 Review
Review

Affiliation:
1Department of Clinical Pharmacy, China Pharmaceutical University, Nanjing 211198, Jiangsu, China
ORCID: https://orcid.org/0009-0004-8209-2478
Affiliation:
2Department of Pharmacognosy, China Pharmaceutical University, Nanjing 211198, Jiangsu, China
Affiliation:
1Department of Clinical Pharmacy, China Pharmaceutical University, Nanjing 211198, Jiangsu, China
ORCID: https://orcid.org/0000-0001-7520-5105
Affiliation:
1Department of Clinical Pharmacy, China Pharmaceutical University, Nanjing 211198, Jiangsu, China
ORCID: https://orcid.org/0009-0007-5157-4337
Affiliation:
3Department of Pharmacy, China Pharmaceutical University, Nanjing 211198, Jiangsu, China
Affiliation:
4Department of Pharmacy, Volta River Authority Hospital, Takoradi 237, Ghana
Affiliation:
1Department of Clinical Pharmacy, China Pharmaceutical University, Nanjing 211198, Jiangsu, China
Email: zhxh@cpu.edu.cn
ORCID: https://orcid.org/0000-0002-6375-4497
Explor Immunol. 2026;6:1003247 DOI: https://doi.org/10.37349/ei.2026.1003247
Received: October 02, 2025 Accepted: March 23, 2026 Published: April 16, 2026
Academic Editor: Calogero Caruso, University of Palermo, Italy
Chronic prostatitis/chronic pelvic pain syndrome (CP/CPPS) is a debilitating condition of the urogenital system, with an elusive and multifactorial pathogenesis. Recent data show that there is a potential interplay between dysregulated autophagy and altered exosomal communication that may contribute to the persistent inflammation and pain characteristic of CP/CPPS. This review synthesizes recent advances to propose a hypothetical model: cellular stress in the prostate may trigger dysfunctional autophagy, which could reprogram secreted exosomes biogenesis and cargo in a series of lipid-raft microdomain-involved mechanisms and secretory autophagy. The outcomes of this process include the release of pro-inflammatory cytokines (e.g., IL-1β, TNF-α) enriched exosomes, damage-associated molecular patterns (DAMPs), microRNAs (e.g., miR-155), and fibrotic mediators (e.g., TGF-β1). These signalosomes are hypothesized to transmit the inflammatory and nociceptive signals and may contribute to coordinating the dysregulation of immune cells (such as the M1 polarization of macrophages), sensitization of neurons, and tissue fibrosis, thereby potentially perpetuating the presence of a chronic disease. We critically assess the available evidence based on human studies, animal models, and in vitro systems, but recognize that there is a present requirement for additional CP/CPPS-specific mechanistic evidence. Furthermore, we explore the translational implications of this axis, discussing its promise for yielding novel exosome-based diagnostic biomarkers and its potential as a therapeutic target, while also highlighting the significant preclinical challenges and risks that must be overcome. Ultimately, the autophagy-exosome axis presents a new, integrative concept of CP/CPPS, which shifts the paradigm to the mechanisms of intercellular communication and provides new possibilities to carry out mechanism-selective research and future treatment options.
Chronic prostatitis/chronic pelvic pain syndrome (CP/CPPS) is a complex clinical syndrome of multifactorial etiology and pathogenesis, involving numerous dysregulated cellular and molecular pathways [1, 2]. The most common but least understood prostate disease, according to the classification by the National Institutes of Health (NIH), is referred to as type III prostatitis or CP/CPPS. Current pathophysiological theories have implicated immunologic dysregulation, neuroendocrine cross-talk, and pelvic floor dysfunction as potential contributors to its development and persistence [3, 4]. Recently, cellular processes like autophagy and intercellular communication have emerged as areas of interest, as their dysregulation may represent an additional layer of complexity in potentially contributing to CP/CPPS pathogenesis [5].
Autophagy is an evolutionarily conserved lysosomal degradative pathway, crucial for cellular homeostasis, playing a context-dependent “double-edged sword” role in inflammation and immunity [6, 7]. Exosomes, one of the major subclasses of extracellular vesicles (EVs), are nano-sized, lipid-bilayer vesicles produced by cells that carry a diverse cargo of proteins, lipids, mRNA, and miRNA [8, 9]. There is an attractive new paradigm that indicates the existence of a dynamic relationship between autophagy and exosome biogenesis and secretion [10, 11]. We propose the hypothesis that dysfunctional autophagy in prostate cells may not only contribute to inflammation and cell damage but also alter the quantity and molecular cargo of released exosomes, thereby potentially dysregulating immune and pain pathways within the prostate microenvironment. This pathway of the autophagy-exosome axis, therefore, represents a plausible contributing pathway in the pathology of CP/CPPS, which warrants further investigation, potentially facilitating the activation of inflammatory cells, the release of inflammatory factors, and cross-cellular communication between epithelial, stromal, and immune cells [10, 12]. In this review, we summarize recent advances in understanding this interplay, critically evaluate its proposed role in CP/CPPS pathogenesis, and discuss its potential for revealing novel diagnostic and therapeutic targets. The exosomes are distinguished by a discrete lipid-separated bilayer structure and certain surface markers, including CD63 and CD81, which make them very stable and biocompatible [13, 14]. The autophagic state of the parent cell can influence the molecular cargo of exosomes, such as autophagy-related (ATG) proteins and miRNAs [15, 16]. This interplay positions the autophagy-exosome axis as a plausible and contributory mechanism in the pathogenesis of CP/CPPS.
For instance, exosomes purified from prostate secretions or urine could yield ATG biomarkers (e.g., LC3-II, p62), aiding in patient stratification and guiding therapy development [17, 18]. Furthermore, elucidating how autophagy regulates exosome-mediated inflammation and pain signaling is key to advancing our understanding of the disease and identifying new therapeutic avenues [12, 19, 20]. Investigating this axis requires a multi-model approach, integrating human clinical data, animal models, and in vitro studies to move from correlation to causality and mechanistic insight [21, 22]. Furthermore, recent mechanistic insights into lipid rafts and membrane microdomains provide a crucial conceptual framework for this interplay. These cholesterol- and sphingolipid-rich platforms are now understood to be physical and functional hubs that link autophagy, intracellular trafficking, and EV biogenesis [23]. Notably, processes such as secretory autophagy and the autophagy-driven enrichment of specific lipid raft components (e.g., GD3, ERLIN1) into EVs offer tangible molecular pathways through which dysregulated autophagy could reprogram exosomal cargo and signaling [24, 25]. Integrating these concepts is essential for constructing a robust pathogenic model of CP/CPPS.
In essence, type III CP/CPPS is a common urogenital disease of multifarious and heterogenous pathogenesis. The interplay between autophagy and exosomal communication represents a significant, integrative facet of its pathophysiology. A deeper investigation of this axis may therefore yield novel insights with the potential to improve diagnostic accuracy, therapeutic efficacy, and patient quality of life.
To capture relevant advances in this emerging field, a comprehensive literature search was conducted for articles published between January 2015 and April 2025. Given the hypothesis-generating nature of this review, we employed a broad, iterative search strategy rather than a formal systematic review protocol. Combined search terms using Boolean operators were used to query the electronic databases PubMed, Scopus, and Web of Science, including the following combinations:
(“chronic prostatitis” OR “chronic pelvic pain syndrome” OR “CPPS”) AND (“autophagy” OR “exosome” OR “extracellular vesicle” OR “microRNA” OR “miR”).
Inclusion criteria encompassed: (1) original research articles and high-impact reviews written in English; (2) studies exploring the molecular mechanisms of inflammation, pain, or intercellular communication as applied to prostate, pelvic, or related chronic inflammatory disease models; and (3) studies investigating autophagy, vesicle biology, or related processes.
Exclusion criteria were: (1) only studies that researched acute or bacterial prostatitis (NIH Category 1/2); (2) non-English articles; (3) conference abstracts with no related corresponding manuscripts of full-length and peer-reviewed publication. Hand screening of the reference lists of the key papers identified was also done to find more relevant studies.
It is necessary to clarify that this review is not intended to give a quantitative synthesis characteristic of systematic reviews or meta-analyses. Instead, this comprehensive yet non-systematic approach has been purposefully selected to gather and integrate the evidence of different directions, such as urology, cell biology, immunology, and oncology, to facilitate the emergence of a new hypothesis-driven conceptual framework of CP/CPPS pathogenesis. The goal is to integrate the emerging ideas into an integrative framework that has the potential of producing testable hypotheses and informing the future mechanistic studies and not to give a final evidence grade to clinical recommendations.
Intercellular signaling and autophagy are integral to maintaining cellular homeostasis within the prostate. Autophagy, one of the most conserved lysosomal degradative processes, is particularly essential for cellular quality control, energy balance, and survival [26–28]. Basal autophagy functions as a housekeeping process known to be present in prostatic stromal and epithelial cells, where it participates in clearing damaged organelles, protein aggregates, and pathogens [26, 29]. This is done via the formation of double-membrane autophagosomes that fuse with lysosomes for degradation [30–32]. This autophagy molecular complex is regulated by a set of otherwise referred to ATG proteins that regulate the procedure of its various stages of its induction, formation of an autophagosome, and its destruction [33–35] (Table 1).
ATG proteins in CP/CPPS–evidence and pathogenic implications.
| Protein | Proposed pathogenic consequence in CP/CPPS | Primary function in autophagy | Evidence of dysregulation in CP/CPPS |
|---|---|---|---|
| ULK1 | Stress signaling via mTOR/AMPK may be dysregulated, impairing autophagy initiation in prostate cells, contributing to DAMP accumulation. | Serine/threonine kinase and mammalian ATG1 homolog; forms the ULK1–ATG13–FIP200–ATG101 complex to initiate autophagosome formation. Its activity is directly inhibited by mTORC1 phosphorylation under nutrient-rich conditions and activated by AMPK phosphorylation during cellular stress [93]. | No direct CP/CPPS data reported. Role inferred from its position as a master stress sensor in other chronic inflammatory models. |
| Beclin 1 (BECN1) | If its expression or activity were reduced in CP/CPPS, it could impair phagophore formation, compromising the clearance of damaged components and potentially promoting inflammasome-driven inflammation. | Core subunit of the class III PI3K (PI3KC3) complex; essential for generating PI(3)P to initiate phagophore nucleation and autophagosome formation [94, 95]. | No direct CP/CPPS data reported. Its role is inferred from its established function as a key autophagy regulator and its dysregulation in other inflammatory conditions. |
| LC3 (MAP1LC3B) | The presence of LC3-II in prostate-derived exosomes suggests that autophagic activity may contribute to exosome biogenesis or cargo selection. It represents a potential candidate biomarker for prostate cellular stress. | Ubiquitin-like protein processed to the lipidated form LC3-II (LC3-PE), which stably associates with autophagosomal membranes and serves as the canonical, gold-standard marker for monitoring autophagic flux [96, 97]. | No direct CP/CPPS data reported. The presence of ATG proteins in prostate-derived exosomes from patients remains an unexplored area. |
| ATG5 | Functional deficiency would halt autophagy, leading to the accumulation of damaged organelles and proteins, triggering oxidative stress and cytokine release. | Forms a conjugate with ATG12; essential for phagophore elongation. | No direct CP/CPPS data reported. Comprehensive reviews of CP/CPPS pathogenesis and biomarker studies (e.g., focusing on S100A12, IL-8, oxidative stress mediators) do not include analysis of ATG5 or core autophagy genes, indicating this remains an uninvestigated area [98, 99]. |
| ATG7 | As a master regulator, its dysfunction would broadly disable autophagy, creating a pro-inflammatory cellular state. | E1-like activating enzyme; essential for ATG12-ATG5 conjugation and LC3 lipidation. | No direct CP/CPPS data reported [99, 100]. |
| p62/SQSTM1 | p62 accumulation acts as a persistent signaling hub, activating pro-inflammatory pathways (e.g., NF-κB, NRF2) and promoting chronic tissue inflammation. | Autophagy receptor that links ubiquitinated cargo to LC3; degraded via autophagy. | Significant accumulation in stromal and epithelial cells of CP/CPPS patient prostate biopsies [101]. |
AMPK: AMP-activated protein kinase; ATG: autophagy-related; CP/CPPS: chronic prostatitis/chronic pelvic pain syndrome; DAMP: damage-associated molecular pattern; LC3: microtubule-associated protein 1 light chain 3; mTOR: mammalian target of rapamycin; NF-κB: nuclear factor kappa B; NRF2: nuclear factor erythroid 2-related factor 2; PE: phosphatidylethanolamine; PI3K: phosphatidylinositol 3-kinase; ULK1: Unc-51-like kinase 1.
Exosomes, a major subclass of small extracellular vesicles (sEVs, 40–160 nm), are generated via the endosomal pathway [36–38]. Their generation commences with endosomal membrane inward budding, creating intraluminal vesicles (ILVs) or multivesicular bodies (MVBs). Such MVBs are then able to merge with the plasma membrane, releasing ILVs as exosomes into the extracellular space [37, 39, 40]. A critical connection lies in the convergence of these pathways [11, 15]. Autophagosomes can fuse with MVBs to form amphisomes, which subsequently fuse with the plasma membrane, directly influencing the secretion and cargo of exosomes [41]. This suggests that exosome release from prostate cells may be intrinsically linked to intracellular autophagic activity.
Lipid rafts, specialized membrane microdomains enriched in cholesterol and sphingolipids, have emerged as functional hubs that facilitate a myriad of cellular processes, including signal transduction, protein sorting, and vesicle trafficking. Recent evidence has highlighted a potential role for lipid rafts in the connection between autophagy and EV biogenesis, a process that is essential to maintain cellular homeostasis and to mediate intercellular communication [23]. They are membrane domains which are typically located in the endoplasmic reticulum (ER) and plasma membrane, and the site where autophagy is initiated, especially at the ER-mitochondria-associated membranes (MAMs). Here, we consider how these lipid microdomains might contribute to autophagic vesicle formation, EV secretion, and subsequent use in signaling in the context of inflammatory diseases like CP/CPPS.
MAMs are specialized subcellular compartments where the ER interacts with mitochondria and have been described as playing a central role in the regulation of autophagy. This interaction between the organelles plays a critical role in the initiation of autophagic process, especially when cells are stressed or nutrients deprivation. MAMs contain proteins that are highly important in ATG (e.g., AMBRA1 and ERLIN1), and these proteins mediate the development of autophagosomes. These lipid raft-binding proteins direct the formation of autophagic membranes and their fusion with endosomal vesicles, thereby forming amphisomes, which may either fuse with lysosomes and undergo degradation or with the plasma membrane for unconventional secretion [42]. The involvement of lipid rafts in these processes suggests that, in addition to structural roles in the make-up of cellular membranes, they may also possess functional significance in the regulation of autophagic activity.
A key facet of autophagy that has often been overlooked in traditional paradigms is its contribution to secretory pathways. Recent studies have introduced the concept of secretory autophagy, where autophagic vesicles, instead of fusing with lysosomes for degradation, fuse with MVBs to form hybrid vesicles called amphisomes, which are then released as exosomes. This process, which is known as unconventional secretion, provides a mechanism for cells to release proteins, lipids, and RNA into the extracellular space [24]. The pathway is specifically relevant in the light of inflammatory diseases, where autophagy is not only involved in the process of eliminating the destructive cellular components but also in the discharge of inflammatory mediators through EVs. By promoting the transfer of modified proteins and signaling molecules, secretory autophagy could theoretically play a crucial role in shaping the inflammatory milieu in conditions like CP/CPPS, though this remains to be directly demonstrated. The cargo carried by EVs is fundamentally altered by autophagic processes, particularly through the regulation of lipid raft components. During autophagy induction, key raft markers such as GD3, a ganglioside, and ERLIN1, an ER membrane-associated protein, become enriched within EVs. This cargo modulation may be relevant to the functional properties of the liberated vesicles, potentially influencing their capacity to affect recipient cells. It has been demonstrated that the dynamic interactions between autophagy and lipid raft components in MAMs favor the selective cargo packaging into EVs. Notably, the association between LC3-II, which is an indicator of autophagosomal membranes, and raft components, such as GD3, indicates that autophagy may promote the inclusion of certain lipid and protein molecules in EVs secreted to the extracellular environment [25]. This process ensures that EVs not only carry intracellular components but also play a central role in propagating cellular signals.
We propose a hypothetical model wherein dysregulated autophagy, when triggered by stress, could lead to alterations in the dynamics of MAMs and lipid rafts. In the context of CP/CPPS, this dysregulation could result in the improper modulation of secretory autophagy and exosome biogenesis, particularly through lipid-modifying enzymes such as ceramide synthases and neutral sphingomyelinase 2 (nSMase2). These enzymes, which are critical for the generation of raft-like membranes, could be altered in CP/CPPS, potentially leading to the reprogramming of exosomal cargo. To be more specific on this, we propose that this reprogramming leads to the enrichment of inflammatory cytokines [e.g., TGF-β1, nerve growth factor (NGF)], damage-associated molecular patterns (DAMPs), and miR-155, molecules known to exacerbate the inflammatory processes characteristic of CP/CPPS. By altering the lipid and protein composition of EVs, this model suggests that stress-induced autophagy may serve as a key mechanism through which inflammation is perpetuated and propagated in CP/CPPS (Table 2).
Key aspects of exosome biogenesis, cargo, and their link to autophagy in CP/CPPS.
| Aspect | Key components/Markers | Function/Significance | Link to autophagy in CP/CPPS |
|---|---|---|---|
| Biogenesis pathway | ESCRT complexes (ESCRT-0, -I, -II, -III), ALIX, TSG101 | Mediates inward budding of the endosomal membrane to form ILVs inside MVBs. | Autophagosomes can fuse with MVBs to form amphisomes, directly influencing exosome secretion and cargo [15, 41, 44]. |
| Key surface markers | Tetraspanins (CD63, CD81, CD9), HSP70, MHC classes | Used for exosome identification and isolation. Indicate endosomal origin, and can influence cellular uptake. | General principle: Cellular stress and altered autophagic flux can remodel the exosome surface proteome, potentially affecting biodistribution [10, 44, 45, 102]. CP/CPPS evidence: This specific link remains unexplored in prostate-derived exosomes. |
| Pro-inflammatory cargo | IL-1β, IL-18, TNF-α, HMGB1 (DAMP) | Drives inflammation and immune cell activation. Central to CP/CPPS pathology. | Impaired autophagy leads to the accumulation of these molecules inside the cell, increasing their loading into exosomes [6, 56, 103]. |
| Immunomodulatory miRNAs | miR-155, miR-21, miR-146a | Key regulators of immune responses (e.g., miR-155 promotes M1 macrophage polarization). | Autophagy regulates miRNA levels. Dysfunctional autophagy alters the miRNA profile of exosomes, shifting the immune response toward pro-inflammation [104–106]. |
| Pain and neural cargo | NGF, BDNF, cytokines | Mediates neuronal sensitization, contributing to chronic pelvic pain. | General principle: Exosomes from stressed cells (e.g., glial, cancer) carry pain mediators that directly sensitize sensory neurons, establishing a role in chronic pain [107, 108]. Specific link to autophagy: The hypothesis that autophagy deficiency in prostate cells enhances this exosomal pain signaling remains to be tested in CP/CPPS models. |
| Pro-fibrotic cargo | TGF-β1, fibronectin, collagen | Promotes differentiation of fibroblasts into myofibroblasts, leading to tissue fibrosis. | Autophagy is frequently induced by and required for TGF-β1 profibrotic signaling [109–111]. Inhibition of autophagy (e.g., ATG5/7 loss) attenuates TGF-β1-driven collagen/fibronectin production [109, 112]. The potential enhancement of exosomal TGF-β1 release under autophagy impairment remains less established. |
ATG: autophagy-related; BDNF: brain-derived neurotrophic factor; CP/CPPS: chronic prostatitis/chronic pelvic pain syndrome; DAMP: damage-associated molecular pattern; ESCRT: endosomal sorting complex required for transport; ILVs: intraluminal vesicles; MHC: major histocompatibility complex; MVBs: multivesicular bodies; NGF: nerve growth factor; TGF-β1: transforming growth factor beta 1; TNF-α: tumor necrosis factor-alpha.
Emerging evidence points to a potential bidirectional yet dynamic interaction between autophagy and exosome biology, which may play a major role in shaping the microenvironment at the prostate. Autophagy has been proposed as a key regulator in exosome composition and exosome release. Under cellular stress, autophagic activity can significantly influence the packaging of proteins, lipids, and nucleic acids into exosomes [10, 15, 43]. As an example, impaired autophagy may result in the formation of certain damage-related molecular patterns (DAMPs) such as HMGB1 or mitochondrial DNA, which can be wrapped in exosomes and released [43, 44]. Furthermore, ATG and miRNAs regulating the autophagic process (e.g., miR-30a, miR-224) are themselves exosomal cargos, and enable a cell to signify its autophagic state to other cells [11, 15].
Conversely, the exosomes may also have the capability of regulating the autophagic process in recipient cells in the prostate tissue. Inflamed or stressed cells can release exosomes containing certain miRNAs, proteins, or signaling molecules, which activate/inactivate the autophagic machinery of the target cells (immune cells or neurons) [15, 45, 46]. This is exemplified by the case of exosomal miR-21, which is capable of suppressing autophagy in target cells, which instigates an inflammatory response [46, 47]. Furthermore, exosomal miR-155, a miRNA that is elevated in the tissues involved in CP/CPPS, is a strong inducer of M1 macrophage polarization by targeting suppressors of cytokine signaling (SOCS1) and SHIP1, thereby amplifying pro-inflammatory responses [48]. This suggests a potential feed-forward or feedback loop: a cell’s autophagic state may influence its exosomal output, which in turn can modulate autophagy in recipient cells, thereby potentially amplifying or resolving inflammatory signals.
Dysregulation of the autophagy-exosome interplay represents a plausible mechanism that could contribute to the pathogenesis of type III CP/CPPS. Autophagy impairment in prostate epithelial cells has been shown to lead to the accumulation of dysfunctional mitochondria and protein aggregates in other cell types, and may similarly occur in the prostate [10, 41]. This not only causes oxidative stress and activation of inflammasomes inside the cell, but also alters. These exosomes produced by these stressed cells are enriched with pro-inflammatory cytokines (e.g., IL-1β, TNF-α), DAMPs, and certain miRNAs, which can polarize resident immune cells, such as macrophages, they polarize them towards a pro-inflammatory (M1) phenotype, potentially contributing to a chronic inflammatory state [49–52].
This dysfunctional exosomal communication could theoretically further exacerbate disease progression, though direct evidence in CP/CPPS is lacking. Such pro-inflammatory exosomes are capable of spreading the message of inflammatory actions to other healthy cells and diffusing the inflammatory response [53, 54]. A critical implication of this axis is its potential role in pelvic pain and neuroinflammation. Pain mediators (e.g., NGF, cytokines) and autophagy-modulating miRNAs released by exosomes because of inflamed prostate tissue may be transferred to prostate sensory neurons. This exosome-mediated cross-talk may contribute to peripheral sensitization, is capable of sensitization, lowered pain thresholds, and the development of chronic pelvic pain of CP/CPPS [13, 14, 55].
The proposed mechanisms through which the autophagy-exosome axis contributes to inflammation, fibrosis, and neuronal sensitization precondition a more in-depth analysis of the contribution of the autophagy-exosome axis to immune dysregulation and its potential as a target for novel therapeutic and diagnostic strategies, which will be further discussed in the following sections.
The interplay between dysregulated autophagy and exosome release could create a pathogenic feed-forward loop that might fuel inflammation in CP/CPPS, as illustrated in our hypothetical model in Figure 1. A key mechanism is the role of autophagy in modulating the NLRP3 inflammasome [56–58]. Impaired autophagy in prostate epithelial cells has been associated with the accumulation of damaged mitochondria (a major source of ROS) and protein aggregates, which are potent activators of the NLRP3 inflammasome in various disease models. This could potentially lead to excessive pro-inflammatory cytokine secretion, such as IL-1b and IL-18, in the context of CP/CPPS [56, 59].
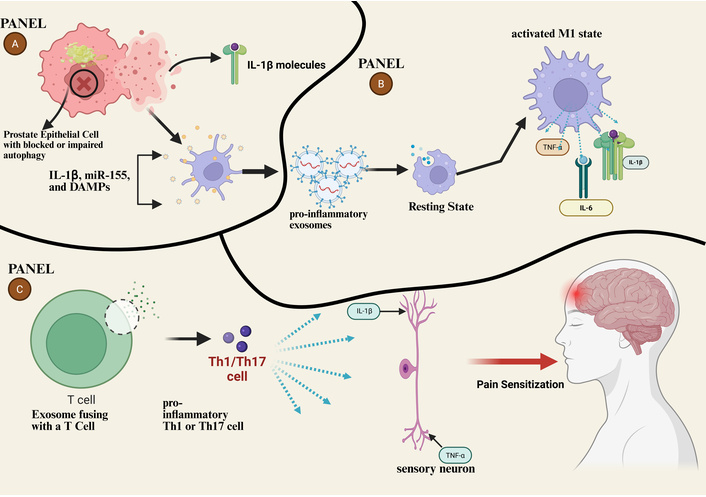
Hypothetical model of the autophagy-exosome axis in CP/CPPS pathogenesis. (A) In prostate epithelial cells, stress or dysfunction leads to impaired autophagic flux (blocked autophagosome-lysosome fusion) and NLRP3 inflammasome activation. This results in the secretion of exosomes loaded with pro-inflammatory cargo (IL-1β, miR-155, DAMPs). (B) These exosomes are taken up by resident immune cells, driving M1 macrophage polarization, which amplifies a local cytokine storm (TNF-α, IL-6, IL-1β) and contributes to pain sensation. (C) Exosomes also fuse with T cells, promoting Th1/Th17 cell responses. The resulting inflammatory milieu (TNF-α) sensitizes peripheral sensory neurons, contributing to neuroinflammation and the chronic pelvic pain characteristic of CP/CPPS. CP/CPPS: chronic prostatitis/chronic pelvic pain syndrome; DAMPs: damage-associated molecular patterns; TNF-α: tumor necrosis factor-alpha; NLRP3: NACHT, LRR and PYD domains-containing protein 3. Created in BioRender. Nyame, D. K. (2026) https://BioRender.com/3339m7v.
Crucially, this inflammatory response is not confined to the originating cell. Exosomes provide a direct link between autophagy deficiency and intercellular signaling. These active cytokines, DAMPs, and even inflammasome components can be packaged into exosomes by autophagy-deficient cells. These exosomes then act as carriers, transmitting inflammatory signals to neighboring cells and recruiting immune cells to the site, thereby amplifying the pro-inflammatory response throughout the prostate tissue [44, 60–62].
Furthermore, exosomes can transfer regulating miRNAs that have the capacity to control autophagy of target cells. As an example, exosomal miR-155. For example, exosomal miR-155, upregulated in CP/CPPS, can suppress autophagy in recipient macrophages, promoting their polarization to a pro-inflammatory M1 phenotype. This occurs through targeting of SOCS1 and SHIP1, exacerbating tissue inflammation [46, 48]. This establishes a potential self-amplifying cycle: autophagy dysfunction promotes the release of pro-inflammatory exosomes, which in turn can suppress protective autophagy in target cells, potentially entrenching a chronic inflammatory state.
The autophagy-exosome axis represents a mechanism that could influence prostate cell fate decisions: survival, death, and fibrosis, in CP/CPPS, though this requires direct experimental validation.
Proliferation and Apoptosis: Under normal conditions, autophagy promotes cell survival by clearing damaged components. In CP/CPPS, it is possible that autophagic flux could be inhibited by sustained stress, potentially resulting in a certain proportion of cells undergoing apoptosis. These cells have the ability to secrete exosomes with pro-apoptotic cargo (e.g., caspase-3, select miRNAs) before dying. These “death exosomes” could, in turn, activate apoptotic mechanisms in neighboring healthy cells, theoretically propagating tissue damage [63–67]. Conversely, compensatory proliferative signals and growth factors (e.g., TGF-β1) may also be delivered via exosomes while further disrupting tissue homeostasis.
Fibrosis is a critical outcome of chronic inflammation in CP/CPPS, which contributes to pain and voiding dysfunctions. In other disease contexts, autophagy has been shown to act abnormally as a check on fibrosis, by degrading key mediators like TGF-β1. Extrapolating from these findings, when autophagy is impaired, TGF-β1 signaling may become enhanced in prostate tissues. Furthermore, the activated cells can package TGF-β1, among other pro-fibrotic signals, into exosomes. These exosomes potently stimulate prostate stromal fibroblasts to differentiate into collagen-producing myofibroblasts and result in tissue scarring, which is one of the major processes of chronicity of the disease [68, 69] (Figure 2).
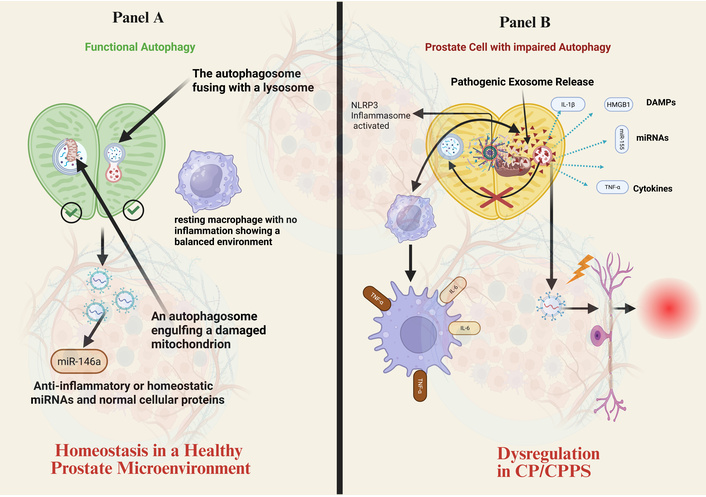
Autophagy dysfunction in prostate epithelial cells alters exosome-mediated intercellular communication. (A) Homeostasis: In a healthy prostate microenvironment, functional autophagy efficiently clears damaged organelles (e.g., mitochondria). Exosomes released under these conditions carry anti-inflammatory or homeostatic cargo (e.g., miR-146a), contributing to immune balance. (B) Dysregulation in CP/CPPS: Impaired autophagy (e.g., blocked autophagosome-lysosome fusion) leads to NLRP3 inflammasome activation and cellular stress. This results in the release of pathogenic exosomes enriched with pro-inflammatory cytokines (IL-1β, IL-6, TNF-α), damage-associated molecular patterns (DAMPs, HMGB1), and specific miRNAs. These exosomes propagate inflammation by activating immune cells (e.g., polarizing macrophages to an M1 phenotype) and contribute to neuronal sensitization, driving the pathophysiology of CP/CPPS. CP/CPPS: chronic prostatitis/chronic pelvic pain syndrome; NLRP3: NACHT, LRR and PYD domains-containing protein 3. Created in BioRender. Nyame, D. K. (2026) https://BioRender.com/iekaor6.
The autophagy-exosome axis plays a proposed key role in the immune dysregulation of CP/CPPS. Stressed prostate cells secrete exosomes in which the cargo is determined by their autophagic state.
Macrophage polarization: As outlined in Linking dysregulated autophagy with exosome-mediated inflammation, exosomes from autophagy-deficient prostate cells, enriched with miR-155 and other cargo, can drive M1 macrophage polarization [70]. These M1 macrophages secrete high amounts of IL-1β, TNF-α, and ROS, causing inflammation. In contrast, exosomes from cells with normal autophagic flux may carry cargo that supports the anti-inflammatory M2 phenotype, promoting resolution. This balance is likely dysregulated in CP/CPPS [71, 72].
T-cell regulation: Exosomes can present antigens and carry immunomodulatory molecules that directly influence T-cell activity. For instance, exosomes from inflamed prostate tissue can carry immune checkpoint proteins like PD-L1. Upon delivery to T-cells, this can induce an exhausted T-cell phenotype, dampening anti-inflammatory responses and permitting uncontrolled inflammation [73–76]. Autophagy also contributes to exosome-mediated immune regulation indirectly, as efficient antigen processing and presentation in professional antigen-presenting cells (APCs) are autophagy-dependent processes.
A novel mechanism: Sterile inflammation through exosomal RNA: Emerging evidence suggests exosomes can deliver specific RNAs that trigger innate immune responses. For example, exosomal TRPM8 mRNA from prostate cells could be transferred to immune cells. If internalized, this RNA might bind with endosomal toll-like receptor 3 (TLR3), which triggers the NF-κB/IRF3 signaling pathway and leads to the secretion of type 1 interferons and other pro-inflammatory cytokines, which ultimately causes a state of sterile inflammation [73–75]. This pathway represents a potential link between the prostate-specific protein (TRPM8) and the activation of immunity by means of exosomes of CP/CPPS.
The inherent biological properties of exosomes, such as stability, biocompatibility, and targeting potential, make them an attractive candidate for novel therapeutic strategies of CP/CPPS. It is crucial to emphasize that these approaches remain largely preclinical (Figure 3).
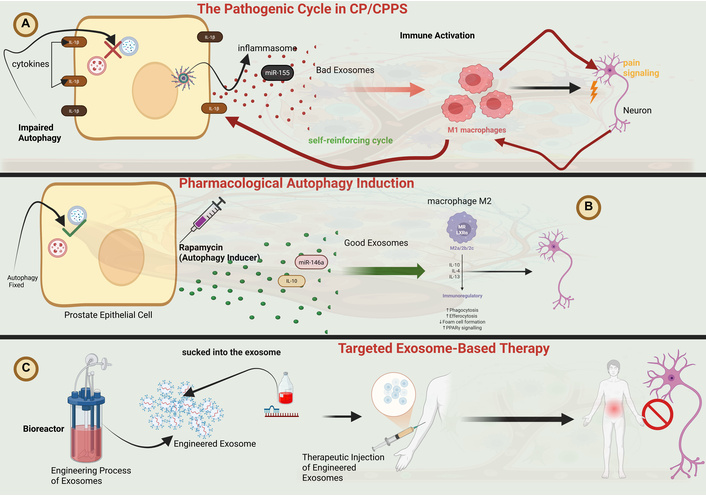
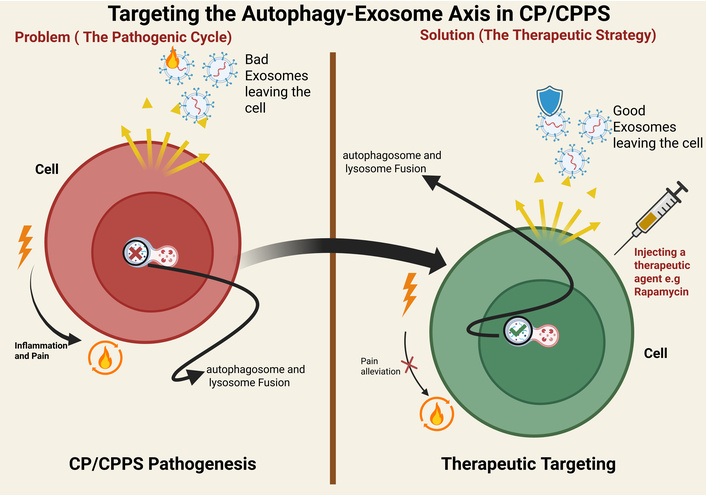
Strategic overview of therapeutic interventions targeting the autophagy-exosome axis in CP/CPPS. The schematic contrasts the self-reinforcing pathogenic cycle (left) with three potential therapeutic strategies (right, panels A–C). The pathogenic cycle: Impaired autophagy in prostate epithelial cells leads to the release of “bad” pro-inflammatory exosomes, which drive immune activation (M1 macrophage polarization), inflammasome signaling, cytokine release, and neuronal pain signaling. Therapeutic strategies: (A) Inhibition of pathogenic exosome release: Pharmacologically disrupting exosome biogenesis/secretion can break the cycle. (B) Pharmacological autophagy induction: Agents like rapamycin can restore autophagic flux, promoting the release of "good" exosomes with anti-inflammatory cargo (e.g., IL-10) that support an M2 macrophage phenotype. (C) Targeted exosome-based therapy: Engineered exosomes loaded with therapeutic cargo (e.g., anti-inflammatory cytokines, miRNA antagonists like anti-miR-155) can be produced ex vivo and injected to directly neutralize inflammatory signals in target cells (e.g., M1 macrophages) and block pain pathways. CP/CPPS: chronic prostatitis/chronic pelvic pain syndrome. Created in BioRender. Nyame, D. K. (2026) https://BioRender.com/e8u0f5o.
Engineered exosomes can be designed as carriers to deliver bioactive molecules that modulate the autophagic process in recipient cells. The multifunctional mechanism of such engineered exosomes is illustrated in Figure 4. For example, mesenchymal stem cells (MSCs)-derived exosomes, which carry anti-inflammatory (e.g., miR-21, miR-146a), have been shown to promote autophagy and induce an anti-inflammatory M2 macrophage phenotype in other inflammatory models, suggesting a potential therapeutic avenue for CP/CPPS [15, 41, 77, 78]. Such exosomes may be delivered intraprostatically or alone, as a systemic (to provide a localized dose of healing cues to the inflamed tissue).
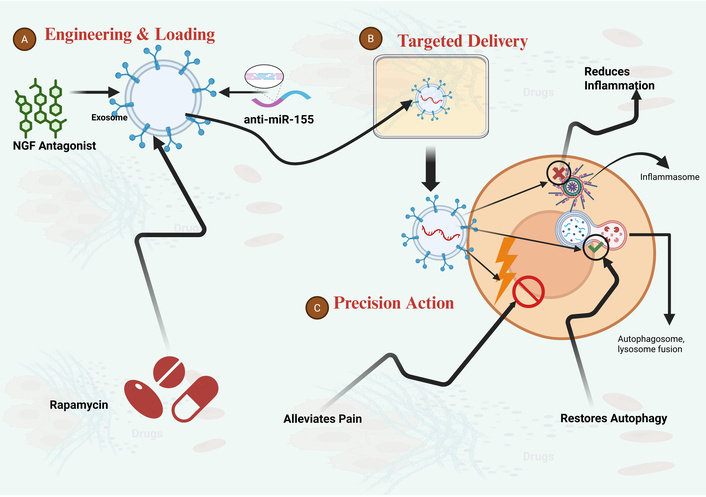
Mechanism of engineered exosomes as multifunctional therapeutics for CP/CPPS. (A) Engineering and loading: Exosomes are engineered in vitro to be loaded with a combination of therapeutic cargoes. This can include autophagy inducers (e.g., rapamycin), anti-inflammatory nucleic acids (e.g., anti-miR-155 antagonists), and pain pathway modulators [e.g., nerve growth factor (NGF) antagonists]. (B) Targeted delivery and anti-inflammatory action: Following administration, these multifunctional exosomes home to the inflamed prostate microenvironment. Upon fusion with target cells, they deliver their payload to suppress pro-inflammatory signaling (e.g., NLRP3 inflammasome) and reduce inflammation. (C) Restoration of autophagy and pain alleviation: The delivered cargo simultaneously restores autophagic flux (promoting autophagosome-lysosome fusion) and blocks pain sensitization pathways, thereby alleviating pain and addressing multiple facets of CP/CPPS pathology. CP/CPPS: chronic prostatitis/chronic pelvic pain syndrome; NLRP3: NACHT, LRR and PYD domains-containing protein 3. Created in BioRender. Nyame, D. K. (2026) https://BioRender.com/yh8pj1s.
Targeted delivery can be enhanced by functionalizing exosome surface with ligands (e.g., PSMA-specific peptides) to improve prostate-specific delivery, potentially increasing local efficacy and reducing systemic side effects [79, 80]. However, achieving consistent targeting efficiency in vivo remains a significant technical challenge (Figure 4).
A complementary strategy involves using small molecules or repurposed drugs to modulate autophagic activity at its source, aiming to normalize dysfunctional exosomal communication.
Autophagy inducers: Drugs like rapamycin (an mTOR inhibitor) or metformin can enhance autophagic flux. By improving cellular clearance, they may reduce the release of pro-inflammatory exosomes, promoting a more homeostatic microenvironment. This approach has shown promise in rodent models of prostatitis but requires clinical validation [81, 82].
Autophagy inhibitors: Given the “double-edged sword” nature of autophagy, transient inhibition (e.g., with chloroquine) might be theorized for contexts where excessive autophagy contributes to cell death. However, this approach is highly problematic due to the critical timing, cell-type specificity, and risk of exacerbating inflammation by impairing a homeostatic process [83, 84].
Targeting exosome release: Inhibitors of exosome biogenesis/release, for example, the nSMase2 inhibitor GW4869, which can disrupt the ceramide-dependent pathway usually linked to lipid rafts, have been effective in the reduction of inflammation and pain in preclinical prostatitis models, which provides evidence-of-concept for targeting the secretory arm of the axis [25, 85–87].
Beyond therapeutics, the autophagy-exosome axis holds significant promise for diagnostics and stratification of patients. Exosomes isolated from accessible biofluids like urine or expressed prostatic secretions offer a “liquid biopsy”, providing a real-time molecular snapshot of the prostate microenvironment.
A diagnostic panel could integrate: (1) ATG markers (e.g., LC3-II, p62) to assess cellular stress status; (2) an inflammatory signature (e.g., exosomal IL-8, miR-155, miR-21) to gauge immune activity; and (3) pain/fibrosis mediators (e.g., NGF, TGF-β1). Validating such a panel in large patient cohorts could enable molecular stratification: identifying subtypes such as “high-inflammatory”, “fibrotic”, or “neuropathic”, moving beyond purely symptom-based classification towards personalized management [18, 88–92].
While the autophagy-exosome axis represents a compelling therapeutic and diagnostic target, translating the concept into clinical practice for CP/CPPS is fraught with significant challenges that must be assessed critically and soberly.
The enthusiasm for exosome-based therapies should be tempered by substantial practical and safety concerns. To begin with, there is a profound inadequacy of effective data on CP/CPPS-specific models. Most supporting data have been extrapolated from oncology, neurodegenerative, or other inflammatory conditions. Positive outcomes within these systems may not translate to the unique pelvic neuroimmune microenvironment of CP/CPPS. Second, safety profiles are to a great extent undefined. The long-term consequences of administering engineered exosomes are unknown. According to the field of oncology, it is valid that exogenous exosomes would enhance tumorigenesis or metastasis, be immunogenic, or lead to off-target effects. Third, formidable technical barriers exist, and the scalable manufacturing of clinical-grade, homogeneous exosome batches is complex and prohibitively expensive. Furthermore, achieving high drug-loading efficiency in the field and consistent in vivo targeting to the prostate remains a major unsolved problem.
The pharmacological modulation of autophagy is also a difficult task due to its context-dependent nature of a “double-edged sword”. Autophagy inducing agent, such as rapamycin, can solve a single cell type or disease stage, but may worsen or cause another cell type to undergo autophagic cell death. Conversely, an inhibitor may be applicable in a hyper-autophagic state but may aggravate the disease by inhibiting an essential homeostatic mechanism. The above paradox requires that specific biomarkers need to be developed to determine the predominant autophagic phenotype in a particular patient prior to any treatment.
For the advancement of the field, a disciplined, sequential research agenda is essential. Immediate priorities must shift from purely conceptual models to foundational validation:
Validate the pathogenic role in human CP/CPPS: Large-scale correlative studies are needed to definitively link specific exosomal cargo signatures (e.g., autophagy proteins, miR-155), and autophagic flux markers of patient biofluids are associated with clinical phenotype, disease severity, and treatment response.
Develop robust preclinical models: More complex animal models that better recapitulate the chronicity and neuroimmune complexity of human CP/CPPS are required to reliably test axis-targeting therapies.
Standardize methodologies: Widespread adoption of MISEV guidelines to isolate and characterize EV/exosomes is non-negotiable to make the process reproducible and to be able to make significant comparisons across studies.
Conduct rigorous early-phase studies: There should be thorough preclinical toxicology and dose-finding studies prior to any advancement to clinical application. Particular attention should be paid to the risks outlined above.
Ultimately, the path forward is not to prematurely champion unproven therapies, but to carefully de-risk the underlying biology. The main goal must be to find conclusive evidence that the modulation of the autophagy-exosome axis in a specific way can safely and effectively alter the progression of CP/CPPS in humans. These strategies can be regarded as high-potential and high-risk avenues in exploratory research until such evidence is available.
The complex and heterogeneous pathogenesis of type III CP/CPPS remains a major barrier to developing effective therapies. This review has synthesized growing evidence pointing to the possibility that the cross-talk between autophagy and exosomal communication may represent a significant, integrative constituent of the inflammatory, immune, and pain pathways characterizing this syndrome. We have outlined a hypothetical model wherein dysregulated autophagy of the cells of the prostate cells destabilizes cellular homeostasis and potentially restructures the molecular content of the secreted exosomes. This proposed feed-forward loop offers a conceptual framework for how an inflammatory microenvironment might become chronic in CP/CPPS pathology. Therefore, targeting the autophagy-exosome axis may represent a compelling, mechanism-based strategy worthy of further investigation, with the potential to inform novel diagnostic and therapeutic approaches for this debilitating condition.
To translate this mechanistic understanding into clinical impact, future research should give precedence to the following directions:
1. Establish causal mechanisms: The existing studies should go beyond correlation to draw a conclusion on causality. To achieve this goal, the implementation of sophisticated experimental models, including cell-specific autophagy knockouts, will be required to map out the role of autophagic activity in discrete prostate epithelial and stromal subsets on exosome biogenesis, exosome secretion, and exosome cargo assembly in inflammatory conditions. Determining the upstream signaling crossroads, and especially mTOR and AMPK, that coordinate this mechanistic crosstalk is essential to discover pharmacologically approachable targets.
2. Advance therapeutic translation with caution: The development of engineered exosomes as delivery vehicles of autophagy modulators, anti-inflammatory agents, or neuroprotective factors must proceed along with the solution of the significant manufacturing, safety, and targeting challenges outlined in Challenges, risks, and future translational directions. Concurrently, carefully designed clinical trials in which approved autophagy modulators (say, metformin) will be repurposed in the treatment of CP/CPPS and exosomal biomarkers will present justified steps.
3. Validate exosomal biomarker panels: One of the key translational goals is the creation of a liquid biopsy based on exosomes in a standard form. This would require the comprehensive validation of potential cargo panels, including autophagy (LC3 II/p62) markers, inflammatory (e.g., miR-155, IL-8) markers, and pain modulators (NGF) in well-modeled patient groups. An effective implementation would support molecular stratification (e.g., inflammatory subtypes versus fibrotic subtypes) to support individualized management.
4. Explore relevance in related urological disorders: Exploring this axis in other conditions associated with interstitial cystitis/bladder pain syndrome (IC/BPS) could help to determine the prevalence of shared pathogenic mechanisms and repurposing opportunities, and also understand disease-specific peculiarities.
In conclusion, sources of lipid raft-regulated autophagy-exosome axis can offer a novel and integrative approach to understanding CP/CPPS. Despite the significant difficulties in translation, there is potential hope that focused efforts to promote mechanistic validation, biomarker development, and cautious investigations of therapeutics include the potential to move beyond treatment of the symptoms of disease, but to address the underlying disease pathways, which ultimately could yield improvements in patient outcomes.
ATG: autophagy-related
CP/CPPS: chronic prostatitis/chronic pelvic pain syndrome
DAMPs: damage-associated molecular patterns
ER: endoplasmic reticulum
EVs: extracellular vesicles
ILVs: intraluminal vesicles
MAMs: mitochondria-associated membranes
MVBs: multivesicular bodies
NGF: nerve growth factor
nSMase2: neutral sphingomyelinase 2
sEVs: small extracellular vesicles
SOCS: suppressors of cytokine signaling
DKN: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Visualization. FG: Formal analysis, Visualization. EDKF: Writing—review & editing. AA: Writing—review & editing, Data curation. BRO: Data curation, Visualization. FA: Data curation, Project administration. XZ: Supervision, Project administration, Writing—review & editing, Resources. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 1322
Download: 17
Times Cited: 0
