 Original Article
Original Article

Affiliation:
Immunoncology and Molecular Theragnostic Lab, Centre for Medical Biotechnology, Amity Institute of Biotechnology, Amity University Uttar Pradesh, Noida, Uttar Pradesh 201301, India
ORCID: http://orcid.org/0009-0003-7597-0802
Affiliation:
Immunoncology and Molecular Theragnostic Lab, Centre for Medical Biotechnology, Amity Institute of Biotechnology, Amity University Uttar Pradesh, Noida, Uttar Pradesh 201301, India
ORCID: http://orcid.org/0009-0006-8830-1677
Affiliation:
Immunoncology and Molecular Theragnostic Lab, Centre for Medical Biotechnology, Amity Institute of Biotechnology, Amity University Uttar Pradesh, Noida, Uttar Pradesh 201301, India
Email: bbondhopadhyay@amity.edu; bbanerjee218@gmail.com
ORCID: http://orcid.org/0000-0002-6679-7791
Explor Drug Sci. 2025;3:1008108 DOI: https://doi.org/10.37349/eds.2025.1008108
Received: January 06, 2025 Accepted: March 19, 2025 Published: April 29, 2025
Academic Editor: Fernando Albericio, University of KwaZulu-Natal, South Africa, Universidad de Barcelona, Spain
Aim: The PI3K (phosphoinositide 3-kinase)-alpha isoform is found upregulated in 30–40% of breast cancer. Currently, there are limited selective and specific drugs that target PI3K-alpha, and no natural therapeutic option is available. This study aims to develop natural hybrid antagonists of PI3K-alpha for breast cancer therapeutics.
Methods: 25 pan-PI3K and PI3K-alpha targeting drugs were obtained from various sources, including the COCONUT (Collection of Open Natural Products) database. On the parent dataset, high throughput virtual screening (HTVS), standard precision (SP) docking, and extra precision (XP) docking were performed to produce Murcko scaffolds and heterogenous fragments. Murcko scaffolds are hybridized with fragments of natural compounds (Category 1) and drugs (Category 2), respectively. Hybrids are docked with HTVS, SP, and XP, followed by induced fit docking and ADME (absorption, distribution, metabolism, and excretion) prediction. MM/GBSA (molecular mechanics/generalized Born and surface area) was performed on the docked poses.
Results: Highest docking scores of –13.354 kcal/mol and –12.670 kcal/mol were achieved by hybrids in Category 1 and Category 2, respectively. MM/GBSA free energy ranged from –51.14 kcal/mol to –72.66 kcal/mol. In terms of binding docking, pharmacological properties, and Lipinski’s rule of five, the natural hybrids outperformed the parent drugs.
Conclusions: PI3K-alpha kinase proteins can be targeted with natural-drug hybrid antagonists for breast cancer treatment. Hybrid molecules, such as NH-01 and NH-06, show better binding with promising ADME properties. Thus, in vivo and in vitro testing is necessary to prove the value of such hybrids.
PI3Ks (phosphoinositide 3-kinases), or phosphatidylinositol 3-kinases, are a family of lipid kinases responsible for regulating fundamental processes such as proliferation, growth, apoptosis, energy metabolism, and survival of the cell [1]. As the name suggests, PI3K influence the signaling cascades by their dual behavior, as they phosphorylate the 3'-OH position of the inositol ring resulting in phosphoinositides in a lipid kinase manner and on the other hand can autophosphorylate in the protein kinase manner [2]. In general, PI3K consists of eight catalytic subunits, with three major classes, class I, class II, and class III, based on their structure and specificity towards their respective substrates. Class I PI3Ks are heterodimeric enzymes that are further classified into class IA and IB respectively, with class IA constituting of p110α, p110β, and p110δ, and class IB being the home to p110γ subunits, along with a p85 subunit for regulation. It is important to note that while p110α and p110β are quite widespread across the body, p110γ and p110δ are solely found to be present in leucocytes. The class I PI3Ks, as their name suggests, typically help in the phosphorylation of PIP2 (phosphatidylinositol (4,5)-bisphosphate), or the phosphoinositide (4,5) biphosphate, to stimulate the production of phosphatidylinositol (3,4,5)-trisphosphate (PIP3).
The p110α subunit of the lipid-protein kinase PI3K is genetically coded by PIK3CA, and the name of the protein is phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha. The mutations in PI3K-alpha lead to a gain of function by the protein kinase leading to an aberrant surge in downstream signaling, primarily through the PI3K/AKT/mTOR pathway, thereby dysregulating hallmarks of cancer such as increased growth and proliferation, rise in hormonal drug therapy resistance, anti-apoptotic behavior, angiogenesis, and cell-cycle disruption [3–6]. It has been implicated as an oncogenic driver in several tumors such as colon cancer, breast cancer, lung cancer, head and neck cancer, renal cancer, endometrial cancer, ovarian cancer, uterine cancer, and cervical cancer with the growing pace of research for therapeutics in those cancers [7–15]. Additionally, according to TCGA PanCancer Atlas studies which comprised 10,967 samples from 32 different cancers, PIK3CA was found to be altered the most in cancers of endometrium, cervix, and breast [16, 17]. Moreover, it is the second most frequently mutated gene in breast cancer after TP53, with about 49% luminal A cells presenting mutations, 7% in triple-negative breast cancer cells, 42% in HER2 positive, and 32% in luminal B, thereby making it the most important therapeutic target in breast cancer drug research [18, 19]. Furthermore, H1047R (histidine to arginine), E545K (glutamic acid to lysine), and E542K (glutamic acid to lysine) are found to be the top three mutations found in majority of the breast cancer patients, which act as putative drivers of aberrant signaling of the PI3K/AKT pathway leading to downstream dysregulation of proliferation, upregulation of cell growth, and metastasis [7, 19–21].
PI3K protein is influenced by several upstream receptors prominent for signaling in breast cancer cells and demonstrates its downstream effect through the PI3K/AKT/mTOR pathway. In breast cancer cells, receptor tyrosine kinases such as HER2, EGFR, IGF1R, and FGFR1, and steroid receptor ER are activated by stimuli. This stimulus in turn activates p85-p110α (PI3K-alpha) heterodimer protein, which phosphorylates PIP2 to PIP3, thereby recruiting PDK1. mTORC2 and PDK1 recruit AKT which in turn activates mTORC1, leading to downstream activation of S6K and 4EBP1 which leads to evasion of apoptosis and cell proliferation [22]. PTEN negatively regulates PI3K by dephosphorylating PIP3 to PIP2. PI3K is also activated by RAS GTPases and VEGFR2, increasing the range of its stimulation. G protein-coupled receptors stimulated by F2, BK, ET1, and Ang-II and toll-like receptors also activate PI3K [23] thereby leading to downstream effects. ITGA and ITGB also activate PI3K downstream via FAK [24]. The significance of PI3K-AKT signaling is further emphasized by the action of AKT as an upstream effector kinase of many pathways such as BAD-Bcl-2 apoptotic pathway, JAK-STAT pathways, TNF-NF-κB pathway, NF-κB-MMP pathway, CASP9, p53 oncogenic inhibition via MDM2, p27 and p21 activation, GSK3-Cyclind1 pathway, all of which lead to aggressive enhancement of hallmarks of cancer such as proliferation, angiogenesis, vascularization, growth, metabolism, inhibition of apoptosis, metastasis, and cell cycle dysfunction [25–27].
Many synthetic approaches have been taken up in order to inhibit PI3K-alpha. Currently, only a couple of drugs exist selectively and solely for p110α inhibition, such as taselisib (phase 3 of trials NCT02457910), alpelisib (FDA-approved drug [28], NCT02437318), and serabelisib (phase 1b of trials NCT04073680) [29]. These drugs, however, inflict adverse damage to the body such as dermatitis, stomatitis, hyperglycemia, peripheral neuropathy, anemia, fatigue, diarrhoea, rash, nausea, and headache [30, 31]. For instance, alpelisib, an FDA-approved drug to be used in combination with fulvestrant specifically for HER2-negative breast cancer patients was revealed to lead to adverse effects such as rashes, hyperglycaemia, and diarrhoea [32–34]. Similarly, pan-PI3K inhibitors like pictilisib, buparlisib, taselisib, copanlisib, sonolisib, are underway clinical trials for breast and other cancers such as lymphoma, head and neck carcinoma but suffer from adverse effects [35–40].
Whereas limited natural therapeutics exist that do not target PI3K directly but target the pathway exhibit lower response but safer alternatives [41, 42]. Thymoquinone has been found to induce apoptosis by inducing cell cycle arrest in the PI3K pathway but not against PI3KCA specifically [41]. Further, isoliquiritigenin, a flavonoid found in licorice, was tested for its inhibition of the PI3K pathway in A549 lung cancer cells and MKN28 gastric cancer cell lines [43, 44]. Apigenin, a common flavonoid found in dietary supplements was found to induce autophagy and apoptosis in HepG2 cell lines by inhibition of the PI3K/AKT/mTOR pathway [42].
Although, synthetic drugs show immense potential for their therapeutic use they suffer from side effects while phytochemicals are safer but do not target PI3K-alpha directly [45, 46]. Therapeutic modalities lack selectively and specifically developed drug candidates against PI3K-alpha isoform in breast cancer. PI3K-alpha is a key player in breast carcinogenesis, making it an indispensable target for therapeutic development for this disease. Therefore, to maximize the best of both worlds, in this study we have adopted fragment-based and combinatorial technology to explore, create, and screen drug-natural hybrid antagonists to PI3K-alpha for breast cancer treatment.
A list of pan-PI3K and selectively targeting PI3K-alpha drugs was prepared and sourced from ChEMBL [47], Guide to Pharmacology [48], and DrugBank [49] databases, and then further removing duplicates to initialize a set of 25 distinct effective drugs. For the natural product dataset, comprehensive database repositories are acquired through the COCONUT (Collection of Open Natural Products) [50] database which had 407,270 natural compounds (Table 1). A molecular weight (MolWt) of 300–600 Da was taken as the size range to work further with the dataset, to eliminate non-druglike larger compounds and smaller compounds with a lack of useful scaffolds, for further processes.
Drugs that target PI3K kinase and selected for the study, with two drug target categories being pan-PI3K and isoform-specific PI3K-alpha
| S.No. | Inhibitor name/Pubchem ID | Type of inhibitor | Molecular weight (g/mol) |
|---|---|---|---|
| 1 | PIK-93 | Pan PI3K | 389.9 |
| 2 | Sonolisib | Pan PI3K | 525.6 |
| 3 | TG-100115 | Pan PI3K | 346.3 |
| 4 | ZSTK 474 | Pan PI3K | 417.4 |
| 5 | Buparlisib | Pan PI3K | 410.4 |
| 6 | Pictilisib | Pan PI3K | 513.6 |
| 7 | GSK-1059615 | Pan PI3K | 333.4 |
| 8 | Izorlisib | Pan PI3K | 377.4 |
| 9 | Taselisib | Pan PI3K | 460.5 |
| 10 | Torin 2 | Pan PI3K | 432.4 |
| 11 | Pilaralisib | Pan PI3K | 541.0 |
| 12 | Alpelisib | Pan PI3K | 441.5 |
| 13 | VPS34-IN1 | Selective PI3K class III | 425.9 |
| 14 | Vps34-PIK-III | Selective PI3K class III | 319.4 |
| 15 | Serabelisib | Pan PI3K | 363.4 |
| 16 | SAR405 | Pan PI3K | 443.8 |
| 17 | PQR-620 | Pan PI3K | 445.5 |
| 18 | SAR-260301 | Selective PI3Kβ | 354.4 |
| 19 | Copanlisib | Pan PI3K | 480.5 |
| 20 | Vps34-IN-3 | Pan PI3K | 276.33 |
| 21 | RIDR-PI-103 | Pan PI3K | 511.5 |
| 22 | Compound 82 | Pan PI3K | 476.9 |
| 23 | Compound 12b | Pan PI3K | 369.4 |
| 24 | Compound 41 | Selective PI3Kδ | 535.6 |
| 25 | 135799939 | Pan PI3K | 448.4 |
PI3K: phosphoinositide 3-kinase; S.No.: serial number
The crystal structure of wild-type PI3K-alpha subunit inbound complexed with an inhibitor taselisib (2-methyl-2-(4-{2-[3-methyl-1-(propan-2-yl)-1H-1,2,4-triazol-5-yl]-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepin-9-yl}-1H-pyrazol-1-yl)propanamide) having a resolution of 1.99 Å [PDB (Protein Data Bank) ID: 8EXL] [51] from the PDB was prepared using Protein Preparation Wizard in GLIDE in Schrodinger Maestro Suite 2023-1 [52], by filling missing side chains, removing water beyond 8 Å and restrained minimization in OPLS4 (Optimized Potentials for Liquid Simulations 4) force field, optimize overlapping hydrogens and replacing them, capping of termini, converting selenomethionines to methionines, filling in missing loops using Prime [53], generating het states at pH of 2.0 with Epik, create disulfide bonds and zero-order binding to metals. This is performed to rectify structural aberrations and optimize the protein structure for subsequent molecular docking studies.
Furthermore, due to the lack of good quality protein structures (parameters such as low resolution, similar co-crystal, similar method of structure characterization) available for mutational models, mutations of single residues were introduced in the copies of the prepared proteins in Maestro (Schrodinger Suite 2023-1). Three mutated proteins were created having mutations—H1047R, E545K, and E542K and then minimized again using Protein Preparation Wizard. Receptor grid generation was performed with a van der Waals scaling factor of 1.0 and partial charge cut-off of 0.25, using GLIDE to prepare the docking grid in the binding pocket of the protein, which facilitates structure-based ligand docking within the prepared protein binding site.
Drug and natural ligand structures were prepared using LigPrep in Schrodinger Suite 2023-1 [54] to generate optimized 3D conformations suitable for molecular docking analyses and virtual screening. A maximum ligand size of 500 atoms, OPLS4 force field, at the target pH of 7 ± 2, using Epik, and generated at most 1 stereoisomer at most per ligand with retention of specified chiralities, during LigPrep. All drugs underwent this preparation process leading to a total of 213,343 prepared natural ligands, while the selected subset of natural ligands from COCONUT underwent this preparation to produce 50 structures. The drugs were then docked against the receptor grid of the PI3K-alpha protein grid, initially using high throughput virtual screening (HTVS) docking, then standard precision (SP) docking in GLIDE on default settings. This was followed by refinement using extra precision (XP) docking in the same settings, for improved accuracy, to determine best poses of the ligand drugs. Simultaneously, natural ligands were made to undergo HTVS with GLIDE, and 153,409 compounds were docked. Those in the top 1% (1,534) with the lowest docking scores from HTVS underwent further refinement through SP docking. Finally, the top 10% (153) of this refined subset based on SP docking scores underwent XP docking for additional precision. This method, thus, ensured a thorough exploration of drug and natural ligand interactions with the PI3K-alpha protein, enhancing the identification of potential candidates with strong binding affinities and therapeutic promise.
In the fragmentation process, the docked drugs and ligands were broken down using the Schrodinger Python [55] utility of fragment_molecule.py with the carbon hetero option. This utility applied specific rules to ensure precise fragmentation and the bonds within the molecules were selectively cut, prioritizing two main types: bonds associated with rings, except for hydrogen bonds and internal ring bonds (with allowances made for bonds between ring-carbon and non-carbon atoms using the -c option), and carbon-carbon bonds that were preserved unless they are a part of a ring structure. The carbon hetero option enabled cutting bonds between ring-carbon atoms and hetero atoms, potentially increasing the variety of generated fragments. Additionally, Murcko fragments of the drugs were generated. This method divides compounds into components such as R-groups, linkers, and ring systems. From each compound, a scaffold or framework is derived by removing all R-groups while retaining core ring systems and linkers. This systematic approach categorizes the atoms of each drug molecule into ring, linker, framework, and side chain components, offering a thorough breakdown of structural data essential for detailed analysis and interpretation. This process resulted in four categories of fragments, drug fragments with carbon hetero setting and Murcko scaffolds of drugs, and natural compounds fragments and Murcko scaffolds similarly. Fragments of natural compounds were further filtered out by keeping fragments below 300 Da MolWt such that the hybridization process is streamlined and does not produce large fragments which are unlike small molecules.
In the hybridization process, fragments extracted from both drugs and natural ligands were merged using the BREED module in Maestro to create novel hybrid entities. The Murcko scaffolds of drugs were combined with fragments from the natural ligands (Category 1), and vice versa, Murcko scaffolds of natural ligands were combined with fragments from drugs (Category 2). This was performed to ensure that the combination of core structures of one category are hybridized with functional groups of the other, thereby ensuring the success of the hybrids by improving on existing features of drugs and natural ligands. The process involved aligning the structures of ligand-bound targets and identifying overlapping bonds across ligand pairs. Fragments on each side of these overlapping bonds were then exchanged to generate a new single generation of molecules, following established methods described in the literature [56] with default parameters which are maximum atom-atom distance of 1.0 Å and maximum 15 degrees angle of distance in bond overlap criteria. Specifically, fragments from one ligand could replace fragments from another if the bond connecting them in the first ligand matched a bond in the second ligand. Constraints were applied to maintain bond order consistency and to ensure that swapped bonds were not part of a ring structure. Each overlapping bond between pairs of input molecules resulted in the creation of a new pair of molecules, with duplicates removed. This iterative swapping process constituted one “generation” of hybridization, ultimately yielding a total of 3,744 unique natural-drug hybrids.
Following the creation of hybrids, they underwent HTVS to identify promising candidates, and 2,190 and 1,479 hybrids were docked from Category 1 and Category 2 respectively. The top 10% of hybrids from Category 1 (219) and Category 2 (147) with the most favorable docking scores were chosen for SP docking. Subsequently, the top 10% of hybrids from Category 1 (22) and Category 2 (15) from SP docking underwent XP docking. The top 5 hybrids from each category were further docked at SP followed by XP in the mutant protein structures (H1047R, E545K, and E542K). The total ten hybrids were analyzed using molecular mechanics/generalized Born and surface area (MM/GBSA) solvation to evaluate their binding affinities with the PI3K-alpha protein wild-type and mutants using the Prime module.
The validation of the docking protocol has been performed by re-docking the co-crystal ligand in the receptor at the same site, measuring the root mean square deviation (RMSD) in the poses between the original co-crystal and the docked molecule [57, 58]. The co-crystal inbound receptor taselisib is docked in the receptor grid of the protein and RMSD value is checked at SP and XP docking. A small RMSD value (< 1.5–2 Å) and superposition indicate that the docking strategy applied is working correctly [59–61].
Induced fit docking (IFD) in the wild-type receptor of top hybrids from MM/GBSA was performed to analyze the binding of the hybrids created. IFD ensures that the docking evaluated binding in a flexible protein-rigid ligand binding conformation, unlike a rigid protein-flexible ligand sampling during molecular docking analyses [62–65]. SP protocol for pose generation was used at 0.5 van der Waals scaling. The hybrids showing comparable scores to the docked ligands should be selected for further studies.
QikProp module in Schrodinger Suite 2023-1 [66], was utilised to predict the pharmacokinetic properties—absorption, distribution, metabolism, and excretion (ADME), of the shortlisted hybrids. It predicts pharmaceutical and physical descriptors of molecules, on the basis of known druglike parameters of compounds. The prediction and calculation of ADME properties are essential due to their importance in the physiological systems. The good druglike parameters, QPlogS (aqueous solubility), QPlogPo/w (octanol/water partition coefficient), hydrogen bond donors and acceptors (donorHB, accptHB), MolWt, polar surface area (PSA), and solvent accessible surface area (SASA) are important parameters, based on Lipinski’s rule of five. The percentage of human oral absorption (%HumOralAbs) was also predicted to determine the absorption capacity of the hybrids.
The danger posed by high expression of the p110α subunit of PI3K has been studied variably in breast cancer, amounting to about 30–40% of breast cancer cells presenting elevated and upregulated expression of PI3K [67]. On studying the binding mechanism of PI3K-alpha, it was revealed that the ligand binding and kinase-catalytic domain of the protein lies in the amino acid residue ranges 765–1,051, wherein key oncogenic driving mutation—H1047R also occurs. The inbound co-crystal ligand displayed binding and interactions with the residues in the ligand binding domain, on docking. Three main loops form the functional structure of the domain, along with the PI3K/PI4K conserved region from 801–815. These are the: G-loop which ranges from 771–777 residues (IMSSAKR), catalytic loop which ranges from 912–920 (GIGDRHNSN) which is conserved, and the activation loop which lies in 931–957 (HIDFGHFLDHKK-KKFGYKRERVPFVLT) [68, 69].
213,343 natural ligands were prepared and 50 structures of the drugs were generated by LigPrep. The best docking scores of –9.697 kcal/mol by alpelisib, –10.471 kcal/mol by taselisib, and –12.121 kcal/mol by GSK-615, were achieved in HTVS, SP, and XP docking of drugs, respectively. On the other hand, the best docking score of –10. 527 kcal/mol by CNP0097211 (3-(3,4-dihydroxyphenyl)-2-({3-[7-(3,4-dihydroxyphenyl)-3,4-dihydroxy-5,6-dihydronaphthalen-1-yl]prop-2-enoyl}oxy)propanoic acid), –11.353 kcal/mol by CNP0419682 (5-acetyl-7-(5-carboxy-7-hydroxy-4-oxo-4H-chromen-2-yl)-2,3-dihydroxy-6-methyl-9-oxo-9H-xanthene-1-carboxylic acid) and –17.190 kcal/mol by CNP0093692 (2-(3,4-dihydroxyphenyl)-5,8-dihydroxy-7-{[4-hydroxy-4-(hydroxymethyl)-3-{[3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}oxolan-2-yl]oxy}-4H-chromen-4-one), were achieved in HTVS, SP, and XP docking of natural ligands, respectively.
On fragmentation of drugs, 25 Murcko scaffolds and 1,372 fragments were generated. For natural ligands, 151 Murcko scaffolds and 49,114 fragments were produced and 4,894 were retrained for further analysis. Hybridization of Murcko scaffolds of drugs with fragments of natural ligands, produced 2,225 hybrids (Category 1), whereas hybridization of Murcko scaffolds of natural ligands and fragments of drugs produced 1,519 hybrids (Category 2). The top 10 hybrids were selected based on further virtual screening (Figures S1–S10).
The best docking scores of –10.614 kcal/mol by CNP0302777 (frag 7) + 23582824 (frag 1) and –11.080 kcal/mol by CNP0171717 (frag 5) + 23582824 (frag 1) were achieved in HTVS and SP docking of Category 1, respectively. On the other hand, the best docking scores of –10.055 kcal/mol by CNP0382214 (frag 1) + 72709209 (frag 22) and –11.112 kcal/mol by 50990924 (frag 15) + CNP0475782 (frag 1) were displayed in HTVS and SP docking of Category 2, respectively. The best scores in XP docking by the two categories were –13.354 kcal/mol by 23582824 (frag 1) + CNP0243567 (frag 44) (NH-01) from Category 1 and –12.670 kcal/mol by 56649450 (frag 59) + CNP0392152 (frag 1) (NH-06) from Category 2. The top 5 hybrids based on docking score from each category were selected for further analysis (Table S1). The molecular docking and MM/GBSA analysis of the mutants of PI3K-alpha showed that the hybrids showed slightly better binding affinity for mutant proteins as compared to wild-type (Table S1). NH-01 performed the best in binding with all three mutant protein receptors, followed by NH-06 similar to the wild-type. However, the ranking of the structures was different when compared with the docking ranking against wild-type receptors.
On docking of the originally bound ligand, taselisib, a docking score of –9.571 kcal/mol on XP was achieved, with an RMSD value of 0.4307, thereby waiving a green flag for the screening strategy. The IFD corroborated the results achieved in XP docking. The IFD docking scores were in a different order from the XP docking but displayed comparable results. MM/GBSA revealed high negative free energy (dG) release on the binding of ligands (Table 2 and Table S1).
The top 2 hybrids from Category 1 and 2 are sorted by the order of best to worst docking scores, and IFD values, IFD RMSD values, and MM/GBSA bind scores are mentioned
| Name | Docking score at XP (kcal/mol) | IFD docking score at SP (kcal/mol) | IFD score (kcal/mol) | MM/GBSA dG bind energy (kcal/mol) |
|---|---|---|---|---|
| Category 1 | ||||
| 23582824 (frag 1) + CNP0243567 (frag 44) NH-01 | –13.354 | –12.040 | –2,079.60 | –54.34 |
| CNP0106688 (frag 9) + 70798655 (frag 1) NH-02 | –12.661 | –12.661 | –2,080 | –51.14 |
| Category 2 | ||||
| 56649450 (frag 9) + CNP0392152 (frag 1) NH-06 | –12.67 | –12.692 | –2,083.46 | –67.81 |
| 50990924 (frag 5) + CNP0475782 (frag 1) NH-07 | –12.327 | –13.022 | –2,082.72 | –72.66 |
IFD: induced fit docking; MM/GBSA: molecular mechanics/generalized Born and surface area; RMSD: root mean square deviation; SP: standard precision; XP: extra precision
The best hybrid molecule from Category 1 (NH-01), displayed several interactions with the binding site residues. Three hydrogen bonds with VAL851, TYR836, and ASP810 are a part of the catalytic domain, while ASP810 additionally is a residue in the conserved domain. A charged interaction with residues like ASP810, LYS802, TYR836, and LEU807 was exhibited by the hybrid, while most of the interaction was hydrophobic with residues lying between ILE848 to GLN859 (Figure 1). In IFD, two more pi-pi stacking interactions were observed with TYR836 and TRP780, with a more polar interaction with residues lying from ASN853 to GLN859 (Figure 2). The hybrid shows similar pi-pi stacking with TYR836 as the drug GSK-615 from which the Murcko scaffold is derived and hydrogen bonding at VAL851. Whereas the hybrid exhibited the interactions of the parent flavonoid-7-O-glucuronide natural derivative (CNP0243567), as a hydrogen bond at ASP810, TYR836, and VAL851 (Figure 3).
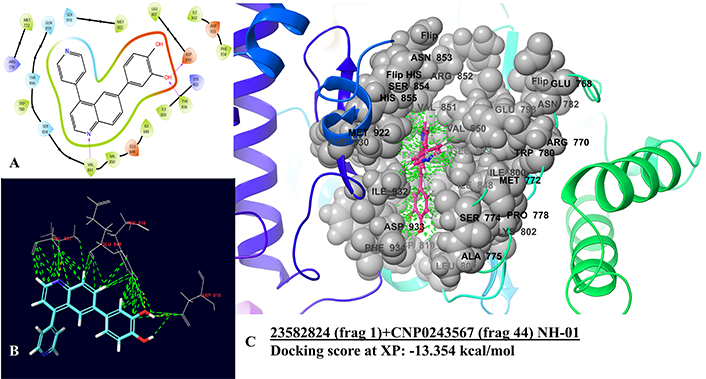
PI3K-alpha and NH-01 docking interactions. (A) 2D interaction diagram of NH-01 with PI3K-alpha, where purple arrows denote hydrogen bonding, green denotes hydrophobic interaction, red denotes negatively charged interaction, and blue denotes polar interaction. (B) 3D interactions with residues in interaction. (C) 3D interactions with binding residues and binding pocket interactions. Green dashed lines are good interactions while yellow denotes hydrogen bonds, and orange denotes bad clashes. PI3K: phosphoinositide 3-kinase; XP: extra precision
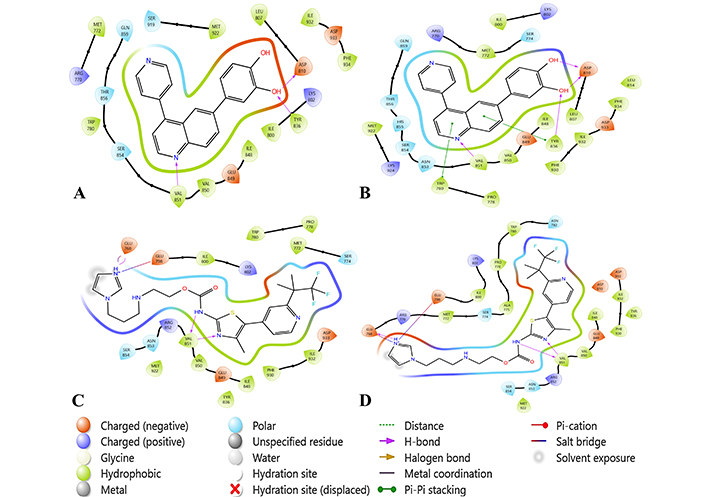
2D interactions of docking at XP vs IFD. (A) and (B) are NH-01 ligand interactions with PI3K-alpha at XP and IFD-SP respectively. (C) and (D) are NH-06 ligand interactions with PI3K-alpha at XP and IFD-SP respectively. IFD: induced fit docking; PI3K: phosphoinositide 3-kinase; SP: standard precision; XP: extra precision
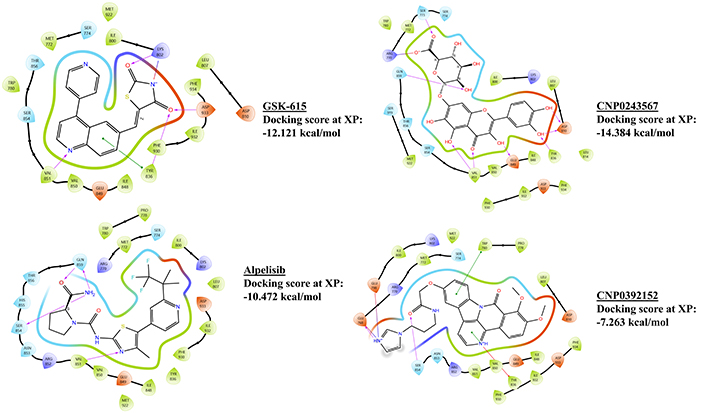
2D ligand interaction diagram of the parent molecules. Where GSK-615 and CNP0243567 are parent molecules for NH-01, and alpelisib and CNP0392152 are parent molecules for NH-06. XP: extra precision
The best hybrid molecule from Category 2 (NH-06) forms a salt bridge with GLU768 and GLU798 from the imidazole ring end of the hybrid, while two hydrogen bonds with VAL851 are observed. These interactions lie in the G-loop domain of the protein. Hydrophobic interactions from ILE848 to VAL851 were observed, while charged positive and polar interactions were seen with SER854 to ASN853, MET772 to SER774, and LYS802 to ILE800 (Figure 4). No changes were observed in interactions in the best IFD pose (Figure 2). The natural scaffold of CNP0392152 displayed a similar salt bridge with GLU768 and GLU798, while the drug fragment is from alpelisib and displays a similar hydrogen bond with VAL851 (Figure 3).
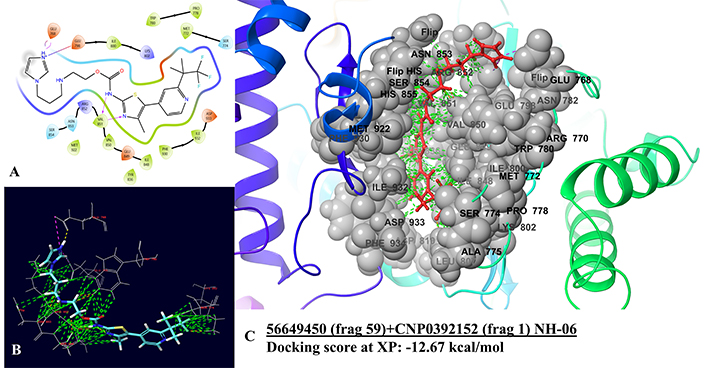
PI3K-alpha and NH-06 docking interactions. (A) 2D interaction diagram of NH-06 with PI3K-alpha, where purple arrows denote hydrogen bonding, pink lines are salt bridges, green denotes hydrophobic interaction, red denotes negatively charged interaction, dark blue denotes positively charged interaction, and blue denotes polar interaction. (B) 3D interactions with residues in interaction. (C) 3D interactions with binding residues and binding pocket interactions. Green dashed lines are good interactions, pink denotes salt brides, while yellow denotes hydrogen bonds, and orange denotes bad clashes. PI3K: phosphoinositide 3-kinase; XP: extra precision
Thus, these interactions indicate a good binding potential of the hybrids with very low bad clashes and no ugly interactions according to Maestro, increasing the affinity and selectivity of natural hybrids and reducing the risk potential of the drugs.
The desired values of the ADME parameters predicted in the pharmaco-analysis should be such that the MolWt lies between 130.0–500.0 Da, donorHB should be between 0–5, accptHB should be between 2–10, the predicted QPlogPo/w should lie in the range of –2.0–6.5 mol dm–3, the predicted QPlogS should be preferably within –6.5–0.5 mol dm–3, the %HumOralAbs should be more than 70%, the desirable range of values of PSA is 7.0–200.0, the SASA should be 300.0–1,000.0 and the violations of rule of five should be a maximum of 4.
The 10 hybrids were predicted to have good druglike parameters. Only one hybrid NH-09 exhibited two violations of rule of five, while all the other hybrids showed 0 violations, therefore, displaying good characteristics (Table S1). This makes it evident that the safety and ADME profile of parent molecules are highly increased when they are hybridized (Table 3 and Table S2).
ADME properties predicted are presented MolWt in Da, predicted donorHB and accptHB values, QPlogPo/w, QPlogS, %HumOralAbs, PSA, SASA, rule of five violations
| Name | MolWt (Da) | donorHB | accptHB | QPlogPo/w (mol dm–3) | QPlogS (mol dm–3) | %HumOralAbs | PSA | SASA | Rule of five |
|---|---|---|---|---|---|---|---|---|---|
| Category 1 | |||||||||
| NH-01 | 314.347 | 2 | 4 | 3.128 | –4.586 | 90.947 | 66.24 | 577.217 | 0 |
| NH-02 | 362.325 | 1 | 7.5 | 2.354 | –4.065 | 64.041 | 110.28 | 631.441 | 0 |
| Category 2 | |||||||||
| NH-06 | 497.567 | 2 | 8.5 | 4.221 | –6.502 | 87.125 | 123.45 | 860.744 | 0 |
| NH-07 | 467.933 | 2.8 | 11.2 | 1.636 | –5.158 | 73.316 | 174.52 | 716.095 | 0 |
%HumOralAbs: percentage of human oral absorption; accptHB: hydrogen bond acceptors; ADME: absorption, distribution, metabolism, and excretion; donorHB: hydrogen bond donors; MolWt: molecular weight; PSA: polar surface area; QPlogPo/w: octanol/water partition coefficient; QPlogS: aqueous solubility; SASA: solvent accessible surface area
In recent times, multiple attempts have been made to develop inhibitors for the PI3K, to inhibit tumor proliferation and develop a therapeutic approach toward the treatment of breast cancer. To address the daunting challenge, of developing selective and safe anti-cancer therapeutics against PI3K, this study aimed to design natural hybrid inhibitory drugs combining the advantages of both. The study utilized combinatorial and fragment-based technologies. This approach sought to mitigate the adverse effects associated with existing drugs while increasing the potency and selectivity of PI3K-alpha, ultimately advancing treatment options for breast cancer.
In silico studies are being rampantly conducted to evaluate the clinical potential of natural and synthetic ligands using the ever-developing computational approaches. Various in silico studies have also been conducted to hypothesize the use of novel PI3K inhibitors for malignant cancers. One such study conducted identified five zinc compounds that could potentially be used as anti-cancer agents, which were triple target inhibitors, potentially inhibiting all three, the PI3K/AKT/mTOR pathways, in a bid to find natural products that could be used for this purpose [70]. Additionally, the potential of apigenin, luteolin, and pinoresinol, which are phenols extracted naturally from Olea europaea has also been studied [71]. Phytochemicals were also evaluated as dual inhibitors of PI3K and mTOR in another study using an in silico approach [72].
In contrast, our study has created novel antagonists of PI3K-alpha by fragmenting existing drugs and natural compounds and combining them. The docking scores in the natural hybrids thus created, displayed an improvement in binding as compared to the parent drug molecules, thereby increasing binding affinity for the target. However, in an unexpected turn of events, the natural compounds displayed better binding to the protein molecule than their synthetic counterparts. This fortified the notion of utilizing phytochemicals for the study’s novel drug design. The druglike properties such as the violations of the rule of five, high number of donorHB, and low %HumOralAbs of the natural ligands were highly improved in the hybrids.
The result shows a promising avenue where the hybrids overperform existing drugs. The hybrids created, possessed the functional scaffold of both natural compounds and drugs, such that the hybrids created in Category 1 (Murcko scaffolds of drugs and fragments of natural compounds) while similar comparable characteristics were observed in Category 2 (Murcko scaffolds of natural compounds and fragments of synthetic drugs) performed better in virtual screening and identification, indicating that by using the ring systems of drugs and natural ligands, we can tap into the interacting potential of the synthetic molecules and functional carbonyl groups of natural compounds, while at the same time, natural compound scaffolds provide safer, more druglike properties and drug scaffolds provided better binding capabilities to the final natural hybrid antagonists.
The study lays the foundation for further studies in the realm of hybrid drugs targeting PI3K-alpha, which shows the highest involvement in breast cancer progression. The study aimed to maximize the features of both drugs and natural products by combining their fragments and producing hybrids that portray better binding, and pharmacological properties. It was observed that the natural hybrid antagonists perform better than natural compounds when it comes to their pharmacological properties, while the hybrids showed better binding scores than the drugs. Furthermore, the hybrids showed a high level of binding with mutant proteins with higher docking scores as compared to the wild-type. Thereby, emphasizing their promising potential to provide the most therapeutic effect in breast cancer as these particular mutations are most frequently reported.
It is not unknown that in silico studies are auxiliary to real-life drug-discovery processes and cannot replace the real-life testing of compounds in in vitro and in vivo systems, because of which these compounds must be considered for further research processes. Only further studies in cell systems and testing in animal models can prove the potential of natural hybrids. Additionally, as revealed in our study, natural compounds outperform hybrids and drugs in terms of binding affinity. Therefore, presenting improved opportunities in natural compound-based lead discovery processes. However, at the same time, currently, the usage of fragment and combinatorial approaches to create and screen natural-drug hybrid antagonists, is still a very novel concept and is under improvement. Thus, this new approach down the line, may potentially prove to be a useful methodology to repurpose existing drugs and naturally available compounds into better medicinal and therapeutic solutions to morbidities like breast cancer.
%HumOralAbs: percentage of human oral absorption
accptHB: hydrogen bond acceptors
ADME: absorption, distribution, metabolism, and excretion
COCONUT: Collection of Open Natural Products
donorHB: hydrogen bond donors
HTVS: high throughput virtual screening
IFD: induced fit docking
MM/GBSA: molecular mechanics/generalized Born and surface area
MolWt: molecular weight
OPLS4: Optimized Potentials for Liquid Simulations 4
PDB: Protein Data bank
PI3K: phosphoinositide 3-kinase
PIP2: phosphatidylinositol (4,5)-bisphosphate
PIP3: phosphatidylinositol (3,4,5)-trisphosphate
PSA: polar surface area
QPlogPo/w: octanol/water partition coefficient
QPlogS: aqueous solubility
RMSD: root mean square deviation
SASA: solvent accessible surface area
SP: standard precision
XP: extra precision
The supplementary Figures and Tables for this article are available at: https://www.explorationpub.com/uploads/Article/file/1008108_sup_1.pdf.
The authors acknowledge Amity University Uttar Pradesh, Noida for providing the facilities for the conduct of the present study. The authors also acknowledge ICMR-DHR (R.12013/21/2023-HR/ E-Office: 8206474). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
NA: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing—original draft, Writing—review & editing. SS: Visualization, Writing—original draft, Writing—review & editing. BB: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Protein structure was retrieved from the RCSB-PDB website (https://www.rcsb.org/structure/8exl), PDB DOI: https://doi.org/10.2210/pdb8EXL/pdb. Natural compounds were accessed and downloaded from COCONUT databases at https://coconut.naturalproducts.net/ and drugs from Pubchem repository http://pubchem.ncbi.nlm.nih.gov/. The data generated and analyzed for this study are included in the manuscript and the supplementary files. Additional data should be requested from the corresponding author which may be provided on reasonable request and with a signed data access agreement.
This work was financially supported by ICMR-DHR [(R.12013/21/2023-HR/ E-Office: 8206474)]. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

