Original Article
Original Article

Affiliation:
1Bioscience Department, School of Human Sciences, London Metropolitan University, N7 8DB London, United Kingdom
Email: a.gerges@londonmet.ac.uk
ORCID: https://orcid.org/0000-0002-1979-9129
Affiliation:
2Independent Scholar, SE4 1JT London, United Kingdom
ORCID: https://orcid.org/0000-0002-0091-9776
Explor Drug Sci. 2025;3:1008109 DOI: https://doi.org/10.37349/eds.2025.1008109
Received: January 09, 2025 Accepted: April 15, 2025 Published: May 09, 2025
Academic Editor: Fernando Albericio, University of KwaZulu-Natal, South Africa, Universidad de Barcelona, Spain
 A Correction to this article was published on 09 January 2026
A Correction to this article was published on 09 January 2026
Aim: This paper investigates two possible treatment targets for neuroblastoma (NB) stage 4 (NBS4), c-Src kinase (Csk) and retinoic acid (RA) signalling pathways as potential candidates for a multi-target drug. Research has demonstrated that many cancer cells overexpress and/or hyperactivate c-Src, a tyrosine that is a member of the Src-family kinases. In the case of NBS4, there are indications that successful inhibition of c-Src could inhibit disease progression. Research into the altered signalling of RA, which preserves the differentiated state of adult neurons, neural stem cells, and NB cells (SH-SY5Y), is also investigated as a potential multi-target drug.
Methods: Using computer-aided technology, including OpenEye Scientific suite, Molegro Virtual docking, Samson suite, and Discovery Studio Visualiser, the results revealed that the receptors for both targets, Csk and RA, share similar amino acid sequencing that ranges from 80–100%, offering the possibility of further testing for multi-target drug use. Work was done to explore possible synthesis routes for each of the four compounds using the retrosynthesis program Spaya. Predictive toxicology was done using the Toxicity Estimation Software Tool (T.E.S.T.).
Results: Four compounds (inhibitors) targeting the Csk tyrosine kinase and RA pathways were identified as potential inhibitors.
Conclusions: Currently, no effective therapeutic agents for NBS4 exist. Immunotherapy which has proven effective in treating various cancers, is currently used to treat NBS4 and has a 40% to 50% survival rate. This paper investigates two possible treatment targets for NBS4, Csk and RA signalling pathways as possible candidates for a multi-target drug. Four potential inhibitors have been identified.
There are five stages in the clinical neuroblastoma (NB) staging system: 1, 2A, 2B, 3, 4S, and 4 [1]. For those diagnosed with NB stage 4 (NBS4) the treatment options are limited with a survival rate of between 40% to 50% [1]. In a review of drug therapies aimed at treating NBS4, Gerges and Canning [2] (2024) examined the potential for creating multi-targeted drugs to treat high-risk NBS4 using computational methods available to medicinal chemistry. Finding novel therapeutic drug targets by this approach usually involves computational techniques like molecular docking, homology modelling, molecular dynamics, and quantitative structure-activity relationships (QSAR) [3]. The work reported here follows on from that review and investigates two different types of inhibitors for the signalling pathways c-Src kinase (Csk) and retinoic acid (RA) signalling pathways[2]. Six targets were originally identified and investigated by the authors [2] and the results of the Protein Aligner revealed that the receptors for four of the six targets shared similar amino acid sequencing that ranged from 80–100%. The four targets that share similar sequencing included histone deacetylase (HDAC), bromodomain (BRD), hedgehog (HH), and tropomyosin receptor kinase (TRK) and are the focus of a separate paper. The remaining two targets of the original six, Csk and RA signalling pathways showed similarity across their receptors of 80–100% are the focus of this paper. Their potential for multi-targeted drug use was investigated further with four compounds (inhibitors) targeting the Csk and RA pathways identified.
With therapeutic agents finding little success in the treatment of NBS4, immunotherapy which has been effective in treating a number of cancers is currently used. This treatment uses monoclonal antibodies and has increased the 5-year survival rate of NBS4 to 40% to 50% [4–6]. Dinutuximab (UNITUXIN), a monoclonal antibody, is an example of cancer immunotherapy and for patients with NBS4, it is used in conjunction with other therapeutic agents. It was approved by the FDA in 2015 for use in the United States [6].
Proliferation, differentiation, migration, and membrane trafficking are just a few of the cellular functions that are critical to the Src family kinases (SFKs) of non-receptor tyrosine kinases (TK) [7]. TK deregulation has been linked to cancer, with pharmaceutical companies and academic institutions now targeting small molecule TK inhibitors (TKIs) as one of the largest drug families for the treatment of cancer (Figure 1) [8]. TKIs may be associated with fewer harmful side effects during anti-tumor treatments because they target particular molecular targets [8]. As a result, numerous TKIs have undergone testing for their anti-proliferative and anti-cancer properties in vitro and in vivo, with some approved for clinical trials and others currently used in cancer treatment [9]. One subclass of non-receptor TKs targeted in the treatment of human cancers is the SFKs. Of these, c-Src promotes angiogenesis, invasion, migration, and cell proliferation (Figure 1) [10]. c-Src hyperactivation results in abnormal cell activity, which aids in cancer development. High expression levels of c-Src, which have been found in several cancers, are typically associated with a poor overall survival prognosis [11]. More than one study has shown that c-Src inhibitors play an important role in antiproliferative activity in SH-SY5Y cells (NBS4 cell line) [12–23].
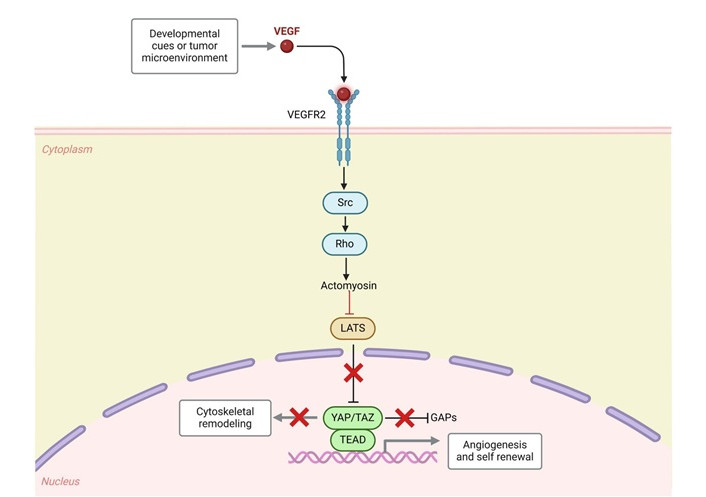
c-Src in cancer. VEGF: vascular endothelial growth factor; VEGFR2: VEGF receptor 2; LATS: large tumor suppressor; YAP: Yes-associated protein; TAZ: transcriptional co-activator with PDZ-binding motif; TEAD: transcriptional enhanced associate domain; GAPs: GTPase-activating proteins. Created in BioRender. Gerges, A. (2024) https://BioRender.com/t65n288
The small lipophilic molecule RA, derived from vitamin A, is essential for axonal growth, neuronal differentiation, neuronal patterning, and embryonic development (Figure 2) [24]. Research on the effect of RA on neuronal development has improved our knowledge of neurodegenerative diseases [25]. It has been noted that the symptoms of neurodegenerative diseases are caused by RA altered signalling, which maintains the differentiated state of adult neurons, neural stem cells, and NB cells in particular (cell line SH-SY5Y) [26–36].
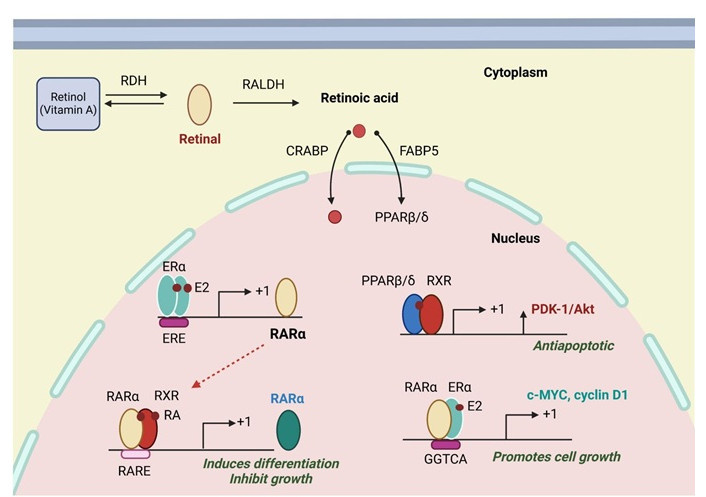
Retinoic Acid receptors and their actions. The three retinoic acid receptor subtypes (RARα, RARβ and RARγ) act as ligand-inducible transcription factors binding to DNA regulatory elements in the promoter regions of target genes by forming heterodimers with the retinoid X receptors (RXRα, RXRβ and RXRγ). RDH: retinol dehydrogenase; PALDH: retinaldehyde dehydrogenase; CRABP: cellular retinoic acid-binding protein; FABP5: fatty acid binding protein 5; PPARβ/δ: peroxisome proliferator-activated receptor beta/delta; ERα: estrogen receptor alpha; ERE: estrogen response elements; E2: estradiol; RARα: retinoic acid receptor-alpha; RXR: retinoid X receptor; PDK-1: phosphoinositide-dependent kinase-1; RA: retinoic acid; RARE: RA responsive element. Created in BioRender. Gerges, A. (2025) https://BioRender.com/v19a099
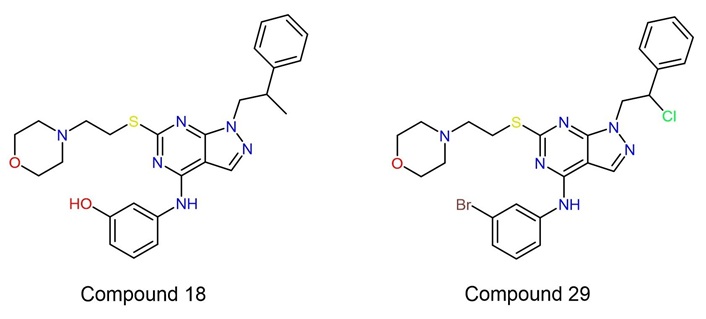
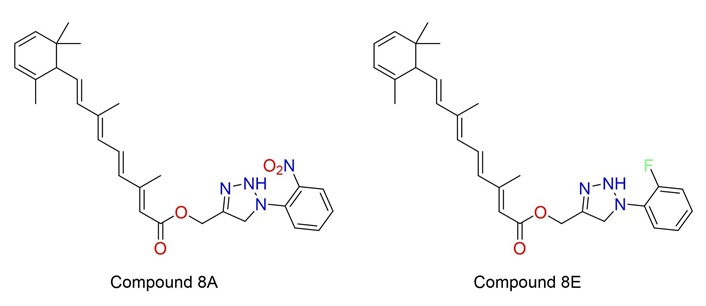
In the search for a multi-target drug for NBS4 using advanced medicinal chemistry software, the approach is to assesses the possibility of different targets (Figures 1 and 2) by assessing receptor similarities and selecting two lead compounds for each target (four compounds in total), see Figure 3 [8] and Figure 4 [29], that have demonstrated an inhibitory effect on NBS4 cells to form the basis of a search for a multi-targeted drug [29].
For the two targets (Csk and RA) two receptors from each target were selected (Table 1) and their amino acid sequence similarity tested using Protein Aligner [37] (see Figure 5), which reported 80–100% similarity. Protein Aligner checks for amino acid sequence similarity between receptors with high similarity indicating their suitability for use in cross-docking. For the purposes of this work docking involves docking a compound to a receptor known for that target (i.e. Csk 4O2P [8] and 5D10 [38]) whereas cross-docking involves docking a compound to a receptor that belongs to a different target (i.e. RA 1CBQ and 1CBS [39]) and vice versa. The aim of cross-docking is to explore the suitability of selected compounds belonging to different targets to determine their suitability for use as a multi-target drug. This is a process that involves selecting known lead compounds to produce hitlists of compounds, and was done using BROOD [40] as part of the OpenEye suite.
Drug targets and their selected receptors
| Drug target (n = 2) | Proteins selected from PDB (n = 4) |
|---|---|
| c-Src kinase (Csk) | 4O2P [8] and 5D10 [38] |
| Retinoic acid (RA) | 1CBQ and 1CBS [39] |
PDB: Protein Data Bank
Several BROOD rounds (Figure 6) were completed on the two lead compounds for each target to produce hitlists using “Shape and Color” and “Shape and Electrostatics”. On completion, BROOD ranked each of the hitlists according to BROOD hitlist parameters (see list below) and the top 25 of each hitlist was selected to run on OEDocking, using FRED (Figure 7) [41], ROCS (Figure 8) [42], AFITT (Figure 9) [43], and VIDA [44]. In addition, the Samson docking suite, including AutoDock Vina Extended (Figures 10 and 11) [45, 46] and Fitted (Molecular Forecaster) [47], with another docking program, Molegro [48], acted as confirmation for both the docking and cross-docking procedures. From this process, eight compounds for the Csk target were identified and six from the RA target, making a total of 14 compounds. All 14 compounds showed improved parameters compared to the original lead compounds (see Table 2). The compounds for each individual target were then cross-docked with the receptors identified for the other targets [43]. From the 14 compounds run, four showed potential suitability for multi-target use (two from each list) see Table 2.
Docking results from AFITT with the top clusters from the hitlists (n = 14)
| Target | Clusters | AFFIT | FRED | AutoDock Vina Extended | Molegro | Fitted |
|---|---|---|---|---|---|---|
| RA (n = 6) | Clusters | 1CBS | 1CBS | 1CBS | 1CBS | 1CBS |
| 1 | Cluster 18, 1 of 1 | 0.6619 | –6.652 | –8.355 | 106.873 | –25.3637 |
| 2 | Cluster 3, 1 of 30 | 0.6612 | –7.42 | –9.364 | 32.95 | –28.3973 |
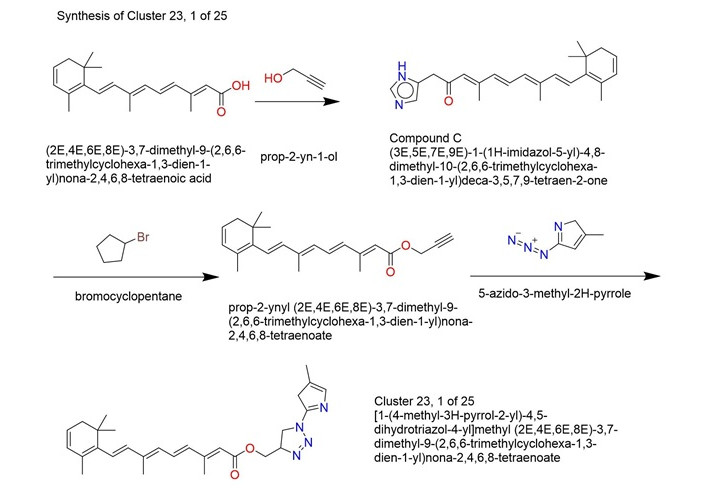
| 3 | Cluster 23, 1 of 25 | 0.6207 | –7.27 | –9.222 | 113.34 | –22.8749 |
| 4 | Cluster 20, 1 of 7 | 0.5474 | –9.061 | –9.472 | 14.94 | –23.4724 |
| 5 | Cluster 21, 1 of 5 | 0.5448 | –4.6477 | –8.952 | 76.28 | –20.8754 |
| 6 | Cluster 22, 1 of 4 | 0.5448 | –4.67 | –8.843 | –10.53 | –25.0819 |
| 7 | 8E (lead compound) | 0.4543 | –4.156 | –8.589 | –94.37 | –23.6039 |
| c-Src (n = 8) | Clusters | 402P | 402P | 402P | 402P | 402P |
| 1 | Cluster 16, 1 of 1 | 0.7654 | –6.03 | –8.384 | –58.58 | –25.413 |
| 2 | Cluster 20, 1 of 5 | 0.7471 | –6.51 | –8.361 | –45.59 | –26.526 |
| 3 | Cluster 7, 1 of 1 | 0.5397 | –6.43 | –8.856 | –60 | –28.785 |
| 4 | Cluster 19, 1 of 5 | 0.5065 | –5.7 | –8.386 | –58.99 | –22.472 |
| 5 | Cluster 1, 1 of 43 | 0.503 | –6.34 | –8.931 | –58.72 | –24.192 |
| 6 | Cluster 17, 1 of 3 | 0.489 | –5.65 | –8.823 | –39.86 | –26.285 |
| 7 | Cluster 4, 1 of 1 | 0.3898 | –6.88 | –8.392 | 58.04 | –28.788 |
| 8 | Cluster 3, 1 of 1 | 0.3717 | –3.63 | –8.655 | –44.07 | –27.369 |
| 9 | Compound 18 (lead compound) | 0.7432 | –6.34 | –8.108 | –45.94 | –24.476 |
Selected compounds in bold. RA: retinoic acid
To check and compare the compounds (clusters) for toxicity and mutagenicity, the Toxicity Estimation Software Tool (T.E.S.T.) was used to provide the prediction mechanisms of the toxic action of the clusters [49]. The results from T.E.S.T. showed some similarity with the original lead compounds (Table 3), which also confirmed their suitability for use. Further work was done to explore possible synthesis routes for each of the four compounds using the retrosynthesis program Spaya [50].
Results from the T.E.S.T. compared to the lead compounds
| Clusters | Bioconcentration factor1 | Ames mutagenicity2 | Oral rat LD503 [–Log10(moL/kg)] | 48-hour T. pyriformis IGC504 (mg/L) |
|---|---|---|---|---|
| 18, 1 of 1 | 19.08 | Negative | 373.49 | N/A |
| 23, 1 of 25 | 18.78 | Negative | 833.23 | N/A |
| 19, 1 of 5 | 39.65 | N/A | 839.23 | N/A |
| 4, 1 of 1 | 21.54 | N/A | 1,714.20 | N/A |
| Compound 8A | 12.25 | Negative | 881.07 | N/A |
| Compound 8E | 15.68 | Negative | 1,193.20 | N/A |
| Compound 18 | 15.64 | Positive | 1,010.18 | N/A |
| Compound 29 | 28.91 | Positive | 457.49 | N/A |
1 Bioconcentration factor: ratio of the chemical concentration in fish as a result of absorption via the respiratory surface to that in water at a steady state; 2 Ames mutagenicity: a compound is positive for mutagenicity if it induces revertant colony growth in any strain of Salmonella typhimurium; 3 Oral rat LD50: amount of chemical (mg/kg body weight) that causes 50% of rats to die after oral ingestion; 4 48-hour T. pyriformis IGC50: Concentration of the test chemical in water (mg/L) that causes 50% growth inhibition to Tetrahymena pyriformis after 48 hours. N/A: not applicable
Computer programs:
OpenEye Scientific programs, which include various applications. The suite comprises BROOD, VIDA, MakeReceptor, FRED, and AFITT;
Molegro Virtual Docker;
The Samson suite includes Protein Aligner, AutoDock Vina Extended, and the Fitted suite by Molecular Forecaster;
T.E.S.T.;
BIOVIA Discovery Studio Visualizer [51];
Spaya-retrosynthesis software.
Identifying drug targets;
Selection of two proteins (receptors) for each target;
Download the Protein Data Bank (PDB) files and their electron density map from the PDB database;
Comparing the similarities of the receptors. Run protein similarity on Samson (Protein Aligner) to determine suitability;
Selection of two lead compounds for each type;
Run the lead compounds on BROOD (from the OpenEye suite) and produce hitlists using Shape and Colour and Shape and Electrostatics;
Receptor preparations using MakeReceptor from the OpenEye suite;
Docking the hit compounds with OpenEye suite (FRED), Molegro, and Samson suite (AutoDockVina and Fitted);
Run cross-docking: each hitlist clusters from one target to the other target (using their protein/receptor);
Run hits with AFITT to rank the compounds according to their fitting probabilities;
Run selected clusters on ROCS;
Run selected clusters on T.E.S.T.;
Run clusters on Spaya to find the best synthesis route.
Figure 5 shows the Protein alignment [37] (similarities) between the selected proteins ranging from 80% to 100%.
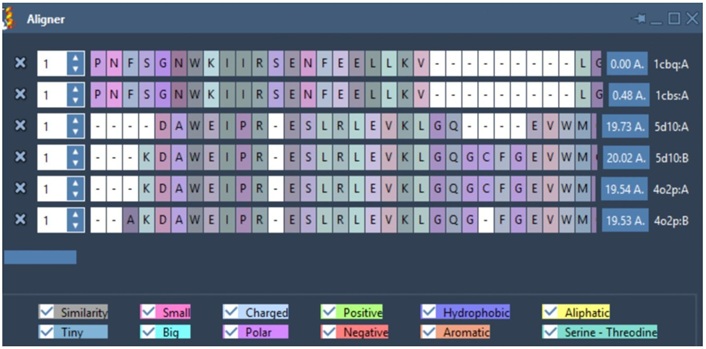
Protein alignment of the selected receptors from two targets. The similarity (in grey) was obtained using Protein Alignment by Samson. Zero indicates 100% similarity, and 100% means no similarity
Figure 6 below shows some of the hitlist from BROOD [40].
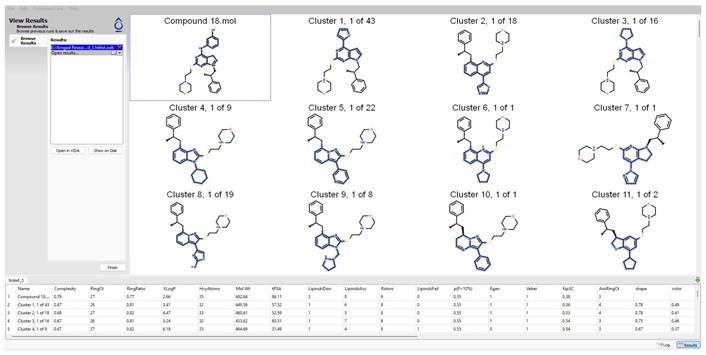
Print screen of BROOD (Shape and Color) outcome when using compound 18 as a lead compound, only showing the first 11 clusters
BROOD Hitlist parameters (Figure 6) include:
AroRingCt: number of aromatic rings in the molecule;
ClusterID/IdeaGroup: cluster ID of the molecule;
Colour: the replacement fragment's colour Tanimoto score in comparison to the query fragment;
Combo: Tanimoto combo score for the replacement fragment's shape and colour in comparison to the query fragment;
Egan: the Boolean indicates if the molecule satisfies the Egan bioavailability model;
Fragment: SMILES string of the replacement fragment;
Freq: the replacement fragment’s frequency;
fsp3C: the molecule’s fraction of sp3 hybridized carbon atoms;
HvyAtoms: number of heavy atoms in the molecule;
LipinskiDon: number of Lipinski donors in the molecule;
LipinskiAcc: number of Lipinski acceptors in the molecule;
LipinskiFail: Boolean specifying whether the molecule fails Lipinski’s rule of five;
Local strain: calculated local strain of the molecule;
Molecular TanimotoCombo: shape + colour Tanimoto combo score of the molecule against the query molecule;
MolWt: molecular weight of the molecule;
p(active): belief score of the molecule;
RingCt: number of ring atoms;
RingRatio: ratio of the number of ring atoms to the total number of heavy atoms;
Rotors: number of rotatable bonds in the molecule;
Shape: compare the replacement fragment’s Shape Tanimoto score to that of the query fragment;
Source Mols: SMILES strings of the molecules the replacement fragment is part of;
Source Mol Labels: labels of the molecules the replacement fragment is part of;
tPSA: calculated topological polar surface area of the molecule;
Veber: Boolean specifying whether the molecule passes the Veber bioavailability model;
XlogP: calculated LogP of the molecule [52].
The next stage was to dock the hitlists with their relevant receptors: Figure 7 shows one of the docking outcomes using FRED (OpenEye suite).
The chosen compounds were also subjected to ROCS, a program that scores and aligns a database of molecules with a query. The score assigns a number to molecules according to their likelihood of having biological characteristics in common with the query molecule (see Figure 8).
AFITT is another crystallographic tool from the OpenEye suite (Figure 9). By combining the shape and MMFF technologies of OpenEye, AFITT creates a new combined forcefield that fits small molecules into crystallographic density while preserving superior chemistry. It also offers an interface to external refinement programs, such as real space correlation coefficient (RSCC) calculation and interactive Ramachandran plots to verify the refinement [43].
AutoDock Vina Extended (Figures 10 and 11) showing the docking of clusters to receptors 402P and 1CBS.
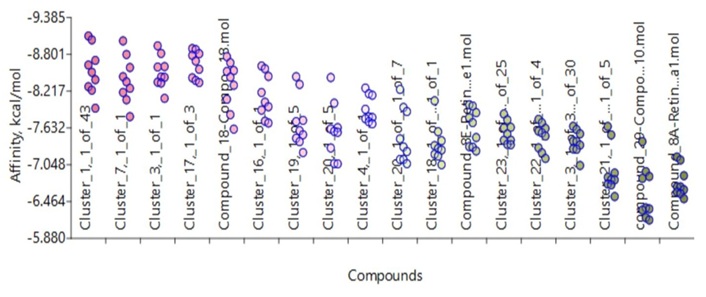
Docking receptor 4O2P with all the clusters from c-Src and RA, including the lead compounds, using AutoDock Vina Extended from Samson suite
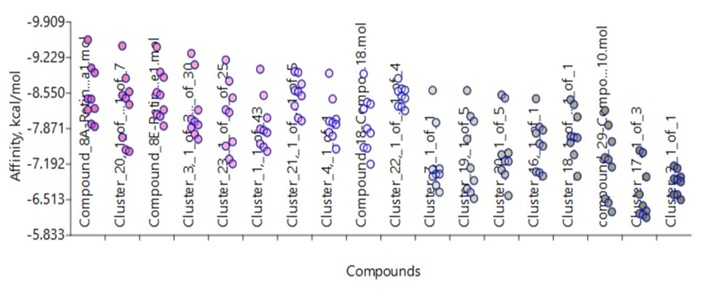
Docking receptor 1CBS with all clusters from c-Src, and RA, including the lead compounds, using AutoDock Vina Extended
The process of developing a multi-target drug is described as taking “a well-established primary target for a particular disease [and adding], secondary activities […] to enhance efficacy and reduce the effect” [53]. Creating such a drug involves taking the inhibitory effects of two or more drugs and combining them into a single molecule [54]. Previous methods used for drug development include “Pharmacophore” and “Screening”. This article employs a computer-aided virtual screening (VS) strategy with some modifications in the pursuit of multi-target drug discovery. Having identified two targets, two receptors from each target were selected (see Table 1) and a Protein Aligner tool from Samson was used to check for their suitability. Protein Aligner checks for amino acid sequence similarity between receptors with high similarity indicating their suitability for use in cross-docking. The aim of cross-docking is to explore the selected compounds for use as a multi-target drug. This is a process that involves selecting known lead compounds to produce hitlists of compounds and was done using BROOD [40] as part of the OpenEye suite.
As part of the OpenEye suite, several rounds from BROOD were completed on the two lead compounds for each target to produce hitlists using “Shape and Color” and “Shape and Electrostatics” with the top 25 of each hitlist selected to run on OEDocking [41], ROCS [42], AFITT [43], and VIDA [44]. The hitlists of compounds (clusters) were docked, and the selected compounds cross-docked. The clusters were docked with more than one docking program as a means to validating the result. The final ranking of the selected clusters was done on AFITT as the receptor preparation with the tool MakeReceptor, giving the user more control over the receptor-creating process. AFITT also has the advantage of real-space fitting of ligands in density, integrated with REFMAC, PHENIX, BUSTER, CNX, and COOT and fragment and cocktail fitting [43]. In AFFIT, it is possible to select more than one ligand to fit generation of high-quality refinement dictionaries for use. This can be done during reciprocal space refinement that includes the use of forcefield (MMFF); semi-empirical (AM1, PM3) methods during reciprocal space refinement for BUSTER and Phenix; real space fitting of protein residues; proper handling of covalently bonded ligands, and proper handling of multiple occupancy ligands.
The ranking is based not only on the best results but also on the ability of the identified compounds to cross-dock on the receptors of other targets, and it was this process that led to the selection of the four compounds. Using the tool ROCS provided cluster validation, which is based on a large database search [42]. The toxicity and mutagenicity of the four compounds were tested using T.E.S.T. to provide the prediction mechanisms of the toxic action of the clusters [49]. The results from T.E.S.T. showed some similarity with the original lead compounds (Table 3), which also provided data on their suitability for use. Further work was done to explore possible synthesis routes for each of the four compounds using the retrosynthesis program Spaya [50].
In conclusion using a computer-aided modified VS approach, four compounds were identified demonstrating the possibility of inhibiting two targets simultaneously: the Csk and the RA signalling pathways. Going forward, future research will concentrate on preparing and evaluating the identified compounds both in vitro and in vivo [55].
Csk: c-Src kinase
NB: neuroblastoma
NBS4: neuroblastoma stage 4
PDB: Protein Data Bank
RA: retinoic acid
SFKs: Src family kinases
T.E.S.T: Toxicity Estimation Software Tool
TK: tyrosine kinase
TKIs: tyrosine kinase inhibitors
VS: virtual screening
Thanks to Dr. Corinne Kay for her advice and support over the years and to Mr. Colin Gaudion for testing some of the programs used for this work.
AG: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Validation, Writing—original draft. UC: Writing—review & editing. Both authors read and approved the submitted version.
The authors’ daughter, Isabella, was diagnosed with NBS4 in January 2003. Isabella relapsed in March 2005 and died in July 2005, a week after her seventh birthday.
Not applicable.
Not applicable.
Not applicable.
The datasets [generated/analyzed] for this study can be found in the [Zenodo database] [https://zenodo.org/records/14620395?preview=1&token=eyJhbGciOiJIUzUxMiJ9.eyJpZCI6IjQ0OGE0YzVkLWU2N2EtNDgwMy04YzJjLTg2MmQ2YWY4ZGE1MiIsImRhdGEiOnt9LCJyYW5kb20iOiI2YTM2Y2RhYzhhOTM5NzI4MzVmZmYyNTZiNmExYTBiZSJ9.wD-fegFi-zj4iOVk6_c0rL8eRIZYsivt9I1zL5gTWy81pUNAp_EjI1XIWawvfZP83Y7ym8Scu4_q2g9F73KFdg].
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 3549
Download: 45
Times Cited: 0