 Review
Review

Affiliation:
1Medical Oncology, Hospital Universitario De Torrejón, 28850 Madrid, Spain
2Facultad de Medicina, Universidad Francisco de Vitoria, 28223 Madrid, Spain
Email: pitucgp@hotmail.com
ORCID: https://orcid.org/0000-0002-3468-3626
Affiliation:
1Medical Oncology, Hospital Universitario De Torrejón, 28850 Madrid, Spain
2Facultad de Medicina, Universidad Francisco de Vitoria, 28223 Madrid, Spain
ORCID: https://orcid.org/0000-0001-8710-2592
Affiliation:
1Medical Oncology, Hospital Universitario De Torrejón, 28850 Madrid, Spain
2Facultad de Medicina, Universidad Francisco de Vitoria, 28223 Madrid, Spain
ORCID: https://orcid.org/0000-0001-7538-1459
Affiliation:
3Department of Pharmacy and Nutrition, Faculty of Biomedical and Health Sciences, Universidad Europea de Madrid, 28670 Madrid, Spain
Affiliation:
1Medical Oncology, Hospital Universitario De Torrejón, 28850 Madrid, Spain
2Facultad de Medicina, Universidad Francisco de Vitoria, 28223 Madrid, Spain
ORCID: https://orcid.org/0000-0003-0141-1306
Explor Immunol. 2025;5:1003196 DOI: https://doi.org/10.37349/ei.2025.1003196
Received: July 08, 2024 Accepted: April 22, 2025 Published: May 20, 2025
Academic Editor: Cunte Chen, South China University of Technology, China
The article belongs to the special issue The Role of Immune Checkpoint Molecules in Cancer and Hematological Malignancies
The introduction of immune checkpoint inhibitors (ICIs) has transformed the landscape of oncology, offering significant improvements in patient survival and achieving remarkable long-term outcomes. Despite these advances, the therapeutic benefits of ICIs are not universal, and existing biomarkers often fall short in accurately predicting patient responses. A comprehensive understanding of the mechanisms underlying resistance to ICIs is essential for the development of strategies to mitigate these challenges and enhance therapeutic efficacy. This review provides a detailed exploration of the resistance mechanisms associated with ICIs, focusing on the role of the tumor microenvironment and intrinsic tumor cell alterations in mediating both primary and secondary resistance. Furthermore, it evaluates emerging strategies to overcome resistance, including combination therapies and innovative therapeutic approaches. By dissecting the molecular and immunological pathways implicated in ICI resistance, this review aims to highlight novel predictive and prognostic biomarkers and outline optimized therapeutic strategies to maximize the clinical impact of ICIs in cancer management.
The emergence of immune checkpoint inhibitors (ICIs) in multiple types of tumors has revolutionized oncology. Monoclonal antibodies targeting the programmed cell death protein 1/programmed death ligand 1 (PD-1/PD-L1) pathway, demonstrate significant clinical efficacy in a range of solid tumors [1–6]. However, many patients eventually develop resistance to ICIs over time, leading to a loss of efficacy.
While numerous studies have sought to identify biomarkers predicting primary non-response to ICIs, the mechanisms underlying acquired resistance remain largely unknown.
Primary resistance mechanisms encompass the absence or loss of tumor antigens [7], alterations in the major histocompatibility complex (MHC) processing pathway [8], low T-cell infiltration, increased expression of vascular endothelial growth factor (VEGF) and immunosuppressive cytokines [9], and mutations in STK11 [10]. PD-L1 expression on tumor cells is regarded as a positive predictive marker [6, 11, 12]; however, many patients fail to respond to ICI treatment despite having tumors with high PD-L1 expression [13].
Mechanisms contributing to acquired resistance include the upregulation of alternative immune checkpoints, the loss of human leukocyte antigen (HLA) expression, and mutations in β2-microglobulin (β2M) and Janus kinase 1/2 (JAK1/2) [14–17]. When patients develop resistance to immunotherapy, there are limited therapeutic options, and a lack of detailed understanding of these resistance mechanisms makes it difficult to guide treatment decisions.
Currently, data on the mechanisms involved in ICI resistance are lacking. Gaining a deeper understanding of the molecular and immunological processes governing ICIs will aid in identifying new predictive and prognostic biomarkers and establishing optimal therapeutic strategies. In this review, we examine emerging evidence that sheds light on novel mechanisms of both innate and acquired resistance to ICIs and offer insights into potential strategies for overcoming these challenges.
The immune system has a key role in the natural history of cancer development, tumor growth and metastasis but tumors have the ability to evade the immune response. Several cell types participate in the interplay of the immune reaction: (1) the “immune synapse” is the physical contact of immune cells by antigen presentation and has helper T cells (the subclasses Th1/Th2 of CD4+ T lymphocytes) and CD8+ T lymphocytes which are able to discriminate non self-antigens vs self-antigens [18–20]; (2) natural killer (NK) cells have inhibitory molecules and killer cell immunoglobulin-like receptor (KIR) subtypes [21], NK cells can exert cytotoxicity over with low MHC class 1 expression because they do not need MHC antigen presentation; (3) there are two different phenotypes of macrophages: M1 macrophages that are capable of liberating interferon (IFN) gamma and M2 macrophages and are also responsible for phagocytosis, that can inhibit inflammatory reactions and promote tolerance by the release of cytokines such as transforming growth factor beta (TGF-β), interleukin (IL)-4 and IL-10 [22]; (4) other types of cells that play a role in autoimmunity and cancer are myeloid derived suppressor cells (MDSCs) and FoxP3+ CD25+ CD4+ Treg that dampen cytotoxic T lymphocyte activity [23, 24] and CD4+ T cells that liberate IL-17, the Th17 cells.
The “immune synapse” refers to the immunological phenomenon based upon the capacity of T lymphocytes to differentiate non-self and self-antigens presented by antigen-presenting cells (APCs) and how the cytotoxic CD8+ T cells are modulated depending on the inhibitory and stimulatory receptors which are, furthermore, regulated by cytokines [20]. The T cell receptor (TCR) complex includes the TCR, the CD4 or CD8 receptor that binds to the MHC and the CD3, macromolecular complex formed by CD3 independent molecules [18], which activates through an intracellular tyrosine-based component that transfers surface signals to intracellular effectors [25]. The TCR needs to adhere to a peptide exhibited by the MHC and there also needs to be a set of costimulatory signals that allow an efficient naive CD8+ T cells activation [20]. Thus, naive CD8+ T cell activation leads to the increase of production of pro-inflammatory cytokines such as IFN gamma and IL-12 via CD3 intracellular signaling [20]. CD28 binds to B7-1 and B7-2 (CD80 and CD86) on the APC and it constitutes in naive T cells the main co-stimulatory signal. Inhibitory signals and co-stimulatory molecules such as OX40, GITR [expressed on T cells, regulatory T cells (Tregs) an DC], ICOS (expressed on activated T cells: Th1, Th2 and follicular helper T cells) are involved in regulating immune responses on the APC and T cells regulate the costimulatory process [20]. Programmed cell death protein 1 (PD-1), Cytotoxic T-lymphocyte antigen 4 (CTLA-4), T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) and lymphocyte activation gene-3 (LAG-3) are types of co-inhibitory, also known as molecules that elicit an immune response. In malignant clones, there could be an exhaustion phenotype characterized by the chronic recognition of an antigen leading to feedback inhibition of effector T cell function [19]. LAG-3 is expressed in exhausted T cells and is an inhibitory receptor that constitutes a novel immunotherapeutic target with more than 20 clinical trials evaluating its possible efficacy in different tumor types [26]. A fixed-dose combination of anti-PD-1 and anti-LAG-3 has demonstrated efficacy in the treatment of unresectable or metastatic melanoma [27].
Systemic therapy focused on immune system response in order to address and control cancer are widely known as immunotherapy and ICIs constitute one of the most relevant therapeutic approaches. The most studied ICIs are antagonistic antibodies that inhibit CTLA-4, LAG3, PD-1 and PD-L1. PD-1 and CTLA-4 suppress immune responses in different ways and the mechanisms of inhibitions in T cell responses are different: (1) PD-1 is upregulated following antigen presentation and PD-1 is a marker for T-cell activation in physiological immune responses which are different from tumor-associated T-cells that express, in later stages, PD-1 and other immune checkpoint molecules. (2) CTLA 4 is expressed early in the activation of T cells and its ligands are usually APCs in secondary lymphoid organs. Therefore, PD-1 and CTLA-4 have different roles repressing progressive immune responses [28]. Table 1 summarizes the ICIs approved by the European Medicines Agency (EMA) for different tumors and indications [2, 29–88].
List of EMA-approved immune checkpoint inhibitors with their approved indication
| ICIs | Author | Approved indication | Special consideration | Predictive biomarker | |
|---|---|---|---|---|---|
| Anti-PD-1 | Nivolumab | Larkin et al. [29] | Advanced melanoma | N/A | N/A |
| Weber et al. [30] | Resected melanoma with metastatic lymph nodes or resected metastases | Adjuvant setting | N/A | ||
| Forde et al. [31] | Resectable NSCLC with high recurrence risk | Neoadjuvant setting in combination with platinum-based chemotherapy | PD-L1 ≥ 1% | ||
| Borghaei et al. [32] | Locally advanced or metastatic NSCLC | Subsequent therapy after chemotherapy | N/A | ||
| Motzer et al. [33] | Advanced renal cell carcinoma | Subsequent treatment | N/A | ||
| Choueiri et al. [34] | Advanced renal cell carcinoma | First line therapy in combination with cabozantinib | N/A | ||
| Ferris et al. [35] | Recurrent or metastatic squamous head and neck cancer | Subsequent therapy after platinum-based chemotherapy | N/A | ||
| Bajorin et al. [36] | Resected urothelial carcinoma with high recurrence risk | Adjuvant setting | PD-L1 ≥ 1% | ||
| van der Heijden [37] | Locally advanced or metastatic urothelial carcinoma | First line therapy combined with cisplatin and gemcitabine | N/A | ||
| Sharma et al. [38] | Locally advanced or metastatic urothelial carcinoma | Subsequent therapy after platinum-based chemotherapy | N/A | ||
| Kelly et al. [39] | Esophageal or gastroesophageal junction carcinoma with residual pathologic disease after neoadjuvant chemoradiation | Adjuvant setting | N/A | ||
| Doki et al. [40] | Advanced unresectable, recurrent or metastatic squamous cell esophageal carcinoma | First line therapy combined with fluoropyrimidine and platinum-based chemotherapy | PD-L1 ≥ 1% | ||
| Kato et al. [41] | Advanced unresectable, recurrent or metastatic squamous cell esophageal carcinoma | Subsequent therapy after platinum and fluoropyrimidine-based chemotherapy | N/A | ||
| Janjigian et al. [42] | Advanced or metastatic gastric, gastroesophageal junction or esophageal adenocarcinoma | First line therapy combined with fluoropyrimidine and platinum-based chemotherapy | - HER2 negative- PD-L1 + with CPS ≥ 5 | ||
| Pembrolizumab | Luke et al. [43] | Completely resected melanoma stage IIB, IIC or III | Adjuvant setting | N/A | |
| Schachter et al. [44] | Irresectable or metastatic melanoma | N/A | N/A | ||
| Reck et al. [45] | Metastatic NSCLC | First line therapy | - PD-L1 + CPS ≥ 50- EGFR and ALK negative | ||
| Gandhi et al. [46] | Metastatic NSCLC and non-squamous cell carcinoma | First line therapy in combination with pemetrexed and platinum-based chemotherapy | EGFR and ALK negative | ||
| Paz-Ares et al. [47] | Squamous NSCLC | First line therapy in combination with paclitaxel and platinum-based chemotherapy | N/A | ||
| Herbst et al. [48] | Locally advanced or metastatic NSCLC | If EGFR or ALK positive, targeted therapy should be received prior to pembrolizumab | PD-L1 + CPS ≥ 1 | ||
| Bellmunt et al. [49] | Locally advanced or metastatic urothelial carcinoma | Subsequent therapy after prior platinum-based chemotherapy | N/A | ||
| Balar et al. [50] | Locally advanced or metastatic urothelial carcinoma | Unfit for platinum | PD-L1 + CPS ≥ 10 | ||
| Burtness et al. [51] | Recurrent or metastatic squamous head and neck cancer | First line therapy in combination with 5-fluorouracil + platinum | PD-L1 + CPS ≥ 1 | ||
| First line therapy, monotherapy | PD-L1 + CPS ≥ 50 | ||||
| Choueiri et al. [52] | Resected renal cell carcinoma (including resected metastases) | Adjuvant setting | N/A | ||
| Rini et al. [53] | Advanced renal cell carcinoma | First line therapy in combination with axitinib | N/A | ||
| Motzer et al. [54] | Advanced renal cell carcinoma | First line therapy in combination with lenvatinib | N/A | ||
| André et al. [55] | Metastatic colorectal cancer | First line therapy or after previous chemotherapy based on fluoropyrimidines | Deficient in mismatch repair (dMMR) | ||
| Marabelle et al. [56] | Recurrent or advanced endometrial carcinoma | Subsequent therapy in progression to platinum-based chemotherapy | dMMR | ||
| Metastatic gastric cancer | Subsequent therapy | ||||
| Metastatic small intestine cancer | |||||
| Metastatic biliary tract cancer | |||||
| Sun et al. [57] | Metastatic esophageal cancer or adenocarcinoma of gastroesophageal junction | First line therapy in combination with platinum and fluoropyrimidine-based chemotherapy | PD-L1 + CPS ≥ 10 | ||
| Schmid et al. [58] | Localized triple negative breast cancer | Perioperative setting | N/A | ||
| Cortes et al. [59] | Metastatic triple negative breast cancer | First line therapy in combination with chemotherapy | PD-L1 + CPS ≥ 10 | ||
| Makker et al. [60] | Advanced or recurrent endometrial carcinoma | Subsequent therapy after platinum-based chemotherapy | N/A | ||
| Monk et al. [61] | Recurrent or metastatic cervical cancer | In combination with or without bevacizumab | PD-L1 + CPS ≥ 1 | ||
| Janjigian et al. [62] | Unresectable or metastatic gastric or gastroesophageal junction adenocarcinoma HER2 positive | First line therapy in combination with trastuzumab + platinum-based chemotherapy and fluoropyrimidines | - HER2 positive- PD-L1 + CPS ≥ 1 | ||
| Rha et al. [63] | Unresectable or metastatic gastric or gastroesophageal junction adenocarcinoma | First line therapy in combination with platinum-based chemotherapy and fluoropyrimidines | - HER2 negative- PD-L1 + CPS ≥ 1 | ||
| Kelley et al. [64] | Unresectable or metastatic biliary tract cancer | First line therapy in combination with cisplatin and gemcitabine | N/A | ||
| Dostarlimab | Mirza et al. [65] | Advanced endometrial cancer (new diagnosed or relapse) | In combination with carboplatin and paclitaxel | dMMR | |
| Oaknin et al. [66] | Primary advanced or recurrent endometrial cancer (relapse or progression after first line therapy) | Monotherapy | dMMR | ||
| Cemiplimab | Migden et al. [67] | Metastatic or locally advanced squamous cell skin cancer | N/A | N/A | |
| Stratigos et al. [68] | Locally advanced or metastastic basal cell carcinoma | Subsequent therapy, in progression or those who not tolerate a Hedgehog pathway inhibitor | N/A | ||
| Sezer et al. [69] | Locally advanced NSCLC not candidates for chemoradiotherapy or metastatic NSCLC | First line therapy, monotherapy | - PD-L1 ≥ 50%- No EGFR, ALK or ROS1 mutations | ||
| Gogishvili et al. [70] | Locally advanced NSCLC not candidates for chemoradiotherapy or metastatic NSCLC | First line therapy, in combination with platinum-based chemotherapy | - PD-L1 ≥ 1%- No EGFR, ALK or ROS1 mutations | ||
| Tewari et al. [71] | Metastatic or recurrent cervical cancer | Subsequent therapy in progression to a platinum-based chemotherapy | N/A | ||
| Retifanlimab | Lakhani et al. [72] | Locally advanced or metastatic Merkel cell carcinoma | First line therapy | N/A | |
| Tislelizumab | Shen et al. [73] | Unresectable, locally advanced or metastatic esophageal squamous cell carcinoma | Subsequent therapy, after progression to platinum-based chemotherapy | N/A | |
| Anti-PD-L1 | Atezolizumab | Powles et al. [74] | Locally advanced or metastatic urothelial carcinoma | Subsequent therapy after platinum-based chemotherapy or first line for unfit for platinum | N/A |
| Rittmeyer et al. [75] | Locally advanced or metastatic NSCLC | After previous chemotherapy. If EGFR or ALK mutation, targeted therapy must have been received prior to atezolizumab | N/A | ||
| Avelumab | D’Angelo [76] | Metastatic Merkel cell carcinoma | N/A | N/A | |
| Powles et al. [77] | Locally advanced or metastatic urothelial carcinoma with no progression after platinum-based chemotherapy | First line therapy-maintenance therapy | N/A | ||
| Motzer et al. [78] | Advanced renal cell carcinoma | First line therapy combined with axitinib | N/A | ||
| Durvalumab | Antonia et al. [79] | Unresectable and locally advanced NSCLC with no progression after chemoradiotherapy | Maintenance therapy after chemoradiotherapy | PD-L1 ≥ 1% | |
| Paz-Ares et al. [80] | Metastatic SCLC | First line therapy in combination with carboplatin/cisplatin and etoposide | N/A | ||
| Burris et al. [81] | Irresectable or metastatic biliary tract carcinoma | First line therapy in combination with gemcitabine and cisplatin | N/A | ||
| Abou-Alfa et al. [82] | Advanced or unresectable hepatocellular carcinoma | First line therapy, in monotherapy | N/A | ||
| Anti-CTLA-4 | Ipilimumab | Hodi et al. [83] | Metastatic melanoma | N/A | N/A |
| Anti-CTLA-4 combined with another ICI | Nivolumab + ipilimumab | Larkin et al. [2] | Metastatic melanoma | N/A | N/A |
| Motzer et al. [84] | Metastatic renal carcinoma with intermediate-high risk | First line therapy | N/A | ||
| Baas et al. [85] | Unresectable malignant pleural mesothelioma | First line therapy | N/A | ||
| Kato et al. [86] | Advanced unresectable, recurrent or metastatic squamous cell esophageal carcinoma | First line therapy | PD-L1 ≥ 1% | ||
| Lenz et al. [87] | Metastatic colorectal carcinoma | Subsequent therapy after fluoropyrimidine-based chemotherapy | dMMR | ||
| Tremelimumab + durvalumab | Abou-Alfa et al. [82] | Advanced or unresectable hepatocellular carcinoma | First line therapy | N/A | |
| Tremelimumab + durvalumab + platinum-based chemotherapy | Johnson et al. [88] | Metastatic NSCLC | First line therapy | No EGFR or ALK mutations | |
ICIs: immune checkpoint inhibitors; PD-1: programmed cell death protein 1; PD-L1: programmed death ligand 1; NSCLC: non-small cell lung cancer; HER2: human epidermal growth factor receptor 2; CPS: combined positive score; EGFR: epidermal growth factor receptor; ALK: anaplastic lymphoma kinase; ROS1: ROS proto-oncogene 1 receptor tyrosine kinase. N/A: not applicable
CTLA-4 functions by being expressed on the surface of CD4+ and CD8+ T cells, where it has a stronger binding affinity for the costimulatory molecules CD80 and CD86 (B7-1 and B7-2) on APCs compared to the T cell costimulatory receptor CD28 [89].
Naive T cells require a stimulation signal through the TCR, derived from an antigen, and a subsequent costimulation signal which passes when the CD28 on T cells engage with B7.1/CD80 or B7.2/CD86 on APCs [90–92]. CTLA-4 is an inhibitory receptor present in T cells that decreases T cell activity and it can be increased when a T cell is activated [93–95]. CTLA-4 can decrease the costimulation process by binding and competing with a higher affinity for the same ligands as CD28, therefore, inhibiting T cell activity [96, 97]. CTLA-4 can suppress T cell activity via the extrinsic and intrinsic pathways. In the extrinsic inhibition, CTLA-4 depresses the optimal level of costimulatory signals required by the T cell by competing with CD28 for ligands [98]. In the intrinsic inhibition, CTLA-4 recruits phosphatases and transcription factors (NFAT, NF-κB and AP-1) are also inhibited, which are associated with the activation of T cells [99–105]. On the other hand, Tregs express CTLA-4 on the surface which control the generation and function of Tregs [106–110], thus, suppressing immune responses.
One of the main ways anti-CTLA-4 functions is by enabling macrophages to eliminate Treg cells from the tumor microenvironment (TME) [89]. The effectiveness of anti-CTLA-4 antibodies relies, at least partially, on reducing tumor-infiltrating Tregs through interactions with human Fcγ receptors (FcγRs) and human immunoglobulin Gs (IgGs). Enhancing antibody-dependent cell-mediated cytotoxicity—either by optimizing the fragment crystallizable (Fc) region or by the presence of high-affinity FcγR variants—boosts therapeutic efficacy, but primarily in highly immunogenic tumors [89]. Anti-CTLA-4 immunotherapy does not eliminate FOXP3+ cells in human tumors, indicating that its effectiveness could be improved by altering the Fc regions of monoclonal antibodies to enhance Fc-driven depletion of Tregs within the tumor [111]. Depleting both intratumoral and nodal Tregs independently of CTLA-4 led to even stronger antitumor effects, indicating that targeting nodal Tregs could be a valuable approach to boosting the effectiveness of current immunotherapies [112].
Anti-CTLA-4 antibodies’ development made the ICIs development possible. The first evidence of the antitumor effects of anti-CTLA-4 activity was in 1996 when mice with colon carcinoma cells treated with anti-CTLA-4 antibodies obtained an objective tumor response [113], and also a protective immunity for the long term was observed. In 2010, the survival benefit of anti-CTLA-4 antibody ipilimumab was demonstrated in a phase III clinical trial over a melanoma-specific peptide vaccine in patients with melanoma.
PD-1 is an immunoreceptor that has two tyrosine residues in the cytoplasmic regions and belongs to the immunoglobulin superfamily [114]. It is a transmembrane protein that regulates programmed T cell death [115] expressed on macrophages, Langerhans cells, dendritic cells (DCs), B cells and T cells, but its expression is highly increased on exhausted T cells [116, 117]. Hematopoietic and tumor cells can express PD-L1 on the surface, whereas PD-L2 is more often present in hematopoietic cells [118, 119]. Monocytes, DCs and NK cells can express PD-1 and/or PD-L1. PD-1 has an inhibitory activity that engages to PD-L1 (B7-H1) and PD-L2 (B7-H2) which downregulates T cell immune responses [118, 119]. Thus, the engagement of PD-1 and PD-L1/2 lead to phosphorylation of its cytoplasmic immunoreceptors and inhibits tumor cell apoptosis, contributes to peripheral T effector cell exhaustion and the transformation of T effector cells to Treg cells [7, 120]. The specific phosphorylation of the immunoreceptor tyrosine-based switch motif (ITSM) recruits the Src homology region 2 domain-containing phosphatase-2 (SHP-2) and represses the activity of some intracellular molecules that propagate signals following the TCR, therefore, mediates the PD-1 inhibitory functions [28, 114]. The dephosphorylation events dull the inflammatory reaction of the affected cells: (1) in T cells, PD-1 signals dephosphorylate ZAP70 and CD3ζ, which transduce signals downstream of TCR engagement [114]; (2) in B cells the recruitment of SHP-2 dephosphorylates effector molecules such as Syk and PI3K [121]. The Ras/MEK/ERK pathway, that controls cell proliferation, survival and growth, can be inhibited via SHP-2 which may contribute to a restrained proliferation after activation [122]. The second key pathway of PD-1 intracellular signaling in T-cells involves enhancing the expression of E3 ubiquitin ligases at the transcriptional level [123]. It has been shown that PD-1 works together with LAG-3 to suppress T-cell activity via the E3 ubiquitin ligase pathway, which plays a crucial role in resistance to PD-1/PD-L1 blockade monotherapies [124]. The function of the PD-1/PD-L1/PD-L2 axis is summarized in Figure 1.
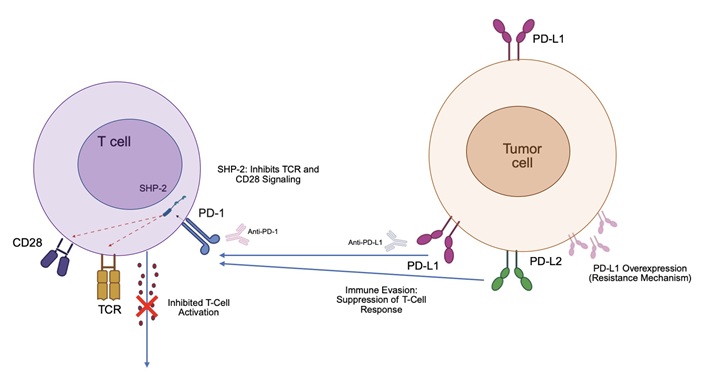
The function of the PD-1/PD-L1/PD-L2 axis. This illustration highlights the PD-1/PD-L1/PD-L2 axis as a critical immune checkpoint in oncology. PD-1, expressed on T-cells, binds to PD-L1 (often overexpressed on tumor cells) or PD-L2 (on tumor cells or antigen-presenting cells), triggering SHP-2 to inhibit TCR and CD28 signaling, thus suppressing T-cell activation and promoting immune evasion. This is a key mechanism of resistance to immunotherapy, as tumors can upregulate PD-L1 in response to IFN-γ from the microenvironment. PD-1: programmed cell death protein 1; SHP-2: Src homology region 2 domain-containing phosphatase-2; TCR: T cell receptor; PD-L1: programmed death ligand 1; IFN-γ: interferon-gamma
Targeting PD-1/PD-L1 has been evaluated in order to boost antitumor activity [125–130]. In mice, the interplay between PD-L1 present in tumor cells and PD-1 present on cytotoxic T cells resulted in an accelerated tumor growth, thus, PD-L1 antibody therapy obtained a contributed to tumor progression [125]. Furthermore, PD-L1 and PD-L2 expression has also been associated to poor disease prognosis in various cancer types [131–135]. Numerous studies have demonstrated the efficacy of anti-PD-1 and anti-PD-L1 antibodies as a systemic therapy for cancer. Blocking PD-L1 inhibits the PD-1/PD-L1 axis as well as the PD-L1/CD80 cis interplay on DCs which ends up liberating more CD80 molecules that are able to increase T cell priming [136]. Ongoing clinical trials are currently investigating the use of monoclonal antibodies targeting other immune checkpoints, such as TIM-3, LAG3, and T-cell Immunoreceptor with Ig and ITIM domains (TIGIT), in combination with PD-1 inhibitors. For instance, studies on TIM-3 have shown that blocking both PD-1 and TIM-3 together significantly boosts the immune activity of effector T cells in animal models of diseases like acute myeloid leukemia and lung cancer, achieving greater effectiveness than PD-1 inhibitors alone [137–140]. Atezolizumab, avelumab and durvalumab are the three main anti-PD-L1 antibodies that have been approved for various cancer types and, although all of these antibodies stop the interaction of PD-L1 with PD-1 and CD80, there are some differences in their mechanisms of action that should be noted. The Fc region has a modification in durvalumab and atezolizumab that prevents exhaustion of T cells that express PD-L1 by the removal of antibody-dependent cellular cytotoxicity. Whereas, avelumab contains the native Fc region that induces antibody-dependent cellular cytotoxicity through the engagement of FCγ receptors on NK cells [28].
PD-L1 is part of the B7 family of molecules that regulate immune responses, acting as either co-stimulatory or co-inhibitory signals. It is expressed by various cell types, including tumor cells [141]. PD-L1 is a membrane-bound protein composed of an immunoglobulin-like extracellular region, a transmembrane segment, and a short intracellular domain and can also bind CD80 as PD-1 [142]. PD-L1 plays a crucial role in preserving peripheral tolerance and supporting DCs in presenting antigens to T cells. In cancer, its expression in tumors is strongly associated with advanced disease and poor outcomes, often signaling resistance to conventional therapies such as chemotherapy and radiotherapy [141, 143]. One of the ways PD-L1 exerts its protective effects is by inhibiting IFN-gamma signaling within cancer cells through its intracellular domains [143]. This also enhances cancer cell resistance mechanisms by disrupting the JAK/signal transducer and activator of transcription (STAT) signaling pathways through mutations [144].
The signaling pathway that drives the adaptive expression of PD-L1 and PD-L2 in response to IFNs is crucial for advancing PD-1 blockade therapies in cancer treatment. When T cells recognize tumor antigens, they release IFNs, which stimulate cancer cells or other cells in the TME to express PD-L1. This suppresses the antitumor immune response in a process called adaptive immune resistance. This mechanism specifically limits T-cell recognition of cancer cells while preserving immune responses to other antigens, preventing widespread immune suppression [145, 146].
PD-L2 (also known as B7-DC or CD273) is a type I transmembrane protein belonging to the B7 family. Its extracellular region consists of an immunoglobulin (Ig)-like V-type domain and an Ig-like C2-type domain [147]. PD-L2 is found not only in tumor cells but also in immune cells, and its elevated expression has been shown to play a key role in cancer development and immune evasion. PD-L1 and PD-L2 are ligands of the PD-1 immune checkpoint, which suppresses immune responses. Abnormal PD-L2 expression plays a major role in cancer development and progression by helping tumor cells evade detection and destruction by the immune system [147]. Their expression can be triggered in tumors by IFN exposure, allowing cancer cells to evade immune attack. This mechanism plays a crucial role in immunotherapies that target PD-1 inhibition. When PD-1 binds to PD-L2, it significantly suppresses TCR-driven proliferation and cytokine release in CD4+ T cells. At low antigen levels, PD-L2-PD-1 interactions weaken the strong signaling from B7-CD28. However, when antigen levels are high, these interactions limit cytokine production but do not prevent T cell proliferation [119].
LAG-3 is a protein found on B cells, certain T cells, NK cells, and tumor-infiltrating lymphocytes (TILs), playing a role in regulating immune checkpoint pathways [148]. LAG-3 is a membrane protein that interacts with MHC class II, promoting Treg function while suppressing T cell proliferation, activation, and balance. Similarly, Programmed Death 1 (PD-1) inhibits T cell activity. Both LAG-3 and PD-1 are temporarily expressed on CD8 T cells following acute stimulation [149]. It enhances the activity of Tregs by interacting with MHC class II molecules, which suppresses T cell differentiation and proliferation [150].
LAG-3 expression on T cells helps to suppress activation and maintain immune balance [151]. Earlier research showed that transgenic CD4 T cells exposed to their specific self-antigen in vivo exhibited increased LAG-3 expression and became anergic. In this model, these tolerized CD4 T cells demonstrated regulatory functions both in vitro and in vivo, and their in vitro suppressive activity was blocked using a LAG-3-specific monoclonal antibody [152]. Naive CD8 T cells initially express low levels of LAG-3, but its expression sharply rises in response to antigen stimulation [153]. Blocking LAG-3 with specific antibodies can restore the function of exhausted T cells, improving their ability to target and destroy tumor cells.
The simultaneous increase in PD-1 and LAG-3 expression on CD4 and CD8 T cells is a key factor contributing to resistance against treatments that target PD-1 or PD-L1 alone [154, 155]. The presence of both PD-1 and LAG-3 on TILs has been proposed as a marker of impaired immune function in non-small cell lung cancer (NSCLC) [156]. An increasing amount of research suggests that the co-expression of PD-1 and LAG-3 in T cells contributes to resistance against anti-PD-1 and anti-PD-L1 treatments [157–160].
Relatlimab (a human IgG4 LAG-3-blocking antibody) combined with nivolumab (anti-PD-1) has been proven to be a safe and effective treatment for advanced melanoma [27]. Analysis of biospecimens from an ongoing study showed that relatlimab plus nivolumab treatment enhanced CD8+ TCR signaling and modified CD8+ T cell differentiation, boosting cytotoxic activity while maintaining characteristics of exhaustion [155]. The co-expression of cytotoxic and exhaustion markers was regulated by PRDM1, BATF, ETV7, and TOX. Furthermore, effector function increased in clonally expanded CD8+ T cells that appeared after treatment with the combination [155].
One of the main contributors to primary resistance to ICIs associated with tumor intrinsic mechanisms is the low availability of neoantigens and low mutational load [161]. Neoantigens are tumor-specific antigens that can derive from tumor genome instability that has previously led to somatic mutations in different cancer types [161]. Tumors can acquire a high neoantigen load through mismatch repair or genomic instability or, as observed in NSCLC and melanoma, the mutation acquisition could be through carcinogen-induced DNA damage or UV-damage, respectively [11, 162, 163]. Neoantigens are able to confer high immunogenicity with more affinity to MHC-II which then derives to a more intense immunological response with an increased antitumor T cell response with ICIs [164, 165]. ICIs can activate tumor infiltrating T cells which are more abundant in patients with mutations in DNA repair as MSH2, MLH1 and ATM [166, 167]. Neoantigen depletion can be associated with primary resistance to ICIs, and in the presence of tumor immune elimination, tumor cells can evade the antitumoral response by the HLA loss of heterozygosity or enhance the loss of tumor-specific neoantigens [161]. Therefore, tumor response to ICIs could fail if there is tumoral neoantigen depletion [168].
IFN-γ is a cytokine that deploys proapoptotic and antriproliferative effects, induces the boost of the MHC I in tumor cells and, furthermore, promotes CD8+ cytotoxic T cell activity uplifting PD-L1 levels, which all together accounts for the interplay of IFN-γ in the initiation and maintenance of antitumor response [169, 170]. Effector T cells release IFN-γ which promotes JAK-STAT signaling cascades, IFN-γ binds to the IFN-γ receptor it activates the receptor-associated kinases JAK1/2 and STAT1/2, that promote tumor cell death by upregulation MHC class I expression [161, 171–173]. PD-L1 functions as an anti-apoptotic signaling molecule in cancer cells by interfering with type I and II IFN signaling pathways, helping tumor cells resist immune-mediated cell death [174, 175]. Tumors who harbor mutations in IFNGR1/2 and JAK1/2 have been identified in non-responders to anti-PD-1 and anti-CTLA-4 ICIs [176, 177]. Therefore, the dysregulation of the IFN-γ pathway in tumor cells are associated with a subdued response to ICIs and the loss of function alterations in the JAK/STAT pathway result in the loss of MHC class I by inducing cell resistance to IFN [69]. In this context, an IFN-γ signature has demonstrated a predictive value in cancer patients treated with anti-PD-1 [178–180]. Inflammatory cytokines and tumor-promoting inflammation, such as IL-6, IL-8 and C-reactive protein, have been linked to ICI resistance and can play a role as potential biomarkers [181]. Ceramide metabolism alterations have also been associated with TNF-induced melanoma cell dedifferentiation and certain ceramide metabolites could be predictive biomarkers of immunotherapy resistance [182].
The recognition of antigens on MHC by APC is one of the mainstays of T cell activation in immunological responses [161]. β2M is a gene involved in the modulation of antigen presentation to cytotoxic CD8+ T cells by MHC class I complex [183–185] and it restores MHC-I/peptide complex formation [186]. β2M loss of function has been associated with MHC class I deficiency in cancer cells which can confer resistance from immune surveillance by reducing T cell recognition [186]. Cancer patients with a β2M loss show dysregulated immune responses because of the impairment of surface MHC class I via the degradation of heavy chain of MHC class I in cytosol. Another gene involved in antigen presentation is MEX3B, which downregulates MHC I expression in tumors, has been observed at higher levels in non-responders to anti-PD-1 cancer patients compared to responders [187].
PD-L1 expression on tumor cells is a well-known biomarker that has been associated with response to ICIs [185–187]. Previously, we have noted the importance of antigen presentation and activated CD8+ T cell responses in the response of ICIs, thus, PD-L1 expression associates with tumoral immune activation at this level [186]. Therefore, PD-L1 low-expression in patients with NSCLC and melanoma has shown up to 10% of response rates compared to 40–50% response rates in patients with increased PD-L1 expression [185, 186]. Nonetheless, anti-PD-1 treatments have shown to be effective in PD-L1 negative tumors, and on the other hand, patients with an increased PD-L1 expression don’t always respond to ICIs [6, 11].
TME compromises non-immune stromal cells (fibroblasts and endothelial cells) and immune cells which includes B cells, T cells, NK cells, MDSCs and DCs [188]. ICIs response may be altered depending on the TME [161].
As previously mentioned, ICIs seek to promote anti-tumor immune responses by T cells [189, 190] and the amount of CD8+ T cells in the TME have been related with an overall increased response to PD-1/PD-L1 blockade [6, 191]. In this setting, the amount of T cells in the TME is crucial for the ICI response [161], but the mere presence is not enough to predict response in patients treated with ICIs [188]. There is an existing high variability of tumor-infiltrating T cells across tumor types [192–200] and by single-cell RNA sequencing differences between lymphocytes have been observed: (1) cytotoxic genes as GZMA has been related with PD-1 blockade response [201]; (2) overall, gene signatures related to T cell cytotoxic activity have been associated with anti-CTLA-4 ipilimumab response [202]. Tissue-resident T cells and new infiltrating T cells can be found in the TME and response to ICIs vary depending on their cytotoxic effect [188, 203]. Evidence suggests that ICIs promote that less-differentiated memory-like CD8+ T cells convert into effector CD8+ T cells, enhancing anti-tumor response [188].
Furthermore, CD8+ T cells play a role in the genesis of immunotherapy adverse events, for example, gastrointestinal adverse events. Future therapeutic strategies based on blocking the JAK pathway could be beneficial in this context and modulate immunotherapy adverse events [204]. Tofacitinib is a pan-Janus kinase inhibitor that has shown to control immunorelated colitis in case reports and small case series, warranting prospective validation [205].
The activation of the WNT/β-catenin pathway downregulates T cell activation and has been associated with a paucity of T cell infiltration in metastatic melanoma [161, 206–208]. The mechanism through which the activation of β-catenin suppresses ICIs can be related to the inhibition of CCL4 transcription, described in mouse models, which results in impaired CD8+ T cell infiltration [19]. On the contrary, CCL4 levels can recover when there is no activation of β-catenin, which facilitates CD8+ T cells response to ICIs [161]. Moreover, PTEN deficient cells are uncapable of IFN signaling pathway activation and that has also been associated with a decrease in the immune response of the TME [209, 210]. PTEN deficiency can also increase T cell suppression in the TME by the increase of downregulating factors and decreasing the destructive functions of cytotoxic T lymphocytes (CTLs) [209, 210].
CD4 T cells play a role in cancer immunity by providing essential helper functions. Additionally, studies have shown that they can also exhibit direct cytotoxic activity in certain cancer patients [211]. Cancer immunotherapy research has largely centered on CD8+ T cells within the TME [116]. However, studies suggest that effective antitumor immunity requires tumor cells to present MHC class II-binding neoantigens, which are recognized by CD4+ T cells [212]. CD4+ T cells likely play a crucial role in sustaining the cancer immunity cycle by ensuring a continuous supply of CTLs to the TME [213].
CD4+ T cells develop into distinct functional subtypes [214]: Th1 cells, which enhance CD8+ T cell responses to eliminate intracellular pathogens, and Th17 cells, which recruit neutrophils to combat fungi and extracellular bacteria. In cancer immunity, Th1 cells are considered key players by producing IFNγ and boosting CD8+ T cell activity. Meanwhile, Th17 cells, known for their stem cell-like characteristics, are thought to support long-lasting antitumor immunity [215].
Zuazo et al. [154]. showed that baseline systemic CD4 immunity serves as a key factor in determining clinical response. All patients who showed objective treatment responses had functional systemic CD4 T cells, which could be identified prior to therapy by their high proportion of memory CD4 T cells. Inomata et al. [216]. indicated that the proportion of peripheral CD45RA– CD4+ T cells was linked to progression-free survival after beginning ICI therapy, independent of multiple clinical variables. Also, neoantigen-specific CD4 T cells are fundamental [217].
PD-1 expression can also be found in regulatory B cells [218] or CTLA [219] and ICIs could have a direct repercussion on B cells. B cell activation and tertiary lymphoid structures have been correlated with tumor response to ICIs in several types of tumors [161, 188]. In melanoma, single-cell studies have observed that a higher number of B cells in the TME were related to an increased tumor response to PD-1 blockade [192] and there is a higher number of memory-like B cells and plasmablast-like cells in responders [220]. High levels of B-cell-secreted antibodies were found in melanoma anti-CTLA-4 or anti-PD-1 responders [221]. Germain et al. [222] observed that patients with lung cancer that had an infiltration of B-cell in tertiary lymphoid structures which served as a defensive immunological marker.
These other immune cells have an impact on tumor progression and can also be present in the TME [188]. Macrophages show two main states of polarization: (1) M1 macrophages which are activated and can promote CD8+ T cells by antigen presentation and the release of cytokines; (2) activated M2 macrophages secrete TGF-β and show pro-angiogenic and immunosuppressive properties [223]. Furthermore, activated M2 macrophages have been related with hyperprogression in NSCLC patients treated with anti-PD-L1 blockade and, the increase in M2 phenotype could be a consequence of the union of ICIs to macrophage Fc receptors [224]. The antitumor M1 phenotype, CD68+ CD16+ activated M1 macrophages, have been observed in melanoma patients who respond to CTLA-4 blockade therapy in contrast to non-responders [225]. The protumor M2 phenotype can express PD-1 more often and it has been observed that PD-(L)1-blockade could return the M2-polarized function into an antitumor M1 phenotype capable of malignant cell phagocytosis [188, 226]. Nevertheless, the M1/M2 macrophages dichotomy could not reflect other macrophage phenotypes that can be present at the TME [227]. Tumor-associated macrophages (TAMs) can promote tumor cell angiogenesis, proliferation and lymphangiogenesis, invasion and metastasis and can have an impact on anti-tumor therapy response [228, 229]. TAMs upregulate the CCL22 levels that promote Treg production in order to indirectly suppress immune function [230]. TAM subsets and subpopulations, TREM2+, SPP1+, MARCO+, FOLR2+, SIGLEC1+, APOC1+, C1QC+, among others, have been described in cancer and have been associated with poor prognosis [231]. More specifically, MARCO+ macrophages are a subset of TAMs that are immunosuppressive and the inhibition of MARCO was associated with pro-inflammatory phenotype and anti-tumoral coverage of T cells and NK cells in experimental models [232–235].
For the immune response led by CD8+ T cells to occur, DCs, among other antigen-presenting cells, present antigens and they can be divided in classical DCs (cDCs) and type I IFN-producing plasmacytoid DCs (pDCs). Moreover, cDCs are classified into cDC1, that promotes exogenous antigen presentation to CD8+ T cells, and cDC2 which induce Th-2 or Th-17 responses and favor antigen presentation to CD4+ T cells [236, 237]. cDC1 produces IL-12 which has been related with effective anti-PD-1 blockade in mouse models [77]. DCs liberate IL-12 in the presence of IFNγ secreted by T cells during anti-PD-1 blockade therapy, therefore, promoting T-cell immune responses [169].
NK cells are innate lymphocytes that execute cytotoxic functions or secrete cytokines when there is a lack of expression of class I MHC [238, 239]. The presence of PD-1+ NK cells with an activated phenotype have been described in the TME and the PD-L1 binding has been associated with an inadequate function of the NK cells [239, 240]. NK cells exhaustion in mouse models have been observed to diminish the PD-1 and PD-L1 effects on inhibition [241].
Enzymes in the extracellular matrix (ECM), such as matrix metalloproteinases and a distintegrin and metalloproteinsases, can modulate the cell-cell and cell-ECM interplay [161]. Furthermore, the ECM accounts for growth factors and cytokines, in this scenario, a rigid ECM promotes hypoxia and angiogenesis with a depletion of anti-tumor responses [242–244].
Metabolism in the TME is regulated by immune and tumor cells that secrete cytokines, such as ILs, IFN and tumor growth factors, which can alter response to ICIs [161, 245–247]. Glucose is a key factor for the activation of cytotoxic CD8+ T cells and cancer cells compete for glucose consumption in a TME characterized by low-glucose levels and hypoxia [161]. Furthermore, glutamine depletion promoted by cancer cells can decrease T cell activity and suppress immune response [161]. In the tumor cell surface, CD73 and CD38 promote adenosine production which suppresses CD8+ T cell function and generates an acquired resistance to anti-PD-1 blockade [248–250]. Also, indoleamine 2,3 dioxygenase (IDO) can degrade tryptophan into kynurenine that can ultimately activate aryl hydrocarbon receptor that favors an immunosuppressive response by favoring T-cell differentiation to Tregs [161, 251, 252].
The primary host-related factors influencing the effectiveness of ICI treatments are diet and the gut microbiome.
Nutrient consumption has the potential to directly or indirectly influence both the immune system and cancer progression. The majority of existing data are derived from human studies rather than experimental animal models [253]. While potential mechanisms have been identified that may impact immune function and, in turn, cancer growth and response to ICI, there is limited understanding of how these mechanisms affect and modify therapeutic outcomes.
However, there is some evidence regarding the effect of the ketogenic diet on anticancer efficacy. It has been shown to amplify the effects of anti-CTLA-4 therapy by downregulating PD-L1 expression, while also increasing levels of IFN and genes involved in antigen presentation [254, 255]. Additionally, obesity and dietary restrictions have been reported to affect the responsiveness to immunotherapy, though findings in this area remain inconsistent [256]. Moreover, research has explored the immunomodulatory role of vitamin D, which impacts the adaptive immune response by activating Th1/Th17 cells, thus influencing ICI therapy [257]. Various studies have examined how different dietary patterns affect immune responses to ICIs. While some dietary approaches have been found to enhance immune activity, the exact mechanisms are not well understood, and detailed documentation on dietary influences is often lacking [161].
Another series of nutrients have been investigated for their potential role in modulating the response to ICIs [258]. Specific dietary elements, such as vitamins, fatty acids, and other nutrients, may enhance the efficacy of anticancer immunotherapies by influencing immune responses. Caloric restriction and fasting-mimicking diets could potentially lower cancer risk and improve immunotherapy outcomes by inducing immunogenic cell death and increasing the sensitivity of tumor cells. Additionally, polyphenolic compounds such as resveratrol, apigenin, and curcumin may affect PD-L1 expression, thus modulating immune responses against cancer cells and possibly boosting the effectiveness of immune checkpoint therapies. Low-protein diets have shown varied effects on different cancers, with evidence suggesting they might significantly inhibit tumor growth by altering immune responses and amino acid metabolism.
Furthermore, dietary habits have emerged as a major factor in modulating the gut microbiome. Research indicates that nutritional status can affect the microbiome, impacting intestinal balance and immune processes. Consumption of fruits and vegetables leads to more profound and intricate changes in the gut microbiome [259, 260].
Future research should focus on evaluating the effects of diet, nutritional status, and gut microbiota in immunotherapy studies, clarifying the relationship between diet and circulating immune cells and/or the TME.
In recent years, extensive research has led to the identification of numerous microbial communities in the human gut and skin (estimated to be around 30 billion microorganisms per person), and changes in these microbial communities may lead to profound changes in health. Their functions include regulating the immune system [261–263].
Mouse models maintained under germ-free conditions have elucidated the pivotal influence of the gut microbiota on the ontogeny and modulation of multiple organs and physiological systems, encompassing the immune and endocrine systems, as well as the blood, liver, and lungs [264]. Within the intestinal milieu, the microbiota sustains epithelial equilibrium and facilitates the establishment of gut-associated lymphoid tissue. Furthermore, it enhances the secretion of epithelial cytokines, which subsequently orchestrate the functions of T and B lymphocytes, macrophages, and polymorphonuclear leukocytes [265, 266]. Pro-inflammatory cytokines, such as IL-1β, TNF-α, IL-2, IL-6, IL-15, IL-21, and IL-23, are capable of initiating an inflammatory cascade, whereas cytokines like IL-10 and TGF-β exert anti-inflammatory effects. The interplay between these pro-inflammatory and anti-inflammatory mediators governs whether the intestinal environment adopts an inflammatory or homeostatic profile [267]. The microbiota contributes to various immunological processes, including the maturation of DCs, T cell differentiation, and cytokine production, all of which help regulate antitumor immune responses. Moreover, the gut microbiome produces a diverse array of antimicrobial peptides (AMPs) that defend the host against various pathogens and can enhance immune responses by boosting innate immunity and reducing inflammation [161]. Certain AMPs have the ability to attract and activate immune cells and trigger inflammatory responses. This functional aspect of AMPs makes them valuable as immunomodulators, potentially explaining how the gut microbiota might improve the effectiveness of ICIs [268, 269].
Research has shown that the gut microbiota’s composition can influence the effectiveness of ICIs. For instance, the efficacy of anti-CTLA-4 agents is impacted by specific microbiota profiles, such as those involving Bacteroides fragilis, Bacteroides thetaiotaomicron, and Burkholderiales, which can affect the TH1 immune response triggered by IL-12 [270, 271]. A study by Peng et al. [272] highlighted a connection between the gut microbiome and clinical responses to anti-PD-1/PD-L1 therapy in gastrointestinal cancers. This research found that a higher Prevotella/Bacteroides ratio (an indicator of positive regulation) is associated with better responses to anti-PD-1/PD-L1 treatments. Additionally, responders exhibited a significantly higher abundance of certain microbiota, including Prevotella, Ruminococcaceae, and Lachnospiraceae.
Fecal microbiota transplantation (FMT), which involves transferring gut microbiota from one individual to another’s intestinal system, has emerged as a potential method for reshaping the structure and function of gut microbiomes in recipients. Extensive research has been carried out on using FMT to alter gut microbiota in various in vivo models and cancer patients, with the aim of overcoming resistance to ICIs [273–275].
Scientific research indicates that probiotics can influence both innate and adaptive immune responses. Klein and colleagues [276] found that daily probiotic supplementation notably enhanced the proportion of granulocytes and monocytes exhibiting phagocytic activity in a healthy cohort, compared to a placebo. Gill’s group [277], also confirmed these findings, noting a significant rise in serum antibody responses to both orally and systemically administered antigens in mice treated with probiotics. Lactobacillus and Bifidobacterium are among the most extensively studied probiotics in both animal models and clinical trials. Their immunomodulatory potential has been demonstrated through research on the prevention of allergic diseases [278]. Unfortunately, to date, there are no studies demonstrating their efficacy as adjuvant therapy for ICIs. Figure 2 summarizes the determinants of ICI effectiveness and strategies to enhance its antitumor activity.
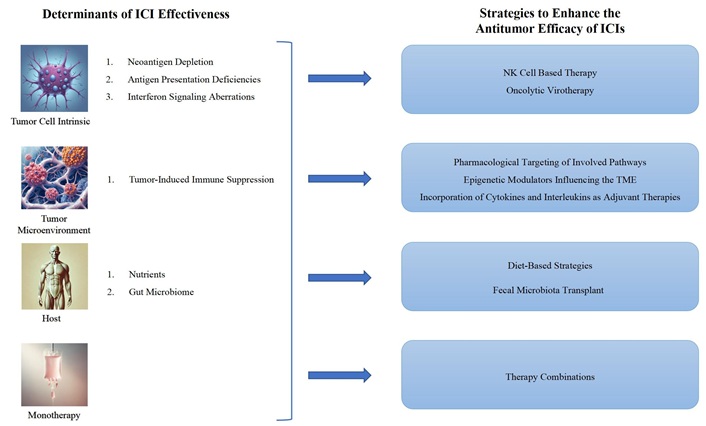
Determinants of ICI effectiveness and strategies to enhance its antitumor activity. The factors influencing ICI effectiveness can be grouped into: (a) Tumor cell-intrinsic mechanisms, including 1. loss of neoantigens, 2. deficiencies in antigen presentation, 3. disruptions in interferon signaling; (b) the TME; (c) host-related factors, such as 1. nutritional status, 2. the gut microbiome; (d) the constraints of monotherapy. To address these, strategies to enhance ICI antitumor activity include: (a) employing NK cell-based therapies and oncolytic virotherapy targeting tumor cell-intrinsic mechanisms; (b) using pharmacological inhibitors, cytokines, interleukins, and adjuvants to counteract immunosuppression within the TME; (c) implementing dietary interventions and FMT; (d) adopting combination therapy approaches. ICI: immune checkpoint inhibitors; TME: tumor microenvironment
Probably, one of the main challenges for the future of oncology will be how to reverse both primary and acquired resistance to immunotherapy. One of the most significant attributes of ICIs is their ability to induce a durable response. Unfortunately, this outcome is only observed in a subset of cancer types and specific patients, owing to the issue of ICI resistance.
Despite the rapid progress in understanding drug resistance to immunotherapy, clinical trials focused on overcoming ICI resistance still face significant challenges. With advancements in medical science, the emphasis has shifted towards ‘personalized medicine’ and ‘precision treatment’ in oncology. Biomarkers play a crucial role in the detection, management, and prognosis of tumors. Currently, PD-L1 remains the most frequently utilized biomarker to forecast immunotherapy effectiveness, though it has notable limitations. Additionally, other indicators such as tumor mutational burden, TILs, and microsatellite instability are also valuable in predicting the success of immunotherapeutic approaches.
Consequently, there is a pressing need to further elucidate the mechanisms underlying resistance to ICIs. This review has examined potential mechanisms of resistance involving tumor cells, the TME, and host-related factors. Additionally, resistance mechanisms may occur concurrently or sequentially. Given the intricate nature of these mechanisms and the TME, it is essential to explore combined strategies to address ICI resistance. Currently, a standardized approach to this issue remains elusive. Integrating immunotherapy with other modalities such as chemotherapy, radiotherapy, and targeted therapy represents a viable strategy for managing a range of cancers.
The following sections briefly discuss different research strategies to overcome ICI resistance (Table 2):
Immune checkpoint inhibitor combination strategies
| Combination | Effects |
|---|---|
| ICI + ICI | Promote T cell activation and the immune response to tumor cells |
| ICI + TT | Increase T-cell infiltration and IFN-gamma expression, besides its specific action on the target |
| ICI + ChT | Reduce the number and activity of immune-suppressive cells and stimulate tumor-specific immune responses by inducing the immunogenic cell death |
| ICI + RT | ICI enhances the immunogenic effects of RT and RT reduces the number and activity of immune-suppressive cells and remodeling of the TME by secreting inflammatory factors |
| ICI + AA | Boost immune cell infiltration, elevate adhesion molecules, promote antigen presentation and reduce Treg immersion |
| ICI + ACT | Increase IFN-gamma-expression |
| ICI + nanomedicine | Transform “deserted tumors” to “inflamed tumors” by boosting T-cell priming and immersion |
ICI: immune checkpoint inhibitor; TT: targeted therapy; IFN: interferon; ChT: Chemotherapy; RT: radiotherapy; TME: tumor microenvironment; AA: anti-angiogenesis; ACT: adoptive cellular therapy
The co-administration of different ICIs has shown to enhance immune responses through their complementary effects. Specifically, agents targeting CTLA-4 and PD-1/PD-L1 operate through distinct signaling pathways. Numerous clinical trials have demonstrated that dual inhibition of PD-1/PD-L1 and CTLA-4 yields superior efficacy compared to monotherapy. Ongoing clinical studies are exploring the efficacy of ICIs in combination with inhibitors targeting additional immune checkpoints, such as TIM-3, LAG-3, V-domain Ig suppressor of T-cell activation (VISTA) and TIGIT [279, 280].
Clinical trials have demonstrated that integrating ICIs with targeted therapies can enhance treatment outcomes. For instance, the addition of atezolizumab to targeted therapy has been shown to improve PFS in melanoma patients with BRAF mutations [281]. Similarly, combining niraparib with pembrolizumab has resulted in higher response rates among individuals with metastatic BRCA-mutant triple-negative breast cancer [282]. However, the effectiveness of this combined approach is currently limited to specific tumor types.
A multitude of studies have explored the effectiveness of combining ICIs with chemotherapy. This approach not only reduces the number of immunosuppressive cells within the TME but also increases the presentation of tumor antigens by inducing tumor cell apoptosis [283, 284]. Such treatment enhances tumor cell clearance and boosts immune system recognition through improved immune surveillance. Presently, extensive research is being conducted on the concurrent and sequential use of ICIs with chemotherapy, revealing significant efficacy improvements across a range of cancer types [285].
Radiotherapy induces cancer cell apoptosis through DNA damage and influences immunogenicity by releasing inflammatory mediators, which can elevate neoantigen expression. It also significantly impacts the TME by secreting these inflammatory factors [286–289]. Nonetheless, challenges remain in optimizing the balance between enhancing immune stimulation and reducing immunosuppressive effects, as well as in managing or preventing potential increases in toxicity, such as pneumonitis.
Tumor angiogenesis is partially regulated by the immune-suppressive environment within the tumor. The activation of T cells results in the production of IFN-γ, which interacts with its receptor on endothelial cells in tumors, leading to the regression of tumor-associated blood vessels [290, 291]. Evidence from murine models shows a synergistic effect between ICIs and anti-angiogenic agents [292, 293]. These agents inhibit the development of blood vessels essential for tumor growth and metastasis and help to restore the TME [292, 294]. In vivo studies have reported an increase in anti-tumor immune cell populations and a decrease in pro-tumor immune cells [295]. Therapies that combine ICIs with anti-angiogenic agents have demonstrated greater efficacy compared to monotherapy [296, 297].
TILs can be utilized to generate immune cells that are less influenced by the suppressive TME. In the context of ICI therapy, T cells may exhibit dysfunction, which can diminish their effectiveness in some patients. Employing TILs has been proposed as a method to address this T cell dysfunction. This approach has demonstrated efficacy in specific cancers, such as metastatic NSCLC that progresses despite ICI treatment [298].
Recently, integrating ICIs with nanomaterials has emerged as a promising strategy to improve the efficacy of ICIs. Research indicates that nanoparticles may enhance the effectiveness of ICIs while mitigating their adverse effects. Notable applications include targeted drug delivery to specific cells and the development of tumor vaccines aimed at DCs. Combining nanomaterials with ICIs has the potential to convert ‘cold’ tumors into ‘inflamed’ tumors by improving T-cell activation and infiltration [299, 300].
Despite advancements in these strategies, challenges persist in selecting the ideal patient population for combination therapies, achieving an optimal balance of synergistic antitumor effects, and elucidating the adverse reactions linked to these treatments.
Immunotherapy is the treatment paradigm against cancer, with resistance to it being one of the most important challenges in oncology. This review summarizes the role of ICIs, as well as their resistance mechanisms and various approaches to reverse it. Much of the research investment in oncology is focused on finding strategies to enhance and overcome both primary and acquired resistance to ICIs. Hopefully, these strategies will soon become a reality in routine clinical practice, exponentially increasing the number of potential patients with advanced cancer who are ‘cured’ or long-term survivors.
AMPs: antimicrobial peptides
APCs: antigen-presenting cells
cDCs: classical dendritic cells
CTLA-4: cytotoxic T-lymphocyte antigen 4
CTLs: cytotoxic T lymphocytes
DCs: dendritic cells
ECM: extracellular matrix
Fc: fragment crystallizable
FcγRs: Fcγ receptors
FMT: fecal microbiota transplantation
HLA: human leukocyte antigen
ICIs: immune checkpoint inhibitors
IFN: interferon
Ig: immunoglobulin
IgGs: immunoglobulin Gs
IL: interleukin
JAK1/2: Janus kinase 1/2
LAG-3: lymphocyte activation gene-3
MDSCs: myeloid derived suppressor cells
MHC: major histocompatibility complex
NK: natural killer
NSCLC: non-small cell lung cancer
PD-1: programmed cell death protein 1
PD-L1: programmed death ligand 1
SHP-2: Src homology region 2 domain-containing phosphatase-2
STAT: signal transducer and activator of transcription
TAMs: tumor-associated macrophages
TCR: T cell receptor
TGF-β: transforming growth factor beta
TIGIT: T-cell Immunoreceptor with Ig and ITIM domains
TILs: tumor-infiltrating lymphocytes
TIM-3: T-cell immunoglobulin and mucin-domain containing-3
TME: tumor microenvironment
Tregs: regulatory T cells
β2M: β2 microglobulin
This manuscript is dedicated to all patients and their families. They are the primary motivation behind our research efforts. We also acknowledge that Figure 2 was created with the help of ChatGPT and appreciate its contribution to the development of this work.
LCG: Data curation, Visualization, Investigation, Writing—review & editing, Writing—original draft. SCC: Conceptualization, Methodology, Software. MPK: Supervision, Investigation. BCO: Methodology, Writing—review & editing, Validation. VPB: Data curation, Validation, Investigation, Writing—review & editing. All authors read and approved the submitted version.
Luis Cabezón-Gutiérrez reports he received payment for presentations of Roche, Astra Zeneca, Brystol Myers Squibb, Merck Serono, Ipsen Pharma, Grunenthal, Kyowa Kirin, Pfizer and Eisai and received support for attending meetings from Roche, Merck, Eli Lilly, Bristol-Myers Squibb and Nutricia. Vilma Pacheco-Barcia reports she received a grant as an award from Merck and FSEOM, payment for presentations of Merck, Eli Lilly, Eisai and Pierre Fabre and received support for attending meetings from Roche, Eli Lilly, Bristol-Myers Squibb, Merck, Amgen, Merck Sharp and Dhome, and Nutricia. Vilma Pacheco-Barcia also reports she participated in an advisory board from advanced accelerator applications, a Novartis company. Sara Custodio-Cabello has received honoraria (outside of this submitted study) from Fresenius, Astellas Pharma, Merck and Abbott and received support for attending meetings from Pierre-Fabre and Amgen. Magda Palka-Kotlowska has received payment for presentations of Pfizer, Devon, Pharmamar, and Esteve and received support for attending meetings from Pfizer and Novartis. Beatriz Chacón-Ovejero have no conflicts of interest to declare. All the authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Raffaele Pellegrino ... Antonietta Gerarda Gravina
Lingli Zhao ... Gaoli Niu
Rawaa AlChalabi ... Ahmed AbdulJabbar Suleiman
Qing Bao ... Hailin Tang
Fakher Rahim ... Issenova Balday
Neha Kannan ... Giuseppe Minervini
