins are epithelial cells, motile cells present in BALF (macrophages, monocytes, basophils, eosinophils, lymphocytes), and structural cells like fibroblasts or smooth muscle cells [58]. Eotaxins can also be released by ECs but there are few studies supporting this statement. One of these showed that CCL11 can be released by both epithelial and ECs to favor angiogenesis. Unfortunately, none of these studies describe the release of eotaxins by ECs in the context of COPD [59]. Similar considerations can be made for neutrophils. Indeed, there is evidence about neutrophil chemoattractant cytokines [C-X-C motif chemokine ligand 1 (CXCL1) and CXCL5)] release by ECs but not in the context of COPD even if they are dysregulated in COPD patients [60, 61]. To study Th2 response is particularly appealing since it defines a specific COPD patient population as shown by the ECLIPSE study reporting that eosinophilic inflammation was present in 37.4% of patients [62]. In these patients, eosinophil count might be relevant to predict their disease progression and to choose the most appropriate pharmacological therapy. Indeed, peripheral eosinophil level from a complete blood count has been evaluated as a surrogate marker for corticosteroid responsiveness and eosinophilic bronchitis [63]. A higher blood eosinophil count appears to indicate a subgroup of COPD patients in which the use of inhaled corticosteroids (ICS) results in reduced exacerbation frequency and when ICS [64] is withdrawn significant increase in peripheral eosinophil count has been associated with a higher exacerbation rate [65]. To deepen these issues could help to improve the discovery of therapeutic targets and to identify patients who are likely to respond to classic or novel treatments.
Neurogenic inflammation is confined to the central and peripheral nervous system and is caused by nerve activation inducing the release of neuropeptides and rapid extravasation. During airway inflammation, neuropeptides are secreted from the airways in response to a multitude of inflammatory mediators, such as exogenous irritants like CS or gas. According to this, neurogenic inflammation may participate in the development and progression of chronic inflammatory airway diseases such as allergic asthma or COPD. Among the large variety of neuropeptides stored in and secreted from airway sensory nerves the most studied are the tachykinins like substance P (SP), neurokinin A (NKA), and the calcitonin gene-related peptide (CGRP) [66]. Tachykinins can bind to three different receptors, called neurokinin receptors (NK). Specifically, SP acts via NK1, NKA acts via NK2 and neurokinin B acts via NK3. The first one is predominantly localized to airway epithelium, submucosal glands, and vessels, the second one is mainly expressed by airway smooth muscle, while NK3 is expressed by airway parasympathetic ganglia. Tachykinins exert various actions on airway functions. They have a direct effect on coronary arteries to decrease or increase tone. Stimulation of NK1 receptors on the endothelium causes vasodilatation mediated by NO. They can vasodilate or vasoconstrict the state of the endothelium depending on the situation; this state appears to be of considerable importance [67]. Tachykinins also enhance eosinophils chemotaxis through the activation of alveolar macrophages and monocytes that release inflammatory cytokines such as IL-6. CGRP is a 37 amino acid peptide that is abundantly expressed in the respiratory tract of several species and is co-stored and co-localized with tachykinins in sensory nerve fibres innervating both upper and lower airways. CGRP is a powerful vasodilator, with a long-lasting effect on bronchial vessels and this is evident in vitro and in vivo. Mapping of the receptors showed that CGRP receptors are predominantly situated in the bronchial vessels, in the epithelium of the human airways, and in the smooth muscles on which it has effects on tone by acting indirectly through the release of other constrictors, such as endothelin. Like tachykinins, CGRP is chemotactic for eosinophils [68]. The molecular mechanisms underlying the different events of neurogenic inflammation in the airways of mammals have been identified thanks to animal models. Based on data obtained from these studies, the existence of a neurogenic inflammatory component in human airway diseases such as asthma and COPD seems very likely. Nevertheless, further studies are required to assess the clinical relevance of this inflammatory process in the onset and progression of COPD [69].
Oxidative stress is another crucial mechanism underlying COPD development and may be a causal link in COPD comorbidities such as CVD [70]. Oxidative stress is caused by an alteration of the prooxidant/antioxidant balance that results in the production and accumulation of ROS. ROS refer to a variety of molecules, either containing unpaired electrons (such as superoxide, O2−) or having a strong ability to oxidize other molecules (such as hydrogen peroxide, H2O2) [71], and are produced by both endogenous and exogenous factors. Inflammatory cells, infiltrated in pulmonary arteries, may be one of the major sources of endogenous ROS generated mainly by xanthine oxidase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and mitochondria. On the other hand, genetic factors, CS, and air pollutants are the main producers of exogenous ROS [72]. More in detail, CS favors the activation of inflammatory cells through the recruitment of transcription factors nuclear factor-kappa B (NF-κB), p38, mitogen-activated protein kinase (MAPK), and post-translational modification of histone deacetylase (HDAC) in macrophages [73]. These processes stimulate the cells to release pro-inflammatory cytokines and activate the immune system and inflammation [74]. Experimental studies performed with cultured rat pulmonary microvascular ECs demonstrated that exposure to tobacco smoke condensate causes increased transcription and activity of the ROS generating enzyme xanthine dehydrogenase/oxidase [75]. Among the causes of oxidative stress, there is also a decrease in antioxidants. Antioxidants, such as superoxide dismutase (SOD), glutathione (GSH), and nuclear factor erythroid 2-related factor 2 (Nrf2) are thought to play very important roles in the lungs since they are continuously exposed to oxidant endogenous and exogenous risk factors. NO released from the vascular endothelium is also an antioxidative factor that neutralizes O2− [76] and impairment of NO synthesis marks the onset of endothelial dysfunction that is mainly mediated by the endothelial NO synthase (eNOS) uncoupling mechanism [77].
Increased oxidative stress and ROS overproduction may cause not only endothelial dysfunction but also programmed ECs death such as ferroptosis, pyroptosis, or necroptosis that are involved in the pathophysiology of many diseases, like COPD [78]. Ferroptosis is an iron-dependent process and is characterized by lipid peroxidation, which is the reaction by which OH– attacks the carbon-carbon double bonds of lipids. It is directed by the glutathione antioxidant system, which is formed by GSH, glutathione peroxidase (GPX), and glutaredoxin (GRX) but most importantly it can effectively prevent ROS overproduction [79]. In recent years, studies have shown that ferroptosis is closely related to ECs death. For example, Qin et al. [80] found that zinc oxide nanoparticles could induce iron and lipid peroxidation in ECs depending on the dose and time. Luo et al. [81] showed that ferroptosis is related to endothelial dysfunction, meanwhile, Sheng et al. [82] showed that lysophosphatidylcholine (LPC) can induce an increase in the amount of intracellular iron and lipid peroxide levels and mitochondrial atrophy in ECs. In a study conducted by Wang et al. [83] they demonstrated, with in vitro and in vivo experiments, that pulmonary microvascular endothelial barrier dysfunction plays an important role in the mechanism by which COPD affects the progression of atherosclerosis and that oxidative stress and ferroptosis are involved in this effect. Another mechanism of programmed cell death is pyroptosis. The molecular features of pyroptosis include the activation of inflammasome, membrane pore formation, and pro-inflammatory cytokine maturation and release. Pyroptosis may require activation of caspase-1, depending on whether it is activated or not, it may be involved in classical or non-classical inflammatory pathways. Among the classical inflammasome pathway, Nod-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome-mediated pyroptosis is the most extensively studied phenomenon and can be activated by different stimuli, first of all, ROS. The NLRP3 inflammasome in ECs can be caused by stimuli such as low-density lipoprotein oxidized, hyperglycemia, and nicotine, thus leading to the death of ECs [84, 85]. For example, Wu et al. [86] found that oxidized low-density lipoprotein (ox-LDL) induced, in a dose-dependent manner, the upregulation of NLRP3, caspase-1, and IL-1β in ECs. Hang et al. [87] found that ox-LDL stimulated in ECs NLRP3 inflammasome activation, increased IL-1β and IL-18 maturation and secretion, increased intracellular ROS and increased lactate dehydrogenase (LDH) release in ECs. Zhang et al. [88] found that nicotine could activate the NLRP3 inflammasome. The team also found that due to the activation of NLRP3 inflammation can promote the destruction of the proteins of the narrow junction between ECs, resulting in increased vascular permeability. Cau et al. [89] found that angiotensin II (Ang-II) induces endothelial dysfunction, vascular remodeling, and hypertension through NLRP3 inflammasome activation. The last cell death program we here describe is necroptosis, a genetically encoded mechanism of necrotic regulated cell death defined by rupture of the plasma membrane. Necroptosis causes the release of damage-associated molecular patterns (DAMPs) into the extracellular environment and for this reason is highly proinflammatory. Indeed, Pouwels et al. [90] discovered that CS-induced necroptosis and the release of DAMPs trigger neutrophilic airway inflammation in mice BAL and this effect was reduced by treatment with the necroptosis inhibitor necrostatin-1. Wang et al. [91] reported a novel regulatory mechanism of necroptosis-mediated inflammation. Endoplasmic reticulum chaperone glucose-regulating protein 78 (GRP78) promoted a CS extract (CSE)-induced inflammatory response and mucus hyperproduction in airway epithelial cells, likely through the upregulation of necroptosis and subsequent activation of the NF-κB and activator protein-1 pathways [91, 92]. Based on data present in literature, necroptosis is increased in the lungs of COPD patients and its inhibition attenuates CS-induced emphysema, airway inflammation, and remodeling making this process an attractive candidate for COPD patients. Nevertheless, there is not enough data about the correlation between necroptosis and ECs, this could be a starting point for new research.
Cellular senescence is now considered an important driving mechanism for chronic lung diseases contributing to the pathophysiology of COPD and CVD.
Cellular senescence is due to telomere shortening (replicative senescence) and oxidative stress (stress-related senescence) with activation of p53 and p16 that lead to activation of p21 and cell cycle arrest, respectively [93, 94]. Patients with COPD showed, accumulated in the lungs, senescent cells, including alveolar epithelial and ECs [95–97]. Exposure to CS is among the most relevant factors that cause oxidative stress and is also an important inducer of senescence in COPD, increases markers of senescence in the epithelial cells of the airways, resulting in endothelial damage. Chronic CS exposure causes pulmonary endothelial dysfunction in animal models, healthy young smokers, and chronic smokers with COPD [98, 99]. Accordingly in a study performed by Paschalaki et al. [100] using circulating endothelial progenitors named endothelial colony-forming cells, accelerated endothelial senescence in smokers and patients with COPD was shown compared with healthy controls. They also demonstrated that patients with COPD on ICS exhibit significantly reduced endothelial senescence [100]. Another very recent study by Zeng et al. [101], documented that CSE averages the ubiquitin-specific protease (USP7)/p300 dependent high expression of p53 and activates the expression of target p53 genes, especially p21. They also showed that the activation of p53, via p21, inhibits cell activity and leads to cell cycle arrest with premature senescence of ECs progenitors [101]. Finally, studies reported that senescent pulmonary ECs release more inflammatory markers and this boosts the inflammatory state in COPD patients [102].
Another mechanism that plays an important role in the pathogenesis of COPD is apoptosis and interestingly, increased apoptotic events correlate with ECs damage [103]. Apoptosis is not a marginal event in COPD. Indeed, recent studies suggest that apoptosis of pulmonary vascular ECs plays a role in initiating and participating in the pathogenesis of COPD [104]. Studies show that the number of dead ECs in the lungs of patients with COPD is increased as assessed by specific nuclei fragmentation, which is a clear feature of apoptotic cells [105]. The importance of apoptosis in COPD, especially in the pathogenesis of emphysema, originates from in vitro, in vivo, and in human studies.
In an in vitro study performed by Farid et al. [106], the authors studied the effect of CSE exposure on VEGF release by human lung fibroblasts demonstrating that the TGF-β receptor/mothers against decapentaplegic homolog 3 (Smad3) pathway mediates CSE-induced VEGF production. VEGF plays important roles by mediating angiogenesis and vascular permeability, inducing cell proliferation, and preventing ECs death. Following CS administration VEGF levels decrease and in parallel emphysematous degeneration increases [106]. In line with this observation, Taraseviciene-Stewart et al. [107] by inducing apoptosis of rat pulmonary vascular ECs, with an intraperitoneal injection of CSE, successfully established a rat emphysema model. To explain this result, they suggested that chronic blockade of VEGF induces alveolar cells apoptosis and emphysema. Along this line, another preclinical study suggests that it is possible to induce emphysema in rodents by deliberately causing endothelial apoptosis through VEGF blockade [108]. The authors treated rodents with a VEGF receptor inhibitor which resulted in EC apoptosis and emphysema. Conversely, when rodents were treated with a caspase inhibitor, apoptosis, and emphysema were reverted, suggesting that apoptosis of ECs may be key in emphysema development [109]. Evidence of increased apoptosis of ECs in humans has also been published. Segura-Valdez et al. [110] were the first to show increased apoptosis in the lungs of COPD patients. They showed that ECs and arterioles exhibited prominent intranuclear staining by in situ-end labeling of fragmented DNA [110]. Moreover, Kasahara et al. [109] described an increased apoptosis of alveolar epithelial and ECs in lung tissue from patients with emphysema. Different and contrasting studies suggest that VEGF and HIF-1α levels change with the severity of pathology. For example, in the study by Yasuo et al. [111], patients with emphysema displayed reduced levels of HIF-1α in lung ECs. On the other hand, a study by Lee et al. [112] showed increased levels of VEGF and HIF-1α in patients with chronic bronchitis. These contrasting findings suggest that the endothelium is variably involved and VEGF is not uniquely implicated in the mechanism of endothelial apoptosis in COPD patients. In this regard, other players could be the cystic fibrosis transmembrane regulator (CFTR) and AAT. CFTR, who participates in the apoptotic process, is necessary for stress-induced apoptosis of human pulmonary ECs as confirmed by the evidence its inhibition, by hydrogen peroxide, attenuates EC apoptosis [113]. AAT has an important role in preventing caspase-3 activation and therefore apoptosis in pulmonary ECs [114]. Overall, endothelial apoptosis may contribute to the destruction of alveoli and consequently, to the onset and progression of emphysema in COPD patients.
Therefore, understanding the mechanisms by which apoptosis is induced in the pulmonary endothelium is important for the development of new therapies to prevent or treat lung diseases characterized by endothelial dysfunction.
Different methods have been developed over the years to assess endothelial damage in COPD patients and some are used in clinical practice. Today, the most widely used method to assess endothelial dysfunction is flow-mediated dilatation (FMD) that is typically performed on the brachial artery. FMD is a non-invasive diagnostic test that measures the ability of blood vessels to dilate in response to increased blood flow. To this aim, arterial diameter is recorded at baseline and at end-diastole and FMD is calculated as a percentage change from baseline [115]. Alterations of FMD, reflect the inability of the endothelium to produce and release substances that help to regulate blood vessels tone and blood flow. FMD is often used as an indicator of cardiovascular risk [116]. In line with this, Barr et al. [117] found that FMD was impaired in a group of former smokers with COPD, especially in those with low FEV1 and advanced emphysema. In two other studies, authors have shown that COPD patients had lower FMD when compared to healthy controls [118, 119]. Marchetti et al. [120], have conducted a study to evaluate endothelial dysfunction in a group of hospitalized patients with COPD using FMD. Patients were 40–80 years old and had more than 20 pack-year history of smoking. The authors demonstrated that patients hospitalized for exacerbation of COPD had severe vascular dysfunction as evidenced by markedly reduced FMD [120]. Since FMD is a valid test to evaluate possible changes in endothelial function in COPD patients, Clarenbach et al. [121] and his team conducted a clinical study using this diagnostic technique. They recruited patients between 45 and 75 years with very severe COPD. Thanks to this simple and inexpensive technique they found a relative annual 5.6% decrease in FMD in COPD patients indicating progressive vascular dysfunction. An annual reduction in lung function tends to be associated with reduced endothelial function over time [121].
Another functional method to evaluate endothelial dysfunction is the venous occlusion plethysmography (VOP), which is a non-invasive diagnostic test that measures venous blood flow in body extremities, typically arm or leg. The test involves the use of a pneumatic cuff that is placed around the body’s extremity and inflated to temporarily stop blood flow. By measuring changes in the volume of the extremity before, during, and after occlusion, VOP can provide information about the ability of the veins to carry blood back to the heart. Thanks to VOP measures, Yang et al. [122] demonstrated that patients with COPD and overweight smokers have impaired endothelial function [122]. However, this technique has important limitations as shown in a study performed by Maclay et al. [123] where endothelium-dependent and endothelium-independent vasodilation, assessed with VOP, did not significantly differentiate COPD patients from healthy controls.
An additional technique is peripheral arterial tonometry (PAT), a non-invasive diagnostic test that measures changes in blood volume in fingers or toes. It is used to assess arterial integrity and to determine CVD risk. Results of the PAT test are used to calculate the PAT index, which is a measure of arteries’ ability to dilate in response to increased blood flow. A low PAT index is associated with an increased risk of CVD. Also, in this case, the literature shows conflicting results about the utility of this test to diagnose endothelial dysfunction [124].
More recent, noninvasive technologies are available to evaluate endothelial damage such as nailfold capillaroscopy, laser speckle contrast imaging, or near-infrared spectroscopy, and can be combined with several reactivity tests and exercises. Currently, these technologies are used in other pathological states and no data are reported about their use in COPD patients. Since endothelium participates in the development and progression of COPD, it would be relevant to expand the clinical utility of diagnostic techniques to improve diagnostic and therapeutic approaches.
CVD is one of the most common risks of morbidity and mortality in COPD. Different studies suggest that endothelial dysfunction is present in a remarkable number of COPD patients and COPD contributes to increasing the cardiovascular risk. The typical COPD patient is just as likely to die from both respiratory and cardiovascular causes [125]. Although these two pathologies undoubtedly recognize CS as the common and principal risk factor, today this statement is restrictive. Indeed, other shared risk factors include physical inactivity, air pollution exposure, diabetes, and hypertension. Those factors are highly prevalent in COPD patients, especially in those who present severe disease [126]. Inflammation, aging, oxidative stress, and hypoxia are the most important mechanisms that can explain the relationship between these pathologies. At the basis of each of them, can certainly found systemic inflammation, foam cell production, and increased production of cell adhesion molecules in ECs, which contribute to the progression of CVD. These events also induce pulmonary vascular remodeling, intimal thickening, and endothelial dysfunction of the pulmonary artery that could be aggravated by the increased presence of inflammatory cells [127]. Accordingly, a review by Ye and others [128] suggests that patients with COPD have increased levels of endothelial dysfunction as well as increased incidence of CVD documented by intima-medial carotid thickening (this finding is reported in 22 different studies). Based on scientific evidence COPD and CVD are closely related and the key shared mechanism is likely inflammation. Considering the clear relationship between COPD and CVD, future studies are needed to deepen our understanding of this aspect to improve the quality of life of patients affected by these diseases.
Accumulated evidence from different works suggests that endothelial dysfunction occurs in COPD and worsens with it. This phenomenon is driven by multiple factors: CS, chemokines, cytokines, ROS, and neutrophils activation. All these factors converge in a common activation of systemic inflammation, which contributes to disease progression and to the onset of other pathologic states such as CVD. To date, there are several tests and biomarkers that allow the evaluation of endothelial dysfunction and damage. However, the mechanisms leading to endothelial damage remain to be clarified, prior to obtain promising new treatment strategies and to understand whether the endothelial injury is primarily involved in the pathology or reflects the consequences of COPD occurrence.
AAT: alpha-1 antitrypsin
CCL11: eotaxin-1
CGRP: calcitonin gene-related peptide
COPD: chronic obstructive pulmonary disease
CS: cigarette smoke
CSE: cigarette smoke extract
CVD: cardiovascular disease
ECs: endothelial cells
FEV1: forced expiratory volume in 1 s
FMD: flow-mediated dilatation
GOLD: Global Initiative for Chronic Obstructive Lung Disease
HIF-1α: hypoxia-inducible factor 1α
ICAM-1: intracellular adhesion molecule-1
ICS: inhaled corticosteroids
IL: interleukin
MAC-1: macrophage-1 antigen
NK: neurokinin receptors
NLRP3: Nod-like receptor pyrin domain-containing protein 3
NO: nitric oxide
PAT: peripheral arterial tonometry
ROS: reactive oxygen species
TEM: transendothelial migration
VEGF: vascular endothelial growth factors
VOP: venous occlusion plethysmography
SS: Conceptualization, Writing—original draft. GN: Writing—review & editing. AMN: Writing—review & editing. MT: Writing—review & editing. CAML: Writing—review & editing. SP: Conceptualization, Writing—review & editing.
GN, SP, and MT are employees of Chiesi Farmaceutici S.p.A. All the remaining authors have not actual and perceived conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Mauro G. Silva ... Walter Manucha
Paolo Giannoni ... Daniela de Totero
Lena B. Kim, Anna N. Putyatina
