5 results in Exploration of Medicine
Latest
Sort by :
- Latest
- Most Viewed
- Most Downloaded
- Most Cited
Open Access
Original Article
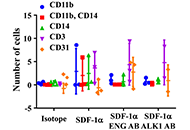
Placental OPRM1 DNA methylation and associations with neonatal opioid withdrawal syndrome, a pilot study
Elisha M. Wachman ... Huiping Zhang
Published: June 29, 2020 Explor Med. 2020;1:124–135
This article belongs to the special issue The Biological Basis of Substance Use Disorders
Open Access
Meta-Analysis
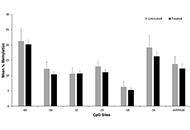
GLP-1 receptor agonists for NAFLD treatment in patients with and without type 2 diabetes: an updated meta-analysis
Alessandro Mantovani ... Giovanni Targher
Published: June 29, 2020 Explor Med. 2020;1:108–123
This article belongs to the special issue Exploring Type 2 Diabetes Mellitus

Open Access
Review
Perspectives of nonalcoholic fatty liver disease research: a personal point of view
Amedeo Lonardo, Stefano Ballestri
Published: June 29, 2020 Explor Med. 2020;1:85–107
This article belongs to the special issue Exploring NAFLD/NASH
Open Access
Review
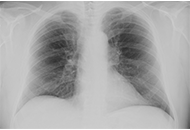
Nonalcoholic fatty liver disease and portal hypertension
Marvin Ryou ... Gyorgy Baffy
Published: June 29, 2020 Explor Med. 2020;1:149–169
This article belongs to the special issue Exploring NAFLD/NASH
Open Access
Original Article
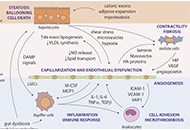
Reduction of endoglin receptor impairs mononuclear cell-migration
Zhenying Han ... Hua Su
Published: June 29, 2020 Explor Med. 2020;1:136–148