 Review
Review

Affiliation:
1Department of Biology, Calibr at The Scripps Research Institute, La Jolla, CA 92037, USA
ORCID: https://orcid.org/0000-0003-3802-5556
Affiliation:
2Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, CA 92037, USA
Email: deborahw@scripps.edu
ORCID: https://orcid.org/0000-0003-2545-1935
Explor Immunol. 2022;2:79–97 DOI: https://doi.org/10.37349/ei.2022.00038
Received: November 13, 2021 Accepted: January 16, 2022 Published: February 24, 2022
Academic Editor: Dieter Kabelitz, Christian-Albrechts University of Kiel and University Hospital Schleswig-Holstein Campus Kiel, Germany
The article belongs to the special issue Interplay of γδ T cells and Tumor Cells
Antitumor immunity relies on the ability of T cells to recognize and kill tumor targets. γδ T cells are a specialized subset of T cells that predominantly localizes to non-lymphoid tissue such as the skin, gut, and lung where they are actively involved in tumor immunosurveillance. γδ T cells respond to self-stress ligands that are increased on many tumor cells, and these interactions provide costimulatory signals that promote their activation and cytotoxicity. This review will cover costimulatory molecules that are known to be critical for the function of γδ T cells with a specific focus on mouse dendritic epidermal T cells (DETC). DETC are a prototypic tissue-resident γδ T cell population with known roles in antitumor immunity and are therefore useful for identifying mechanisms that may control activation of other γδ T cell subsets within non-lymphoid tissues. This review concludes with a brief discussion on how γδ T cell costimulatory molecules can be targeted for improved cancer immunotherapy.
γδ T cells are a unique lymphocyte population that has important roles in tissue homeostasis, infection, and cancer. Although γδ T cells are a minor population within lymphoid tissues (typically 1–5% of T cells in peripheral blood), they are present in larger numbers in both mouse and human epithelial tissues (10–100% of T cells) such as the skin and gut [1]. Within these tissues, γδ T cells detect signals of cellular stress that are increased following tissue damage or malignant transformation, and upon activation, they produce cytokines, chemokines, and growth factors that promote tissue repair and eliminate developing tumors [1]. The critical role of γδ T cells in antitumor immunity is highlighted by the fact that mice lacking these cells T cell receptor delta-deficient (Tcrd−/−) are more susceptible to tumorigenesis in a variety of preclinical mouse tumor models [2–4]. Although some protumor roles of γδ T cells, often associated with interleukin-17 (IL-17) production, have been described in both mouse models and human disease, γδ T cells are generally associated with potent antitumor immunity [5]. For example, the presence of γδ T cells within solid tumors has been identified as a strong prognostic factor for overall survival in patients in both meta-analyses [6] and recent, individual case studies of solid tumors such as Merkel cell carcinoma [7], breast cancer [8], and ovarian cancer [9]. Similar to their αβ T cell counterparts, the antitumor functions of γδ T cells include direct lysis of tumor targets and secretion of inflammatory cytokines such as interferon-γ (IFNγ) that promote broader antitumor immunity. However, γδ T cells recognize antigens independent of major histocompatibility complex (MHC) presentation and detect self-stress molecules associated with tissue damage and malignancy, which allow them to provide rapid, innate-like responses within non-lymphoid tissues [5]. This unique feature of γδ T cells is exemplified by the ability of both mouse and human γδ T cells to lyse a broad range of tumor targets in vitro [10]. The study of γδ T cells is complicated by the evolutionary divergence of T cell receptor (TCR) genes between mouse and human (Table 1) and the limited understanding of bona fide γδTCR antigens, which has recently been reviewed elsewhere [11]. Despite these challenges, the study of γδ T cells in mice has led to the identification of key molecules important for their maintenance of tissue homeostasis and regulation of antitumor immunity that may have relevance for the treatment of human disease. Here, activating receptor-ligand interactions important for the activity of γδ T cells within non-lymphoid tissues are reviewed, with a specific focus on mouse dendritic epidermal T cells (DETC), and their potential roles in antitumor immunity, concluding with a brief discussion on how this knowledge could be leveraged for improved cancer immunotherapy.
Most common γδ T cell subsets in mouse and human
| Circulating subsets | Tissue-resident subsets | |
|---|---|---|
| Mouse | Vγ1.1+ Vγ2+ | Vγ3+ (epidermis) Vγ4+ (dermis, lung, uterus, adipose, liver) Vγ5+ (gut) |
| Human | Vδ2+ | Vδ1+ (epidermis, dermis, adipose, gut, liver) |
Note. Adapted from “γδ T cells in cancer [12]” by Silva-Santos B, Serre K, Norell H. Nat Rev Immunol. 2015;15:683–91 (https://pubmed.ncbi.nlm.nih.gov/26449179/). © 2015 Macmillan Publishers Limited.
DETC are a prototypic tissue-resident γδ T cell population in mouse skin that has critical roles in the maintenance of tissue homeostasis, wound healing, and tumor immunosurveillance [13]. Under homeostatic conditions in the mouse epidermis, DETC display a characteristic dendritic morphology that allows them to survey neighboring keratinocytes [14]. DETC express an invariant, canonical Vγ3+Vδ1+ TCR (Garman nomenclature) that recognizes an unidentified self-stress ligand [15]. Upon activation, DETC retract their dendrites and round up presumably to allow for migration towards signals of tissue insult where they secrete inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and IFNγ as well as growth factors such as keratinocyte growth factor-1 (KGF-1) [16, 17]. The critical role of DETC in the maintenance of skin tissue homeostasis is highlighted by the fact that mice lacking DETC are more susceptible to carcinogen-induced skin tumor formation and exhibit delayed wound healing [2, 14].
Like other γδ T cell populations in both mice and humans, DETC do not recognize antigens in the context of MHC antigen presentation and lack CD4, CD8, and CD28 co-receptors that are critical for the priming of αβ T cells [18]. As a result, the exact mechanisms required for DETC activation are less well-characterized. Investigators have sought to identify the key molecules important for the function of DETC. These studies have identified natural killer group 2D [NKG2D; encoded by killer cell lectin like receptor K1 (Klrk1)], junctional adhesion molecule-like protein (JAML), CD100 ([encoded by semaphorin 4D (Sema4d)], lymphocyte function-associated antigen 1 (LFA-1), and CD316 as activating co-receptors that are critical for DETC function (Figure 1). Each of these molecules has important role in antitumor immunity and is potential therapeutic target for improved cancer immunotherapy.
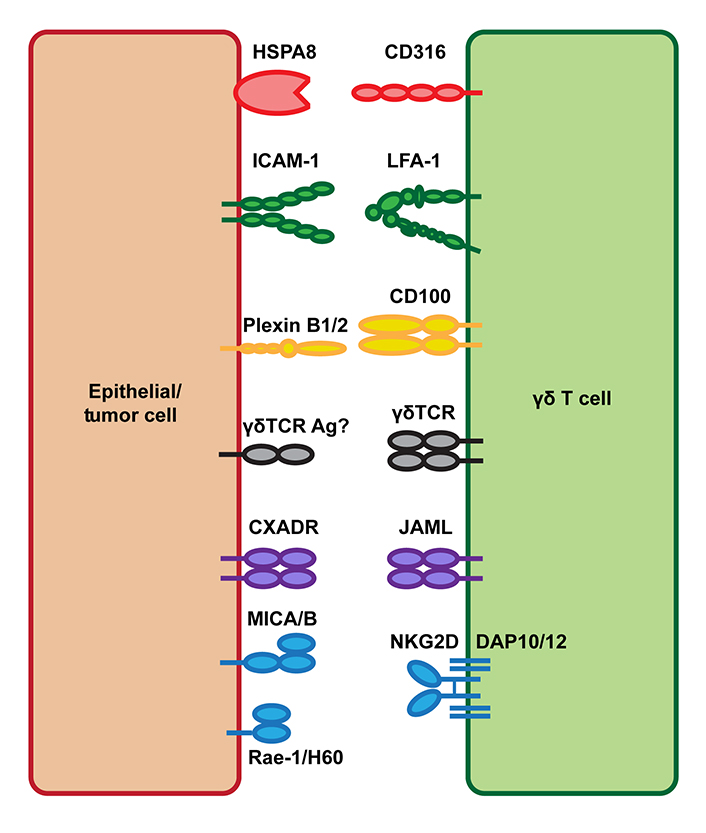
Tissue-resident γδ T cell costimulatory ligand interactions. Tissue-resident γδ T cells express the costimulatory molecules NKG2D, JAML, CD100, LFA-1, and CD316. Upon tissue damage or malignant transformation, epithelial cells upregulate expression of both γδTCR antigens and the costimulatory ligands Rae-1/H60 [MHC class I polypeptide-related sequence A and B (MICA/B) in human], CXADR, plexin B1/2, ICAM-1, and HSPA8 allowing for γδ T cell activation and their promotion of tissue homeostasis. HSPA8: heat shock protein family A member 8; ICAM-1: intercellular adhesion molecule-1; Ag: antigens; CXADR: coxsackie and adenovirus receptor; Rae-1: retinoic acid early inducible 1; H60: histocompatibility 60; DAP10/12: DNAX-activating protein 10/12
NKG2D is a C-type lectin-like activating receptor expressed by natural killer (NK) cells, γδ T cells, and CD8 T cells that are critical for the recognition and elimination of damaged, infected, and cancerous cells. NKG2D interacts with the stress-induced ligands Rae-1, H60, and mouse unique long 16 (UL16)-binding protein-like transcript (Mult-1) in mouse and MICA/B and UL16-binding proteins 1-6 (ULBP1-6) in humans [19]. NKG2D associates with the adapter protein DAP10 in humans and either DAP10 or DAP12 in mice, which stabilizes the receptor complex and mediates intracellular signal transduction upon NKG2D ligand binding. Upon NKG2D activation, DAP10 recruits a p85 phosphoinositide 3-kinase (PI3K) and Vav-1 signaling complex, whereas DAP12 contains a canonical immunotyrosine-based activation motif (ITAM) that binds spleen tyrosine kinase (Syk) and Zeta-chain-associated protein kinase 70 (ZAP70) tyrosine kinases [20].
The function of NKG2D as an activating receptor important for antitumor cytotoxicity was initially discovered based on the observation that human Vδ1+ T cell clones recognize MICA/B expressed on a wide range of epithelial tumors including lung, breast, kidney, prostate, and colon cancers [21, 22]. MICA was found to bind to human NK cells and CD8, but not CD4, T cells and was identified as a ligand for NKG2D [21]. Shortly after this discovery of NKG2D-MICA/B interactions in humans, similar findings were reported in mouse tumor models. This seminal work showed that Tcrd−/− mice were more susceptible to both growth of transplantable squamous cell carcinomas and formation of skin tumors induced by the chemical carcinogens demonstrating a key antitumor role for γδ T cells [2]. The authors additionally went on to show that this antitumor activity of γδ T cells was mediated by DETC, which killed tumor cells via NKG2D recognition of the mouse homologs of human MICA/B, Rae-1, and H60 [2].
Since these initial reports identifying NKG2D as a critical receptor for the antitumor activity of γδ T cells, many other groups have described important roles for NKG2D in γδ T cell biology with a number of studies focused specifically on NKG2D in the function of DETC in murine skin [23–27]. Together, these studies have highlighted NKG2D recognition of self-stress ligands such as Rae-1 and H60 as a key component of tissue-resident γδ T cell activation in response to a wide variety of tissue insults. Transgenic overexpression of Rae-1 in the epidermis of mice was found to result in DETC activation within a 5-day timeframe, as measured by DETC rounding, upregulation of CD69, and downregulation of γδTCR expression [23]. This result indicated that DETC are actively involved in tumor immunosurveillance and rapidly responds to stress signals in murine skin. In addition to roles in antitumor immunity, an important role for NKG2D in DETC function has been described in the homeostatic maintenance of skin tissue [24], atopy [25], cutaneous wounding [26], and contact hypersensitivity [27].
Unlike classical costimulatory molecules which only function in combination with TCR engagement, NKG2D has been proposed to activate DETC independent of TCR signals [28]. In the absence of TCR signals, NKG2D binding induces PI3K-dependent signaling that results in the DETC lysis of target cells in vitro. In contrast, IFNγ production is driven more potently by Syk/ZAP70 signaling downstream of TCR engagement [29]. Similar TCR-independent antitumor cytotoxicity through NKG2D by human Vγ9Vδ2 T cells has also been described. However, depending on the tumor target, Vγ9Vδ2 T cells can kill tumor targets via TCR engagement alone or require co-engagement of TCR and NKG2D [30]. Although the relative importance of TCR versus NKG2D signaling for γδ T cell activation remains controversial, it is clear that NKG2D is an important regulator of γδ T cell cytotoxicity against a broad range of both mouse and human tumor cell lines [31].
In addition to a critical role for DETC function, NKG2D has been shown to have a critical role in the antitumor activity of other mouse γδ T cell subsets. Mouse lymphoid Vγ2+ T cells possess antitumor activity against B16 melanoma cells in vivo, which was associated with inhibition of B16 cell growth, but not direct cytotoxicity, in vitro. Tumor suppressive activity of Vγ2+ T cells was dependent on both TCR and NKG2D signals, which regulated their production of IFNγ. This antitumor activity of Vγ2+ T cells can be further enhanced by treatment with the mTOR inhibitor rapamycin in vitro, which sensitized cells to IL-2 signals resulting in increased expression of both NKG2D and TNF-α and higher cytotoxicity. Vγ2+ T cells expanded in vitro with rapamycin had improved antitumor activity against B16 melanoma in vivo upon adoptive cell transfer (ACT) compared with cells expanded with TCR and CD28 signals alone [32].
JAML (also known as AMICA1 in humans) is a member of the junctional adhesion molecule family, a class of molecules that facilitate tight junction assembly, regulate leukocyte-endothelial interactions, and have diverse roles in development, angiogenesis, inflammation, and cancer [33, 34]. JAML binds to the CXADR, a cell adhesion molecule expressed by non-hematopoietic cells including epithelial cells within the skin and gut tissue [35–37]. JAML-CXADR interactions have been identified as a novel costimulatory mechanism for the activation of DETC. Upon binding to CXADR, JAML induces a PI3K signaling cascade, which promotes cytokine production and proliferation by DETC. CXADR is expressed at low levels in mouse skin under homeostatic conditions, but following cutaneous wounding, keratinocytes increase expression of CXADR allowing for increased JAML costimulation of DETC and their subsequent production of soluble factors such as KGF-1, which promote the wound repair process [37].
Based on this initial characterization of JAML-CXADR-mediated costimulation of tissue-resident γδ T cells and the known roles of γδ T cells in antitumor immunity, the importance of JAML-CXADR interactions for γδ T cell responses within the tumor microenvironment (TME) was investigated. Mice lacking JAML protein expression (Jaml−/−) were more susceptible to both B16F10 melanoma tumor formation and growth, which was associated with decreased activity of γδ T cells. During the early stages of tumor growth, Jaml−/− mice had fewer numbers of IFNγ-producing γδ tumor-infiltrating lymphocytes (TIL) demonstrating that the γδ T cell response to B16 tumor growth is mediated, at least in part, by JAML-CXADR interactions. Importantly, this γδ TIL response consisted of Vγ1.1+, Vγ2+, and Vγ1.1−Vγ2− infiltrating subsets, but not DETC, demonstrating a broader role for JAML-CXADR interactions in γδ T cell biology. Unlike DETC, which constitutively express JAML, naive lymphoid γδ T cells did not express high levels of JAML. However, lymphoid γδ T cell subsets upregulated JAML upon ex vivo stimulation and within the TME after which JAML engagement further enhanced activation and cytokine production. Therefore, JAML is also an important costimulatory ligand for lymphoid γδ T cell subsets after initial T cell priming [38].
CXADR expression is dysregulated in many human cancers [39]. For example, loss of CXADR expression is associated with increased epithelial-mesenchymal transition in breast cancer [40] and increased gastric cancer metastasis [41]. A similar association with decreased CXADR expression and increased malignancy was also observed in both mouse and human melanoma where CXADR expression was higher in benign lesions and during early stages of tumor growth but was decreased upon tumor growth and progression [38]. Furthermore, both mouse and human melanocytes express very low levels of CXADR suggesting that CXADR expression is linked to melanoma tumorigenesis [38]. During the early stages of the disease, this pattern of CXADR expression mirrors findings in cutaneous wound healing where expression is low under homeostatic conditions but is increased following tissue damage or malignant transformation. Together, these results suggest that CXADR is an important stress ligand that functions as a signal to tissue-resident γδ T cells to mediate both tissue repair and antitumor immunity.
In contrast to impaired antitumor immunity in the absence of JAML, treatment of tumor-bearing wild type (WT) mice with an anti-JAML agonistic antibody [42] significantly limited tumor growth and extended median survival. Furthermore, the anti-JAML treatment improved the efficacy of programmed cell death 1 (PD-1) blockade when used in combination. This effect of anti-JAML treatment was associated with improved markers of both CD8 and γδ TIL immunity in WT mice. Critically, in both Tcrd−/− mice and mice treated with CD8 T cell depleting antibodies, the antitumor effect of anti-JAML treatment was not observed [38]. These results point to a key role of JAML-mediated γδ T cells responses in antitumor immunity and the necessary cooperation between CD8 and γδ T cells, which involves JAML-CXADR interactions.
CD100 (also known as SEMA4D) is a group IV semaphorin expressed by T cells, B cells, platelets, NK cells, and monocytes and binds with low affinity to CD72 on other immune cells and with high affinity to plexin B1/2. Semaphorins are a large family of molecules that interact with plexins to regulate cytoskeleton dynamics and were initially discovered to have critical roles in neuronal development [43]. Studies of mice lacking CD100 (Cd100−/−) have since demonstrated key roles for CD100 in the regulation of both humoral and cellular immunity [44]. These studies have shown that CD100-CD72 interactions are critical for both T cell priming by antigen-presenting cells (APCs) and B cell proliferation and antibody production [45, 46]. CD100 can function as both a membrane-bound protein on the surface of immune cells and as a soluble molecule either following proteolytic cleavage by matrix metalloproteinases (MMPs) or cell-intrinsic shedding [47, 48]. In B cells, binding of either membrane-bound or soluble CD100 to CD72 displaces Src homology 2 domain-containing protein tyrosine phosphatase 1 (SHP-1) from its intracellular immunoreceptor tyrosine-based inhibitory motif (ITIM) domain allowing for increased activation [46]. On the other hand, CD100 binding to plexin B1 on epithelial and endothelial cells results in the recruitment of small GTPases, which facilitate changes in cytoskeleton dynamics [49].
Plexin B2 has been identified as a novel ligand for CD100, which is expressed by skin epithelial cells and regulates DETC function in mice [50]. Similar to the findings on JAML-CXADR regulation of DETC function, it was found that plexin B2 is expressed by keratinocytes under homeostatic conditions, is further upregulated upon cutaneous wounding, and interacts with CD100 on DETC to promote their wound healing activity. Upon binding to plexin B2, CD100 induces extracellular signal-related kinase (ERK) and cofilin signaling resulting in DETC rounding and increased integrin expression [50]. Additionally, CD100-plexin B2 regulation of γδ T cell-mediated tissue repair is critical in the dextran sodium sulfate (DSS) mouse model of colitis. In this model, activation of γδ intraepithelial lymphocytes (IEL) in the intestine by CD100-plexin B2 binding resulted in the production of KGF-1, which was required to limit the severity of colitis [51]. In addition to pro-wound healing functions in the skin and gut, CD100-plexin B2 interactions have been shown to be involved in psoriasis in both patients and mouse models [52]. Importantly, in the mouse model of psoriasis induced by treatment with the toll-like receptor 7 (TLR7) agonist imiquimod (IMQ), knockdown of plexin B2 in mouse keratinocytes in vivo limited inflammation and epidermal infiltration by dermal γδ T cells, which are known to be a critical source of IL-17 and drive inflammation in this model [52]. This result, together with the colitis model data, suggests that CD100-plexin B2 interactions may be more generally important for the regulation of γδ T cell function in response to tissue damage and inflammation.
Although a role for CD100 in γδ T cell antitumor immunity has not been described, CD100 does play an important role within the TME. In mouse tumor models, both genetic deletion (Cd100−/−) and antibody-mediated blockade of CD100 limit tumor growth highlighting a pro-tumorigenic role for CD100 signaling [53]. In humans, high CD100 expression has been associated with worse patient outcomes in colorectal [54], ovarian [55], sarcoma [56], and cervical [57] cancers. Because both tumor cells and immune cells can express CD100 and CD100 ligands, the signaling pathways involved in their promotion of tumor growth are complex. CD100 is highly expressed by tumor-associated macrophage (TAM) and interactions with plexin B1 within the TME promote tumor angiogenesis [58]. Tumor cells themselves can also secrete CD100, which polarizes myeloid cells towards an immunosuppressive myeloid-derived suppressor cell (MDSC) phenotype [59]. This promotion of MDSC is limited by treatment with a blocking anti-CD100 antibody, which improves CD8 T cell antitumor activity and synergizes with immune checkpoint blockade (ICB) in mouse tumor models [60].
Although most studies have focused on the roles of CD100 in myeloid cell and B cell function, CD100-CD72 interactions are also critical for CD8 T cell activation and proliferation. CD8 T cells upregulate both CD100 and CD72 upon activation after which CD100-CD72 interaction drive further CD8 T cell activation. In the context of cancer, a recent study of patients with non-small cell lung cancer found that CD8 T cells isolated from bronchoalveolar lavage fluid were activated by CD100 stimulation ex vivo and that decreased shedding of membrane-bound CD100 from T cells and the resulting decreased levels of circulating soluble CD100 may impair CD8 T cell function in vivo [61]. Therefore, CD100 interactions may be important for antitumor T cell responses, and the extent to which CD100 interactions regulate CD8 or γδ T cell function within the TME deserves further investigation. Given the data on the critical role of CD100 in DETC function, it is possible that plexin B2 may function as a tumor stress signal that is important for γδ T cell tumor immunosurveillance and promotion of antitumor immunity. Conversely, the clear antitumor effects of anti-CD100 blocking antibodies could involve inhibition of tumor-promoting IL-17+ γδ T cell function [62]. Parsing apart these roles for CD100 on myeloid cell and T cell function within the TME will help inform if CD100 is a viable therapeutic target for the treatment of human cancers.
LFA-1 is an integrin heterodimer composed of CD11a and CD18 and has important roles in regulating T cell activation and migration. LFA-1 binds ICAM-1 allowing for prolonged T cell-APC interactions, increased tissue migration, and contact with target cells for cytolytic activity [63, 64]. LFA-1 binding also lowers the threshold required for T cell activation by signaling through cytohesin-1, which results in mitogen-activated protein kinase (MAPK) pathway activation, and through Jun activation domain-binding protein 1 (JAB-1), which promotes c-Jun phosphorylation. Together with TCR and CD28 signaling, these LFA-1 driven events promote cell proliferation and IL-2 production [65]. Although most work on LFA-1 has focused on its role in the regulation of αβ T cell responses, LFA-1 has long been known to be important for γδ T cell biology. LFA-1 has been shown to be important for γδ T cell cytotoxic activity against human melanoma [66], ovarian [67], lymphoma [68], myeloma [69], pancreatic [70], and lung cancer cell lines [71]. In this context of tumor cell killing, LFA-1 is critical for adhesion and immune synapse formation with target cells, which enhances γδ T cell cytotoxicity [71]. Although direct cytotoxicity was not measured, LFA-1 has also been shown to be critical for the adhesion of human Vδ1+ γδ T cells to esophageal squamous cell carcinoma cells [72]. Separately, a study using Icam1−/− mice also identified a key role for LFA-1-ICAM-1 interactions in the migration and pro-wound healing function of tissue-infiltrating γδ T cells upon damage to the cornea epithelium [73].
In recent work, a role for LFA-1-ICAM-1 interactions in the function of DETC in response to skin wounding has been identified [74]. Using an unbiased ribonucleic acid (RNA) sequencing-based approach, ICAM-1 was identified as a keratinocyte stress ligand that was significantly upregulated in both mouse and human skin upon wounding. DETC were found to constitutively expresses LFA-1 and could be co-stimulated in vitro with either ICAM-1-fragment crystallizable (Fc) protein or agonist anti-LFA-1 antibodies. Importantly, ex vivo wound closure, a measure of DETC-mediated wound healing function independent of additional infiltrating immune cells, was impaired in skin tissue isolated from Icam1−/− mice [74]. These findings are consistent with prior studies that reported delayed wound closure in both skin and gut tissue in Icam1−/− mice. Upon wounding of both skin and gut tissue, ICAM-1 expression on epithelial cells was shown to be important for cell proliferation and infiltration of polymorphonuclear cells, which supported wound healing [75, 76]. Together, these results suggest that ICAM-1 on epithelial cells acts as a tissue stress ligand in a manner analogous to CXADR and plexin B2 allowing for tissue-resident γδ T cell activation and their maintenance of tissue homeostasis. Although LFA-1 is clearly important for γδ T cell lysis of tumor cells [66–71], additional characterization of LFA-1-ICAM-1 interactions in the regulation of γδ T cell migration, retention, and function within the TME would be of value. Given that LFA-1 and JAML function similarly as both adhesion and costimulatory molecules and studies demonstrating the antitumor activity of JAML agonism in vivo, it is interesting to speculate that agonist anti-LFA-1 antibodies may have a similar therapeutic effect for the treatment of cancer.
CD316 is an immunoglobulin superfamily (IgSF) protein, which associates with CD9 and CD81 within cell membranes [77], and has also been shown to localize to T cell immune synapses and interact with intracellular cytoskeleton elements to regulate T cell function [78]. CD316 has also been characterized as an early marker of dendritic cell (DC) activation and was shown to bind HSPA8, which enhanced CCL21-dependent DC migration [79]. In recent work on the function of LFA-1 for DETC function, HSPA8 was also identified as a tissue stress ligand-induced by wounding of both mouse and human skin tissue [74]. Surface expression of HSPA8 on keratinocytes was significantly increased in wounded mouse skin, and in vitro costimulation of DETC with anti-CD3 and recombinant HSPA8 significantly increased DETC proliferation, CD25 expression, and IL-2 production [74]. Conversely, siRNA-mediated knockdown of HSPA8 in keratinocytes resulted in decreased DETC activation after co-culture [74]. Additionally, DETC constitutively express CD316 and can be co-stimulated by an agonist anti-CD316 antibody. Therefore, CD316-HSPA8 interactions are a novel receptor-ligand pair that activates DETC in mouse skin.
Although a role for CD316-HSPA8 interactions in antitumor immunity has not been described, heat shock proteins (HSPs) have known roles in cancer and have previously been implicated in γδ T cell activation [80, 81]. HSPs are molecular chaperones that mediate protein folding and stability with diverse roles in response to cell stress associated with tissue damage, infection, and cancer [81]. In cancer, HSPs are important regulators of cell proliferation, apoptosis, migration, and metastasis [81]. Inhibitors of both HSP70 and HSP90 can inhibit the growth of a wide range of tumor cell lines in vitro and have shown efficacy in preclinical mouse tumor models [81, 82]. As a result, these inhibitors have been tested in human clinical trials but with limited success to date [82]. More recently, the large HSPs HSP110 and glucose-regulated protein 170 (GRP170) have been tested in cancer vaccine formulations with promising results in mouse tumor models. These HSPs effectively bind tumor antigen-derived peptides and increase APC activity and promotion of antigen-specific T cell responses [83, 84]. Tumor vaccines incorporating HSPs are the subject of ongoing human clinical trials for the treatment of a wide range of both solid and hematological malignancies [85].
In addition to their diverse roles in the maintenance of protein stability and function, HSPs have been shown to activate γδ T cells in a variety of contexts and have been suggested to be γδTCR antigens [80, 86]. In human, Vγ9Vδ2 γδ T cells have been shown to kill both Daudi B cells and primary oral tumor cells in vitro, which can be blocked with anti-Vγ9 or anti-HSP60 antibodies [87]. HSP70 has also been implicated in γδ T cell killing of Eppstein Barr virus-transformed B cells [88]. Mouse lymphoid γδ T cells have been shown to respond to peptides derived from HSP60 in vivo [89] and full-length HSP65 protein in vitro [90]. Additionally, mouse gut IEL γδ T cell clones proliferate and secrete cytokines after co-culture with HSP71 derived from Mycobacterium tuberculosis in vitro [91]. HSP60 has also been implicated in mouse γδ T cell killing of inflammatory macrophage during bacterial infection, which limits excessive inflammation [92, 93]. Together, these studies point to a model where HSPs act as a cellular stress signal in response to infection and cancer that activates γδ T cells to maintain host tissue homeostasis [80]. Despite these findings, the exact role of HSPs in γδ T cell biology has remained controversial given the ability of HSPs to bind endotoxin and studies that have shown removal of endotoxin from HSPs can limit γδ T cell activation in certain cases [94, 95]. However, given increased expression of HSPs on the surface of cancer cells compared to normal cells, it is possible that interactions such as CD316-HSPA8 are also important for γδ T cell function within tumors.
Due to their inherent antitumor cytotoxicity and activity independent of MHC antigen presentation, γδ T cells are a promising target for improved cancer immunotherapy. To date, the clinical use of γδ T cell-based immunotherapies has revolved around targeting Vγ9Vδ2 T cell activation via phosphoantigen sensing. Phosphoantigens such as endogenous isopentenyl pyrophosphate (IPP) and the microbial metabolite (E)-4-hydroxy-3-methyl-but-2-enyl-pyrophosphate (HMB-PP) drive conformational changes in the accessory molecules butyrophilin subfamily 3 member A1 (BTN3A1) and BTN2A1, which are then recognized by Vγ9Vδ2 T cells [96, 97]. The roles of butyrophilin molecules in humans and butyrophilin-like molecules in both humans and mice for γδTCR antigen recognition have been reviewed in detail elsewhere [98–100]. Although the precise mechanism of Vγ9Vδ2 T cell by butyrophilins is still an open area of investigation, Vγ9Vδ2 T cells have clear roles in antitumor immunity [101]. For immunotherapy approaches, Vγ9Vδ2 T cell activation can be targeted in vivo using amino-bisphosphonates, such as zoledronate, which are a class of osteoclastic bone resorption inhibitors used for the clinical treatment of osteoporosis. These drugs additionally inhibit the mevalonate pathway resulting in increased intracellular concentrations of IPP allowing for Vγ9Vδ2 T cell activation and expansion in vivo. Alternatively, Vγ9Vδ2 T cells can be expanded ex vivo using either amino-bisphosphonates or often phosphoantigens such as IPP or HMB-PP directly for ACT approaches [96, 102]. Vγ9Vδ2 T cells expanded in this way have shown enhanced antitumor activity both in vitro and in preclinical mouse tumor xenograft models [103]. Clinical trials using expanded Vγ9Vδ2 T cells have had limited success to date at most inducing partial responses [104], but much work is still devoted to improving ex vivo expansion methods for the ACT. For example, the addition of vitamin C (L-ascorbic acid) derivatives to bisphosphonate-driven Vγ9Vδ2 T cell expansion methods enhances their cellular expansion, cytokine production, and metabolic function [105]. Direct targeting of butyrophilin molecules in vivo, without the need for ex vivo γδ T cell expansion, is also being explored as a therapeutic strategy [97, 106, 107].
Although there is much promise in targeting γδ T cells for antitumor immunity, these therapeutic approaches will additionally have to consider the potential immunosuppressive roles of γδ T cells that can occur after activation through TCR signals. For example, phosphoantigen-activated γδ T cells have been shown to limit αβ T cells responses via expression of programmed cell death ligand 1 (PD-L1) and in the context of cancer vaccination with IL-12-secreting DCs [108, 109]. Furthermore, immunosuppressive FOXP3+ Vδ2+ T cells can be generated in vitro following TCR activation with IPP in the presence of IL-15 and transforming growth factor-β1 (TGF-β1) [110]. Together with other studies that have demonstrated immunosuppressive roles of γδ T cells [111] and potential protumor functions such as IL-17 production [62], these results highlight a critical limitation to overcome in the design of γδ T cell-targeted cancer immunotherapies.
Despite the potential to generate immunosuppressive γδ T cell responses, several other approaches that do not rely on phosphoantigen sensing to exploit γδ T cells for cancer immunotherapy have shown promise in preclinical studies. A protocol for the rapid expansion of highly functional Vδ1+ T cells from human peripheral blood has been developed [112]. These cells, termed Delta One T (DOT) cells, have high expression of NK cytotoxicity receptors, such as NKp30 and NKp44, and exhibit enhanced killing of both chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML) cells [112, 113]. The potential roles for these NK cell receptors on γδ T cell activity have been reviewed elsewhere [31]. Because of their potential use as an allogeneic, “off-the-shelf” ACT therapy, other groups have explored γδ T cells as chimeric antigen receptor (CAR)-T cell vectors [114–116]. Additionally, bi-specific antibodies engineered to co-engage Vγ9 and tumor antigens, such as human epidermal growth factor receptor 2 (HER2) for solid tumors and CD123 for AML, are also in preclinical development [117–119].
Targeting γδ T cell costimulatory molecules is potentially a novel approach to build upon these current strategies being explored to leverage the antitumor activity of γδ T cells. Of note, many groups have developed agonist monoclonal antibodies targeting other T cell costimulatory molecules such as 4-1BB [CD137, tumor necrosis factor superfamily (TNFRSF) member 9], OX40 (CD134, TNFRSF4), CD28, and ICOS (CD278, inducible T cell costimulator), and many of these agents are currently being tested in human clinal trials [120]. Unlike antagonistic anti-PD-1 and anti-cytotoxic T lymphocyte associated protein 4 (anti-CTLA-4) that limit T cell inhibition by block receptor-ligand interactions, these agonist antibodies mediate their effects by clustering costimulatory molecules on the surface of T cells to induce intracellular signaling events that potentiate T cell activation [120, 121]. Despite much work in this area, T cell agonist monoclonal antibodies have had limited success in clinical trials to date, which may be associated with the limited ability of typical bivalent monoclonal antibodies to mediate this receptor clustering [120]. Similar challenges would likely apply to the clinical development of agonist antibodies against the γδ T cell costimulatory molecules discussed above. However, novel engineering approaches such as the use of highly multivalent ligand-antibody fusions and Fc receptor engineering approaches to enhance cross-linking of therapeutic antibodies in trans may overcome these challenges [120, 122, 123]. Additionally, agonist monoclonal antibodies against γδ T cell targets are potentially a useful way to improve the efficacy of either γδTCR x tumor antigen bi-specific antibodies or γδ T cell ACT therapies when used in combination (Figure 2A).
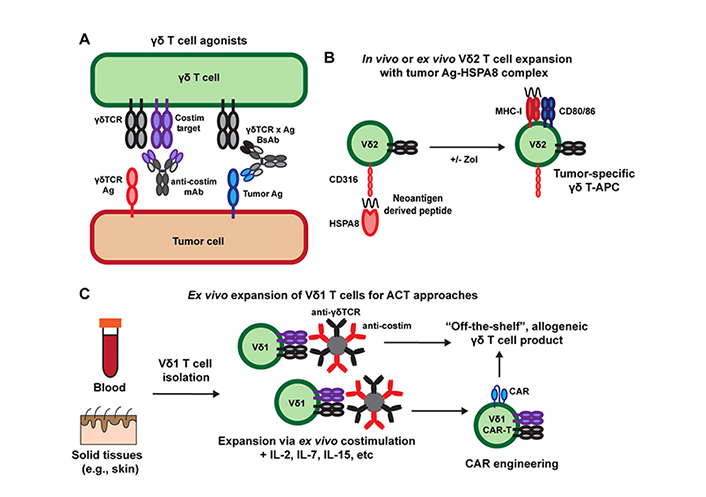
Targeting γδ T cells for cancer immunotherapy. A) γδ T cell agonists include monoclonal antibodies (mAb) and γδTCR x tumor antigen (Ag) bi-specific antibodies (BsAb) that can be used alone, in combination, or after γδ ACT therapy; B) HSPs such as HSPA8 can be used to target tumor neoantigens for uptake by Vγ9Vδ2 T cells to promote their APC function and promotion of broader antitumor immunity. HSP-based vaccines can be combined with Vγ9Vδ2 T cell expansion using bisphosphonate drugs such as zoledronate (Zol) either ex vivo for ACT therapies or directly in vivo; C) addition of costimulatory signals (e.g., with antibody-coated beads) during ex vivo expansion of Vδ1 T cells can be used to generate more functional allogeneic T cell products for the ACT and CAR-T cell approaches
In addition to representing potential targets of agonist monoclonal antibody therapies, CD100 and CD316 have unique roles outside of γδ T cell costimulation that may represent intriguing targets for immunotherapy. Although CD100 has been described as a costimulatory ligand for DETC, its role in cancer appears to be mostly pro-tumorigenic as demonstrated by the antitumor activity of blocking anti-CD100 antibodies in mouse tumor models [53, 60]. Based on these studies, an antagonistic anti-human CD100 antibody (pepinemab) is currently being tested in phase II clinical trials for the treatment of human cancers [124, 125]. If successful in clinical trials, the effect of blocking anti-CD100 antibodies on inhibition of pro-tumor IL-17-producing γδ T cells versus antitumor IFNγ-producing γδ T cells will be of interest and may help inform which solid tumor indications are most suitable for treatment [62]. Similarly, CD316 is not specific to γδ T cell function and has been shown to induce DC activation upon binding to HSPA8 [79]. Given that HSPs have been used in tumor vaccine formulations and that human Vγ9Vδ2 T cells possess antigen-presenting capabilities, it is interesting to speculate that HSPA8-derived tumor vaccines, delivered directly in vivo or in combination with ex vivo expanded Vγ9Vδ2 T cells for ACT therapy, may be a novel approach to activate Vγ9Vδ2 T cell APC function and stimulate endogenous antigen-specific αβ T cells responses [81, 126–128] (Figure 2B). Importantly, recent evidence suggests that Vγ9Vδ2 T cells can both kill tumor targets and cross-present tumor antigens [129]. Whether the use of HSPs and targeting CD316 is a relevant strategy to exploit this activity remains to be explored.
As discussed above, the use of γδ T cells in ACT therapies has garnered much interest because of their potential use as an “off-the-self”, allogeneic T cell therapy without the need for donor-recipient MHC matching [96]. The use of bisphosphonates to expand Vγ9Vδ2 T cells and novel protocols to expand Vδ1+ T cells may prove to be effective immunotherapies, but there is likely much room for improvement in the engineering of these T cell products. One challenge with the development of γδ T cell-based ACT therapies is the limited ability to activate and expand cells ex vivo with more standard T cell costimulatory molecules such as CD28, which are critical for the manufacturing of αβ T cell-based cellular therapies [130, 131]. Vγ9Vδ2 T cells typically express much lower levels of CD28 compared to αβ T cells and decrease expression upon activation, which may limit the use of CD28 costimulation in Vγ9Vδ2 T cell expansion [132–135]. Furthermore, tissue-resident Vδ1+ T cells in humans may be the best population to utilize in ACT therapy based on their intrinsic ability to sense signals of self-stress due to tissue damage and malignancy and function within non-lymphoid tissues. However, research on mouse tissue-resident γδ T cells, such as DETC, has found that these subsets typically lack expression of CD28 and therefore require other signals for activation, which may also apply to human Vδ1+ T cells [1]. Thus, costimulation of either Vγ9Vδ2 T cells or tissue-derived Vδ1+ T cells via NKG2D, JAML, CD100, LFA-1, or CD316 may be novel approach to expand these cells ex vivo (Figure 2C). Similar to the development of DOT cell expansion protocols [112], this work would have to be done empirically and may require sequential activation steps utilizing one or more of these costimulatory ligands at a time. If such a strategy is viable, γδ T cells expanded in this way may be ideal candidates for ACT approaches including CAR-T engineering.
Basic research interrogating γδ T cell biology in mice has identified an important role for γδ T cells in tumor surveillance and led to the identification of novel costimulatory ligands that control their activation within non-lymphoid tissues. Despite the evolutionary divergence of mouse and human γδ T cells, these findings in mice have potential implications for the treatment of human cancers. Most notably, γδ T cell-mediated tumor surveillance and cytotoxicity mediated by NKG2D has clear parallels in mouse and human, and studies of NKG2D’s function on γδ T cells have helped lead to the development of agents targeting NKG2D that are currently being tested in human clinical trials [136]. Although less well characterized, other γδ T cell costimulatory ligands such as JAML, CD100, LFA-1, and CD316 may also represent important targets for cancer immunotherapy. Future studies of these costimulatory molecules and the identification of additional novel mechanisms of γδ T cell activation will be important to leverage the unique properties of these cells for successful cancer immunotherapy interventions.
ACT: adoptive cell transfer
APCs: antigen-presenting cells
CAR: chimeric antigen receptor
CXADR: coxsackie and adenovirus receptor
DAP10/12: DNAX-activating protein 10/12
DC: dendritic cell
DETC: dendritic epidermal T cells
H60: histocompatibility 60
HSPs: heat shock proteins
HSPA8: heat shock protein family A member 8
ICAM-1: intercellular adhesion molecule-1
IFNγ: interferon-γ
IL-17: interleukin-17
IPP: isopentenyl pyrophosphate
JAML: junctional adhesion molecule-like protein
KGF-1: keratinocyte growth factor-1
LFA-1: lymphocyte function-associated antigen 1
MICA/B: major histocompatibility complex class I polypeptide-related sequence A/B
MHC: major histocompatibility complex
PI3K: phosphoinositide 3-kinase
NK: natural killer
NKG2D: natural killer group 2D
Rae-1: retinoic acid early inducible 1
TCR: T cell receptor
Tcrd−/−: T cell receptor delta-deficient
TIL: tumor-infiltrating lymphocytes
TME: tumor microenvironment
The authors thank Travis Young (TSRI) and Nelson Bruno (TSRI) for their helpful comments on the scope and clarity of the manuscript.
JMM and DAW wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by NIH grants AI144825 and AI154918 (DW). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Xiangjin Zhang ... Wei He
Huidi Wang ... Jia Yin
Chang Liu ... Wei He
Yue Wang ... Wei He
Rupert Handgretinger ... Manon Queudeville
Gabrielle M. Ferry, John Anderson
Juan-Pablo Cerapio ... Jean-Jacques Fournie
Alessandro Poggi, Maria Raffaella Zocchi
Navdeep Jhita, Sunil S. Raikar
Jessica Da Gama Duarte ... Andreas Behren
