 Review
Review

Affiliation:
Department of Translational Research, College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
Affiliation:
Department of Translational Research, College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
Affiliation:
Department of Translational Research, College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
Affiliation:
Department of Translational Research, College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
Affiliation:
Department of Translational Research, College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
Email: vrai@westernu.edu
ORCID: https://orcid.org/0000-0001-6286-2341
Explor Asthma Allergy. 2026;4:1009116 DOI: https://doi.org/10.37349/eaa.2026.1009116
Received: December 26, 2025 Accepted: February 10, 2026 Published: March 10, 2026
Academic Editor: Uday Kishore, University of Oxford, England
The article belongs to the special issue Innate Immune Mechanisms in Allergic Diseases
Allergic conjunctivitis (AC) is an inflammatory disorder of the ocular surface caused by allergic reactions to environmental substances. It presents with symptoms such as itching, redness of the eye, excessive tearing, and swelling/irritation in the eyes and eyelids. While many AC episodes occur on their own and go away, some forms of this disease are present in a chronic fashion or have the potential to cause serious loss of vision. In recent years, AC has been viewed as primarily an episodic irritative condition to a mucosal inflammatory condition in which the ocular surface provides an environment for the initiation and perpetuation of local immune responses. The molecular basis of AC represents a phase-linked inflammatory cascade: an immediate (minutes) mediator-driven response followed by a late (6–12 hours) cytokine/chemokine-driven cellular recruitment phase that can sustain symptoms and, in severe phenotypes, contribute to tissue remodeling. The initial response is due to the activation of mast cells via IgE-dependent pathways, producing the early phase response. The sustained response seen in the late phase of the disease is mediated by the action of lipid mediators and cytokines/chemokines involved in the recruitment of eosinophils and Th2-associated leukocytes. This narrative review synthesizes evidence on epithelial “alarmins” (TSLP, IL-33, IL-25) as upstream signals that may amplify type-2 inflammation in a phenotype-dependent manner, particularly in more severe or chronic disease, alongside established IgE/mast-cell biology. Further, we discuss neuroimmune mechanisms implicated in histamine-independent itch and symptom persistence, while noting that their clinical contribution likely varies across AC phenotypes. Finally, we will discuss how the mechanistic pathways relate to current limitations and to developing new therapeutic approaches.
The term “allergic conjunctivitis” (AC) describes an inflammatory reaction of the conjunctival layer caused by contact with an environmental allergen. Typical presentations of this condition include a strong desire to rub the eyes, redness (hyperemia), increased tear flow, and swelling and/or irritation of the eyelids [1, 2]. Typical environmental allergens include seasonal aeroallergens (e.g., pollen) and perennial allergens (e.g., dust mites, animal dander). Individuals experiencing AC frequently suffer from concurrent allergic rhinitis (AR) when they have allergic rhinoconjunctivitis (ARC) [2, 3]. Ocular allergy exists within a spectrum, ranging from mild to potentially vision-threatening conditions such as vernal keratoconjunctivitis (VKC), which has the potential for chronic inflammation extending beyond the conjunctiva to involve the cornea [3].
In terms of public health significance, AC is important due to its high prevalence, frequent recurrences, and functional disruption of activities, particularly in children and adolescents [3, 4]. Epidemiological assessments of AC indicate that regional variability exists and is influenced by the presence of concomitant allergic diseases (e.g., AR) and environmental factors such as climate and air pollution [3]. Studies demonstrate that ocular symptoms negatively impact health-related quality of life and can impair productivity in both work and school settings, thereby suggesting that AC is not merely a cosmetic or trivial annoyance [4, 5]. Epidemiologic estimates suggest AC affects a substantial fraction of the global population, with wide regional variability influenced by climate, allergen burden, air pollution, and comorbid atopic disease [3]. Additional practical burdens of AC, including concerns regarding the recurrence of AC symptoms and perceptions that management of AC is cumbersome, support the necessity for developing long-term approaches for managing AC beyond the episodic suppression of AC symptoms [6].
Current management of AC is typically multi-layered and consists of environmental avoidance of the allergen(s) responsible for the AC, supportive measures (e.g., cool compresses, artificial tears), and pharmacotherapy using topical antihistamines, mast cell stabilizers, dual-action agents, and topical corticosteroids or other adjunctive treatments as required [1, 7]. Recent clinical consensus guidelines (ICON) and reviews of AC indicate that dual-action antihistamine/mast cell stabilizer formulations are effective in reducing itch and are a common first line of therapy for AC. Topical corticosteroids are usually reserved for more severe cases due to adverse effects associated with prolonged use [1, 7]. Despite numerous available therapeutic options for managing AC, real-world control is often imperfect because AC is a highly heterogeneous condition and is often coexistent with dry eye disease. Recurrent AC with continued exposure to the offending allergen(s) results in diagnostic and treatment complexities in daily ophthalmology and optometry practice [3, 4, 8].
Mechanistically, AC should be viewed as a phase-linked inflammatory cascade rather than a single mediator problem. During the early phase of AC, mast cell activation and histamine release mediated by IgE are predominant and drive the rapid onset of itch and redness. The late phase of AC is characterized by lipid mediators and cytokines, leading to the recruitment and activation of various inflammatory cells that can prolong symptoms beyond the time frame in which histamine blockade alone is sufficient [7, 9]. Importantly for the focus of this review, research into allergies has increasingly emphasized that epithelial-derived “alarmins”, thymic stromal lymphopoietin (TSLP), interleukin (IL)-33, and IL-25 serve as upstream signals to initiate type-2 immunity and amplify downstream effector circuits, including group 2 innate lymphoid cell (ILC2) responses [10–12]. Understanding these pathway components is important as they may reveal the cause of ineffectiveness of the current therapy [7, 9]. Antihistamines primarily address histamine-mediated symptoms; however, late-phase inflammation can persist through non-histaminic mediators (lipid mediators, cytokines/chemokines) and tissue recruitment programs described in consensus guidelines and immune response as key contributors to persistent disease activity [7, 9]. The disconnect between early symptom control and inadequate suppression of upstream/type-2 amplification and late-phase recruitment provides a clear justification for mechanism-based approaches to act earlier in the cascade or at convergence points [10, 11].
The purpose of this review is to (i) distinguish between mechanisms that have been shown to be supported by evidence derived from human clinical and/or tear biomarkers vs. those that are based on evidence obtained from animal and/or experimental models, (ii) create maps of pathways to therapeutic failure modes as they relate to each stage of disease (for example, the fact that patients experience continued symptoms after H1 histamine blocking), and (iii) integrate the data regarding epithelial alarmins, mast cell mediators of non-histamine activity, recruitment biology, and neuroimmune signaling related to itch with an understanding of how these may provide upstream and convergent targets to achieve steroid sparing [7, 13, 14]. By relating epithelial alarmins, type-2 cytokine networks, and phase-specific effector biology to treatment performance and limitations, we intend to identify areas where current approaches fail mechanistically and provide the most plausible “next-step” targets for safer, longer-lasting disease control.
The surface of the eye is far more than a simple protective window. It functions as a highly active immunological barrier. The focus of this complex system is the conjunctiva, a thin, transparent mucous membrane that lines the inner eyelids and covers the white part of the globe. The conjunctiva protects and lubricates the eye, lining the inside of eyelids (palpebral conjunctiva) and covering the white part of the eyeball (sclera, called the bulbar conjunctiva), connecting them at the fornix and preventing irritants from entering. Structurally, it consists of a stratified epithelial layer resting upon a vascularized connective tissue known as the substantia propria. This tissue houses the conjunctiva-associated lymphoid tissue (CALT), which acts as a local immune security force for the eye, containing a high density of mast cells, lymphocytes, and dendritic cells [1]. While the epithelium and its goblet cells produce a protective mucus layer to trap debris, this expansive surface area also serves as a primary landing space for environmental aeroallergens, including but not limited to pollen and animal dander.
The most common and typically milder forms of AC are seasonal AC (SAC) and perennial AC (PAC) (Figure 1). The SAC and PAC mechanism of immune response begins with a sensitization phase to an initial, novel exposure of the conjunctival mucosa to environmental allergens [7, 15]. For SAC, these include tree and grass pollen during the spring and summer months, while PAC is induced by house mites, dust, pet dander, and mold, throughout the year (Figure 1). These allergens activate innate immune cells, such as neutrophils and macrophages, using toll-like receptors (TLRs) [7]. Once TLRs are activated, they trigger antigen processing and presentation, for which antigen-presenting cells (APCs) activate naive T lymphocytes. The T cells can then interact with B lymphocytes exposed to similar allergens, releasing IL-4 to direct B cells to class switch to synthesize allergen-specific IgE antibodies. These antibodies bind to high-affinity IgE receptors (FcεRI) on conjunctival mast cells and basophils. These cells become sensitized and primed for future allergen exposures [7, 15].
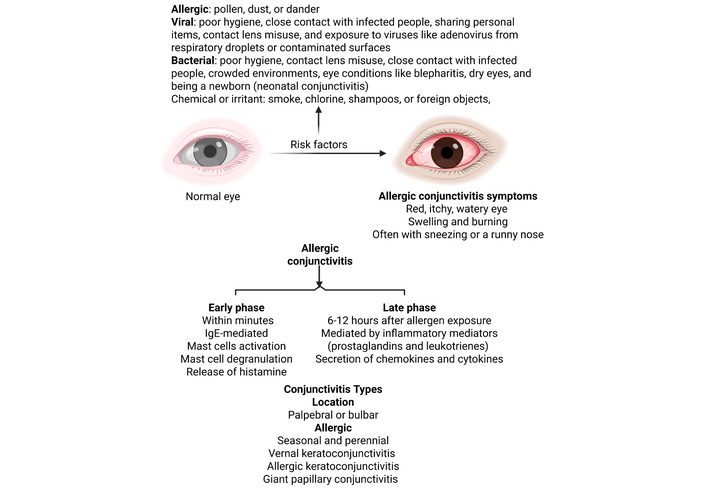
Risk factors, types, and types of conjunctivitis and allergic conjunctivitis. Created in BioRender. Rai, V. (2026) https://BioRender.com/gofy7z2.
The pathophysiology of AC provides a classic example of a Type I hypersensitivity reaction [16], which typically occurs in two waves. When an allergen contacts the eye of a sensitized individual, it cross-links IgE antibodies bound to FcεRI that are already “primed” on the surface of conjunctival mast cells [15, 17]. This event triggers mast cell activation and subsequent degranulation. Mast cell degranulation releases pre-formed inflammatory mediators, most importantly histamine, driving the early phase of SAC. Histamine acts through H1 receptors in the conjunctiva and, upon activation, increases intracellular inositol phosphate and calcium to induce itching, vasodilation leading to redness, and increased microvascular permeability leading to tearing and edema [1, 7, 14, 15]. This early response happens within minutes, creating the immediate discomfort that patients frequently report.
The reaction does not conclude once the initial surge of histamine fades. The late-phase response, which typically occurs 6–12 hours after allergen exposure. Following mast cell activation, a second wave of inflammation develops due to the de novo synthesis of inflammatory mediators such as prostaglandins and leukotrienes in tear fluid [1, 7, 14, 17] (Figure 1). Prostaglandins increase microvascular permeability, leading to worsening conjunctival hyperemia, increasing histamine-induced itching, and stimulating goblet cells, which increase mucus secretion. Leukotrienes work in tandem with histamine and prostaglandins to increase vascular permeability and promote oedema formation. The late phase is sustained by way of cellular infiltration into the conjunctiva [1, 7, 14]. Tumor necrosis factor α (TNF-α) up-regulates adhesion molecules such as intracellular adhesion molecule 1 (ICAM1), which facilitates leukocyte adhesion and migration into conjunctival tissue [7]. Furthermore, Th2 lymphocytes mature during this phase, which release IL-4, IL-5, and IL-13 cytokines. IL-4 and IL-13 specifically form giant papillae through conjunctival fibroblast stimulation and IgE overexpression. IL-5 acts on eosinophils to cause persistent inflammation and may increase long-term conjunctival damage [1] (Figure 1). These chemical messengers act as beacons, recruiting reinforcements from the bloodstream, specifically calling for eosinophils and Th2 cells [1, 2, 16, 17]. In chronic or severe clinical phenotypes, the persistent presence of these cells can lead to significant tissue remodeling and corneal complications. Recent research suggests that the conjunctival epithelium is not merely a passive barrier; it functions as a more active barrier. When stressed or damaged by allergens, it releases alarmins (such as TSLP, IL-33, and IL-25) that serve as master switches [16]. These proteins kickstart the innate immune pathways that sustain the inflammatory cycle and drive persistent disease.
Another form of AC includes VKC (Figure 1). Occurring primarily in male children during the first 10 years of life, VKC is a chronic conjunctival and corneal inflammation due to exposure to airborne environmental allergens [18]. While symptoms can occur perennially, they typically onset in spring and exacerbate in the summer. VKC is a severe ocular allergic disease of childhood characterized by chronic or recurrent conjunctival and corneal inflammation with prominent type-2 skewing and eosinophil-rich infiltration [18–20]. Sensitization and IgE involvement are variable across patients; while many exhibit systemic atopy and evidence of allergen sensitization, disease activity can persist beyond classic episodic IgE-driven mechanisms [18, 20]. As allergen exposure induces IgE production, it binds to FcεRI receptors on mast cells, basophils, and dendritic cells. This results in IgE cross-linking, which triggers mast cell degranulation to mediate the release of histamine, tryptase, and TNF-α to further amplify inflammation [19, 20]. Overall, VKC is driven by a strong type-2 inflammatory program in which IgE-dependent pathways may contribute but are not uniformly dominant across all patients. This establishes VKC as a chronic allergic disease influencing IL-4, IL-5, and IL-13 cytokines, and eosinophilic recruitment similar to SAC and PAC. Specifically for VKC in relation to eosinophils, they also release cytotoxic granule proteins such as eosinophil cationic protein (ECP) and major basic protein, which damage corneal epithelial cells, cause punctate keratitis, and drive the formation of vernal shield ulcers [18, 21]. Lastly, VKC tends to show increased involvement of pattern recognition receptors in conjunctival tissues to sustain inflammation even when allergen exposure fluctuates [18].
Allergic keratoconjunctivitis (AKC) (Figure 1) is a severe form of AC. It is a chronic, noninfectious conjunctival and corneal inflammatory disease clinically and immunologically distinct from SAC and PAC. Most notable is its atopic model involving body systems outside of the eye. It is more than merely an IgE-mediated response and is driven mostly by T-cell responses [22, 23]. The defining features include persistent inflammation, progressive tissue injury, fibrosis, and poor response to antihistamine therapy [22]. Although similar to the previously stated ocular allergies, such as the IgE pathway, eosinophils, and mast cells, individuals with AKC typically exhibit reduced innate immune responses. These include a reduction in keratinocyte antimicrobial peptides and reduced tear deficiency, which can induce recurrent ocular infections to increase inflammation and injury [24]. Centrally, AKC is induced by T-cell immune responses where Th1 delayed hypersensitivity leads to elevated interferon-γ (IFN-γ), increased IL-2, and IL-12. As a result, conjunctival scarring, fibrosis, and notably the poor anti-allergic therapy response make AKC most notable [24, 25].
One unique case of AC includes giant papillary conjunctivitis (GPC) (Figure 1). The disease is an ocular surface inflammation disorder typically associated with contact lens wear, such as soft contacts and extended-wear lenses [26]. GPC has also been noted in ocular protheses, exposed sutures, corneal grafts, and post-surgical foreign material. It is centrally induced by mechanical irritation. This allows contact lens surfaces to quickly accumulate mucus, proteins, bacteria, and airborne particles, increasing conjunctival hyperemia and injection. The immune response is mediated by IgM antibodies in tears, which indicate a local immune reaction rather than a systemic reaction [27]. Mast cell degranulation occurs secondary to mechanical stimulation, which does not induce IgE cross-linking. ECP levels and Th2 responses are also elevated but lower than VKC [27].
AC is increasingly viewed as a mucosal inflammatory disorder of the ocular surface, rather than just a “transient irritant” conjunctivitis. This is because the conjunctiva is located at an extremely high exposure site and has developed its own immune structure, which allows it to respond to antigens in a highly organized manner [1, 28]. Human studies have shown that CALT exists as organized lymphoid tissues containing intraepithelial lymphocytes, subepithelial follicles, and surrounding lymphatic and blood vessels. These findings suggest that ocular surface immunity is built into the eye [28, 29]. The presence of CALT and drainage-associated lymphoid tissue in histological and ultrastructural studies of the human conjunctiva further supports that the conjunctiva can sample antigens and regulate local immune responses [30, 31]. Modern frameworks of mucosal immunology position the ocular surface as part of a larger mucosal immune network. This helps explain why there is a commonality among ocular allergies and other upper airway allergic diseases [28, 32].
During AC, the inflammatory response is generally mediated by IgE-dependent mast cell activation, leading to an influx of inflammatory cells, a model which has been repeatedly reviewed and discussed in both clinical and translational research [1, 13, 32] (Figure 2). In sensitized individuals, the cross-linking of IgE on mast cells results in the rapid degranulation and release of various mediators, resulting in symptoms such as itching, redness, tearing, swelling of the eyes, and chemosis [1, 13]. One major point of convergence in the literature is that the early phase of AC does not occur in isolation from the later phases of inflammation. Early mediator release promotes the subsequent generation of lipid mediators (such as prostaglandins and leukotrienes), chemokines, cytokines, adhesion molecules, and chronic mast cell activation that contribute to the chronic inflammation found in more sustained forms of AC [13]. This connection is important clinically since therapies that primarily reduce early-phase mediator levels may not completely suppress the ongoing cellular response found in chronic forms of AC [13, 32] (Figure 2).
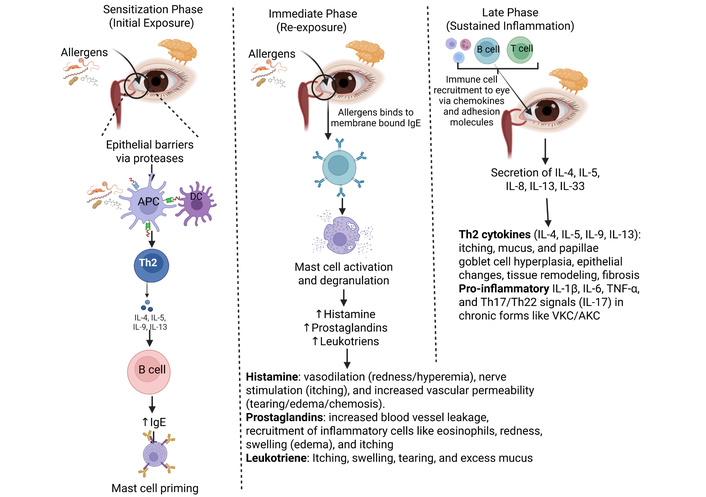
Molecular mechanisms of inflammation of the conjunctiva in allergic conjunctivitis. Allergic conjunctivitis is an immune response, primarily IgE-mediated, involving two phases: an immediate release of histamine from mast cells after allergen exposure, causing itching/redness; and a later phase with inflammatory cells (eosinophils, T-cells) infiltrating, driven by cytokines (IL-4, IL-5, IL-13) from Th2 cells, leading to persistent inflammation, mucus, and tissue changes. This cascade involves allergen cross-linking IgE on mast cells, degranulation, and signaling pathways that recruit more immune cells, causing symptoms like tearing, redness, and swelling (chemosis). IL: interleukin; DC: dendritic cells; APC: antigen-presenting cell; VKC: vernal keratoconjunctivitis; AKC: allergic keratoconjunctivitis. Created in BioRender. Rai, V. (2026) https://BioRender.com/fs6ssw1.
The late phase of AC typically occurs due to recruitment and retention of inflammatory cells in the eye. This is achieved through the action of chemokines and their associated adhesion pathways, converting the initial mediator burst into an inflammatory cell occupation of the eye [13, 33]. Chemokines are specifically identified as critical for organizing the late phase of allergic responses. They recruit and activate leukocytes (including neutrophils, eosinophils, and Th2 lymphocytes) to the conjunctival mucosa [13, 33] (Figure 2). Eotaxin [C-C motif chemokine ligand 11 (CCL11)], IL-8, monocyte chemoattractant protein (MCP-1, MCP-2, MCP-3, MCP-4), Regulated upon Activation, Normal T cell Expressed and Secreted (RANTES, also known as CCL5), macrophage inflammatory protein-1 alpha (MIP-1α/CCL3), and CCL17 are common chemokines involved in the later phase of AC. Activation of leukocytes contributes to the sustained inflammation and symptoms characteristic of the late-phase reaction and thus represents an important mechanism for prolonging symptomatology beyond histamine [17, 34, 35]. The detection of eotaxin and IgE in the tears of human patients suffering from SAC suggests a relationship between chemokines and the pathophysiology of ocular allergic disease. The study proposed that these two biomarkers represent separate aspects of the allergic process, and not redundant signals [35]. Up-regulation of eotaxin-1 in tears during seasonal disease periods, as observed in previous studies, suggests a common link between tear chemokines and symptomatic ocular allergy [36] (Figure 2). Expression of chemokines in more severe ocular allergic disease (VKC) demonstrates elevated expression of RANTES (CCL5), eotaxin, and MCPs in the conjunctiva of VKC patients. This provides further evidence that environments rich in chemokines are associated with chronic inflammation and ocular surface damage [37].
While chemokines provide a gradient for the migration of leukocytes into the inflamed tissue, the literature clearly demonstrates that the adhesion molecules [like ICAM-1, VCAM-1 (vascular cell adhesion molecule 1), E-selectin, and VAP-1 (vascular adhesion protein-1)] expressed on the surface of vascular endothelium and other cells within the eye act as “entry permits” for leukocytes to migrate into the inflamed eye alongside anti-adhesive molecules like endomucin that maintain vascular quiescence [37, 38] (Figure 2). For example, examination of the level of ICAM-1 on the conjunctival epithelial layer and the levels of ECP in the tears of children suffering from AC demonstrated significantly higher levels of ICAM-1 and ECP than in healthy controls. This study demonstrated a direct association between epithelial adhesion biology and eosinophil activation in the ocular surface microenvironment [38]. Experimental studies examining the role of adhesion molecules in murine models of AC have also provided strong evidence for a causal role for the interaction of very late antigen-4 (VLA-4) and VCAM-1 in eosinophil recruitment into the conjunctiva after allergen challenge. Specifically, studies using either antibodies against VLA-4 or VCAM-1 have shown that blocking these molecules can inhibit eosinophil recruitment into the conjunctiva after allergen challenge [39]. More broadly, classical immunology studies have shown that IL-4 can induce VCAM-1 expression on the surface of endothelial cells and promote adhesion of eosinophils and basophils through the interaction of VLA-4. This represents an important cytokine-to-adhesion pathway that links type 2 cytokine signaling to leukocyte recruitment and activation. This is consistent with the established understanding of type 2 cytokine signaling in allergic disease [40].
A key refinement to current mechanistic discussion of ocular allergy is that “histamine biology” alone cannot account for ocular allergy in chronic itch [13, 41]. Histamine-independent mechanisms are associated with allergic ocular itching, including evidence from animal models that TRPA1 neuronal signaling is necessary to develop allergic ocular itching. This indicates that non-histaminergic neural mechanisms may generate symptoms in some subsets of disease [41]. The neuro-immune model is consistent with the translational clinical observation that simply managing the early mediator phase will not necessarily resolve later inflammatory events that maintain late phase symptoms [13, 32].
The “innate immune pathway” model is being increasingly used to characterize the initial signals that predispose ocular surface inflammation to type-2 responses before the typical Th2 effector cascade responses [42, 43]. A recent review of cytokines related to AC summarizes the role of IL-4, IL-5, IL-13, and epithelial-derived cytokines such as IL-33 and TSLP, often viewed as the initial triggers of allergic inflammation [42]. There is also evidence from experimental studies supporting the pollen/TLR4 to IL-33/ST2 pathway in AC models caused by short ragweed pollen, demonstrating an association between an innate receptor pathway and increased type-2 cytokine production due to IL-33/ST2 signaling [43]. Observations in humans also support the importance of epithelial “alarmin” biology by demonstrating the presence of TSLP on the ocular surface and downstream type-2 cytokines (IL-4, IL-5, IL-13) in conjunctival scrapings and tears of patients representing all types of AC [44].
AC effector biology is evolving into a system where epithelial “alarmins” initiate type-2 inflammation; type-2 innate and adaptive effectors amplify it; and neuroimmune pathways convert it into itch. Recent cytokine-centric synopses have identified IL-4, IL-5, and IL-13 as primary cytokines responsible for ocular allergic inflammation, and upstream epithelial cytokines such as IL-33, TSLP (and in broader type-2 contexts, IL-25), influence how disease manifests in the ocular surface [42]. This view is consistent with a papain-soaked contact lens model demonstrating that ocular surface type-2 immune responses dependent upon IL-33 and TSLP signaling are mediated by basophils and ILC2s. These findings support an epithelial-alarmin to ILC2/basophil pathway in experimental animal models of ocular surface inflammation [45]. However, much of the causal evidence for an IL-33/TSLP to ILC2 axis derives from animal models; direct human ocular-surface defining ILC2 abundance, activation state, and correlation with AC severity across phenotypes remain limited. Within this circuit, ILC2 activation is a proximal event since activated ILC2s can rapidly produce IL-5 and IL-13, cytokines which are commonly referred to as type-2 cytokines that recruit and sustain eosinophils/basophils. They also promote mucosa-type programs in tissues relevant to chronic ocular surface disease [42, 45].
Traditionally, mast cells were viewed as effector cells of the early phase in allergic contact dermatitis. However, mast cells release a variety of other inflammatory mediators besides histamine that can contribute to chronic inflammation of the late phase. Histamine is released by activated mast cells along with several proteolytic enzymes such as tryptase; lipid mediators, cytokines, and chemokines [46, 47]. Studies of tear biomarkers have shown that patients with active ocular allergies have elevated levels of tryptase and that tryptase increases in the tears after conjunctival antigen challenge [48]. In addition, tryptase has also been demonstrated to be elevated in severe VKC, where levels correlated with the clinical severity of VKC and decreased with treatment [49]. The non-histamine mediators produced by mast cells are not adequately blocked by H1 blockers, which may explain why some patients may respond poorly to antihistamines. One clinically relevant example is prostaglandin D2 (PGD2) signaling through DP2/CRTH2; a DP2 receptor antagonist topically applied in a murine allergen-challenge model was able to reduce ocular inflammation, thereby demonstrating the PGD2-CRTH2 (also known as DP2) axis as a targetable late-phase mediator [50].
The feed-forward recruitment loops for the persistence of the late phase are supported by both type-2 cytokines and chemokines. The role of chemokines and presence of eotaxin and RANTES in tears of individuals with SAC suggests the existence of an interrelated inflammatory network as opposed to a single inflammatory mediator acting independently of one another [33, 35, 36, 51]. Chemokines mediate their effects through adhesive pathways to facilitate recruitment of the leukocyte to the site of inflammation. Inhibition of conjunctival eosinophil migration and reduced symptoms of AC by blocking VLA-4/VCAM-1 pathway in mice provides proof that blocking the leukocyte’s ability to traffic into the tissue can impede the accumulation of the late phase effectors [39].
Increasingly, neuroimmune communication is viewed as a component of the effector circuit, as it represents an alternative mechanism for itch generation (neuronal output) through cytokine-mediated cooperation between immune cells and neurons, as opposed to histamine alone. Increased conjunctival itching with IL-33 activation-mediated CGRP-producing memory Th2 cells cooperating with somatosensory neurons identifies IL-33-ST2-CGRP-RAMP1 pathway as an itch-generating pathway during severe allergic conjunctival inflammation [52]. IL-33 signaling in sensory neurons promotes chronic itch states, providing biological evidence for IL-33 as a neuroimmune itch signal [53]. Additionally, ocular sensory neuron studies using mouse models demonstrate regulation of ocular inflammatory responses by sensory neuron-associated TRP channel pathways. TRPV1 and sensory nerve innervation regulate mast cell degranulation and eosinophil infiltration through neuropeptide signaling, indicating bidirectional immune-neural regulatory relationships on the ocular surface [54]. Mechanistically, persistent ocular itching can be histamine independent. Conjunctiva innervated sensory neurons have segregated TRP channels associated with itch pathways, which suggests that while blocking histamine will not eliminate itch in all patients [41].
Despite optimized dual-acting antihistamines, many patients have continued to experience suboptimal symptom relief from their disease, indicating that some of these patients may be experiencing prolonged or more intense inflammatory processes that are non-histamine related [7, 16, 31]. Studies utilizing human tear and conjunctival sampling have found increased levels of non-histamine mediators such as proteases and cytokine/chemokine during active disease, suggesting that even though histamine blockade has been achieved, there continues to be significant inflammatory activity occurring within the eye [33–36, 44, 48]. As it stands today, there are limited patient-based stratifications that directly correlate a particular neuro-immune pathway to non-response to antihistamine therapy. Therefore, this is a significant translational gap for future research [34, 42].
The effector-circuit model shows why antihistamines are often insufficient: they block H1 receptors on cells stimulated by histamine to produce early symptoms but do nothing about the other products from activated mast cells (tryptase), prostaglandin signaling (PGD2 to CRTH2), or the cytokine/chemokine networks responsible for the later phase of inflammation for cellular infiltration [33, 46, 48, 50]. Importantly, many future therapeutic options that target the same effector circuitry are conceptualized as part of this model. Chemical amplification of signals upstream can be targeted with reproxalap and was significantly better than placebo for all typical symptoms of AC in the phase 3 INVIGORATE study [55]. Cytokine signaling hubs can be blocked using topical JAK inhibitors. Taficinitib can inhibit experimental AC and mast-cell degranulation through its effect on the JAK3/STAT pathway [56]. The PGD2 to CRTH2 arm can be targeted using DP2 antagonists and has demonstrated direct ocular efficacy in models of allergen challenge [50]. Late-phase cell influx can be decreased by inhibiting trafficking machinery such as VLA-4/VCAM-1 [39]. Inhibiting intracellular effector mechanisms upstream of mediator release is another approach. PF-431396 [focal adhesion kinase (FAK) inhibitor] suppressed IgE-mediated mast-cell activation and decreased allergic inflammation [57]. Finally, refining the steroid pathway aims to decrease the classic steroid liabilities and the late infiltrate. Mapracorat controls late-phase experimental ocular allergy by modifying eosinophil biology and has undergone clinical evaluation for AC in NCT01289431 [58]. As practical precision medicine links between mechanism and therapy choice, tear biomarkers corresponding to the effector arms are repeatedly identified by human studies and can be used to stratify “histamine dominant” vs. “infiltrate dominant” disease when designing and evaluating trials of therapies targeting pathways [33, 35, 49].
Most discussions of ocular allergy focus on IgE and mast cells, but the complement system is increasingly recognized as a critical amplifier of the inflammatory response. Complement proteins are not just present in the tear film as a passive defense; they are also synthesized locally by conjunctival cells. With respect to ocular allergy, the complement system functions as an early-warning bridge, detecting environmental allergens and rapidly amplifying inflammatory signaling at the ocular surface to engage the broader immune response. This amplification occurs primarily through the generation of anaphylatoxins, specifically complement component 3a (C3a) and C5a [59, 60]. These small protein fragments are highly potent mediators of inflammation [61]. Notably, they are capable of directly inducing mast cell degranulation and release of histamine and tryptase even in the absence of allergen-specific IgE [61]. This mechanism represents an alternative innate pathway for the early-phase allergic response, explaining why some experience severe clinical symptoms despite low IgE levels on allergy blood testing or skin prick testing.
Beyond the initial symptoms of pruritus and erythema, the complement system also organizes and sustains the late-phase inflammatory response. C5a functions as a strong chemoattractant, establishing a chemical gradient that recruits eosinophils and Th2 lymphocytes from the circulation into conjunctival tissue [60]. This results in a late phase reaction, typically beginning 6–12 hours after the initial exposure and can last for days. This is mediated by increased secretion of cytokines and chemokines (e.g., IL-4, IL-5, IL-8, and IL-13), recruiting other inflammatory leukocytes, especially eosinophils and Th2 lymphocytes [62]. Eosinophils release cytotoxic proteins (e.g., major basic protein) and other pro-inflammatory factors that can cause sustained inflammation and potential tissue damage. Complement activation could act to amplify inflammation locally at the ocular surface through anaphylatoxins (i.e., C3a/C5a), which increase vascular permeability and also activate innate effectors and recruit leukocytes to tissues [59, 61]. Direct confirmation of a complementary-alarmin amplification loop for human AC is currently limited and should be considered as an evolving model as opposed to a proven mechanism. By linking initial allergen exposure to broader innate and adaptive immune signaling pathways, the complement system sustains a self-reinforcing inflammatory cycle. This mechanism provides a clear explanation for why ocular inflammation can become persistent, self-sustaining, and refractory to treatment with antihistamines alone in chronic disease.
Mechanisms for acute complement activation exist across a range of heterogeneity in terms of evidence [61]. The strongest evidence for the mechanisms exists at the level of human clinical and biomarkers for IgE-dependent mast cell activation in the immediate response to allergens [7, 15], as well as tear/conjunctival enrichment of various inflammatory mediators including proteases and chemokines [33–36, 48]; as well as human clinical evidence for the efficacy of dual-acting antihistamines, corticosteroids and calcineurin inhibitors when used in appropriate phenotypes [7, 32]. Moderate human clinical evidence also exists to support the expression of epithelial cytokine/alarmins on the ocular surface and associated type-2 signatures, although additional research is needed to clarify both causation and the relationship of these mechanisms to specific phenotypes [10, 42, 44]. On the other hand, several pathway-specific, putative causal mechanisms—namely, ILC2-mediated circuits and TRP channel-defined itch circuits—exist primarily through evidence from animal and experimental studies [41, 45, 52–54].
Existing therapies for managing AC follow a stepwise approach depending on the severity of the condition and the risk to the ocular surface. The primary goal of current treatment is to reduce the early phase mast cell-mediated response to relieve symptoms and to manage the late phase cellular inflammation caused by eosinophils, Th2 lymphocytes, cytokines, and chemokines in more severe cases [1, 32].
Avoidance of the allergen responsible for triggering AC is a fundamental aspect of the management. Exposure to airborne allergens, such as pollen, dust mites, pet dander, etc., is responsible for flaring of the disease [1, 32]. Behavioral modifications to minimize exposure to the allergen are regularly recommended and include avoiding eye rubbing and being outdoors during peak pollen seasons. Cold compresses help reduce hyperemia (redness) and edema (swelling) of the conjunctiva through vasoconstriction. Preservative-free artificial tears will dilute the allergens on the ocular surface and stabilize the tear film in patients who suffer from co-existing dry eye syndrome [1, 32]. Treatment for AC reflects the diversity of phenotypic expressions of the disease. SAC and PAC are typically treated with avoidance of the allergens involved, lubrication of the eyes with artificial tears and/or other lubricating drops, and topical dual-acting agents. Corticosteroids are used for short-term emergency relief of symptoms [1, 32]. In contrast, VKC and AKC, which are characterized by chronic inflammation, recruitment of leukocytes by chemokines, and potential damage to the ocular surface, require more intensive and prolonged immunomodulatory treatments [1, 37]. The strategy of treatment by disease phenotype illustrates a major limitation of the current treatment options. While most treatments may effectively inhibit the histamine-mediated early responses, they are unable to completely manage the cytokine-, chemokine-, and adhesion molecule-driven late phase inflammation characteristic of more severe and chronic forms of AC [32, 33].
First-line therapy for most patients suffering from SAC and PAC is topical dual-acting antihistamines/mast cell stabilizers [1, 13]. Antihistamines block the H1 receptors of histamine during the early phase of the allergic reaction, providing immediate relief of symptoms. The mast cell stabilizers prevent the release of mediators from mast cells after degranulation with continued use [1, 13]. Antihistamines/Mast cell stabilizer products available include olopatadine, ketotifen, alcaftadine, bepotastine, and azelastine. Clinical trials comparing these products demonstrate a statistically significant reduction in symptoms such as itching, redness, and tearing among the treated populations. However, there are variations between products regarding the time required to begin providing relief, duration of action, and frequency of application, which may impact physician decision-making [13]. For patients with seasonal allergies who have a predictable pattern of disease flare-ups, prophylactic therapy initiated before expected allergen exposure is often recommended, as mast cell stabilization is most effective when used consistently [1].
A combination of topical vasoconstrictors/decongestants may provide temporary relief of redness associated with the conjunctiva, but they do not address the root cause of the disease and lead to rebound hyperemia when used continuously, thus limiting their utility in long-term management [1]. Topical nonsteroidal anti-inflammatory drugs (NSAIDs) may provide relief from ocular discomfort and inflammation but are generally less effective than antihistamine-containing products in treating the itching of AC. Therefore, they are rarely used as monotherapy [1]. Systemic antihistamines may be helpful to patients with coexisting AR; however, systemic antihistamines are known to exacerbate ocular surface dryness in some patients and increase the symptoms of AC [32].
Topical corticosteroids are used to treat patients with moderate to severe AC, acute flare-ups of the disease, and/or corneal involvement, including VKC and AKC [1, 32]. Corticosteroids provide broad inhibition of the inflammatory processes and rapidly reduce the conjunctival edema, hyperemia, and cellular infiltration. However, their use is limited by side effects, including elevated intraocular pressure, cataracts, impaired wound healing, and increased susceptibility to infections [1]. Therefore, “soft” steroids, including loteprednol and flurometholone, are often preferred for this purpose, and their use is usually limited to short courses of therapy under ophthalmologist supervision [32].
Chronic and steroid-dependent forms of ocular allergy, especially VKC and AKC, benefit from the use of topical immunomodulators as part of long-term disease management [1]. Cyclosporine A inhibits T cell activation and cytokine production, thereby reducing chronic inflammation without the steroid-induced side effects. Studies have confirmed its efficacy in alleviating the symptoms and ocular surface manifestations of severe allergic eye disease [1]. Tacrolimus, a second calcineurin inhibitor, is effective in treating recalcitrant inflammation, corneal involvement, and symptomatology [1]. These agents are being increasingly used as long-term maintenance therapies following initial steroid-induced remission of disease to prevent recurrence [1].
There is an overwhelming consensus among mechanistic reviews over the last few years to push “next generation” therapies for AC upstream and away from just controlling histamine to disrupting the early cascade, blocking the type-2 immune response, and suppressing the late phase recruitment of cells into the eye. This is because the persistence of AC is due to the cytokine and chemokine networks and trafficking of cells, and not a single mediator [13, 33, 42]. Cytokines (such as IL-4, IL-5, IL-13, and epithelial alarmins, including IL-33/IL-25/TSLP) involved in the type-2 immune response have repeatedly been referred to as mechanistic “control knobs” that determine the level of disease and chronicity of AC. Therefore, these control knobs would be logical targets for steroid-sparing treatment strategies [16, 42]. Chemokines have also been identified as playing a major role in the formation of the late-phase inflammatory infiltrate, particularly in terms of eosinophils, and therefore represent a potential target for therapeutic intervention, specifically to address the clinical problems of prolonged symptoms and tissue damage seen in the more severe forms of AC [16, 33].
An additional upstream strategy includes inhibiting the reactive aldehyde species that enhance the inflammatory signaling process. In the phase 3 INVIGORATE allergen chamber study, the reactive aldehyde species modulator reproxalap was found to meet both primary and key secondary endpoints of reducing ocular itching and redness compared to vehicle, providing support for the idea that early chemical mediators can be therapeutically targeted in AC. A second phase 3 program (ALLEDIVIATE) has been registered for AC as well, indicating continued late-stage interest in this upstream mechanism as a steroid-free option. The practical “proposition” suggested by these data is that aldehyde modulating therapy may provide a bridge between antihistamines (rapid symptom relief) and immunosuppressants (widespread control), to reduce the late phase recruitment of cells that is dependent on prior amplification, while avoiding the side effects of steroids [55].
Since many cytokines relevant to AC act through the JAK/STAT pathway, topical JAK inhibition represents a potentially useful strategy to inhibit multiple cytokine inputs (signaling associated with IL-4, IL-5, IL-13) with a single class of small molecules [42, 63]. While direct AC evidence primarily exists as preclinical/experimental data, as opposed to late-stage clinical data specific to AC, tofacitinib has been demonstrated to suppress mast cell degranulation and decrease allergic inflammation in experimental AC models [56]. Ophthalmic exposure data that will inform the feasibility of delivering tofacitinib topically come from studies of topical ophthalmic tofacitinib in other ocular surface inflammatory diseases (i.e., dry eye disease), which demonstrated immunomodulatory effects. Thus, while AC-specific trials are limited, these studies demonstrate the plausibility of using topical ocular delivery for AC [63]. Therefore, a reasonable near-term direction is translation/re-purposing of other drugs or small molecules. Prioritizing well-designed AC studies (both allergen challenge and field trials) to assess whether topical JAK inhibition can decrease the late phase signs and persistent itch beyond what dual-acting antihistamines can achieve is needed [13, 42].
Prostaglandin signaling, particularly PGD2 acting via DP2/CRTH2, has been experimentally demonstrated as an AC-relevant effector pathway and is continually referenced in therapeutic target overviews as a logical target [46, 50]. In a mouse model, decreased ocular inflammation post-allergen exposure with topical DP2 antagonism as a new therapeutic option for AC, specifically associating CRTH2 blockade with the late phase, not with the immediate histamine blockage of the early phase [50]. Systemic CRTH2 antagonism has also shown clinical relevance to AC by decreasing ocular and nasal symptoms in allergic individuals challenged with allergens; thus, demonstrating that the pathway is involved in generating ocular symptoms [64]. The potential future implication is that CRTH2-directed agents (preferably topical) could be assessed as add-on or alternative therapies for patients who have persistent itch/redness despite optimization of their antihistamine regimen [13, 50].
Chemokines are repeatedly referred to as the “cell traffic controllers” of late-phase AC. The effects of chemokine concentration gradients in recruiting eosinophils and Th2 lymphocytes and the presence of chemokines in tears, as discussed above, provide a link between chemokine activity, disease phenotype, and a rationale for chemokine inhibition in both a mechanistic and clinical sense [16, 33, 34]. Involvement of chemokine receptors (e.g., CCR6) in allergic conjunctival inflammation in experimental models provides evidence for receptor-level targeted approaches compared to CCR3-centric eosinophil-based concepts alone [33, 65]. A practical proposal stemming from a two-step strategy: (1) identify a patient’s dominant recruitment signature via tear biomarkers (e.g., eosinophil-attracting chemokines) and (2) match this signature to a receptor/ligand antagonist-based approach to reduce late-phase signs generated by the infiltrates [33, 34].
If chemokines serve as the “GPS”, adhesion molecules are the “hardware” that leukocytes need to access tissue, thereby allowing recruitment blockade to serve as a complementary approach to chemokine antagonism [46, 62]. Mice lacking the ability to interfere with the VLA-4/VCAM-1 interaction had reduced eosinophil invasion into the conjunctiva and reduced experimental AC, establishing adhesion pathways as a causal mechanism as opposed to mere correlations [39]. Therapeutic-target syntheses explicitly include integrins and associated recruitment machinery (e.g., ICAM-1-linked interactions) as potential future areas of investigation for AC because they operate at a critical convergence point for multiple inflammatory triggers [46].
Blocking the cellular signal of mast cells using FAK inhibitors as a “mast-cell” suppressant. The “hub strategy” of convergence inhibits the cellular signaling involved in mast-cell effector functions instead of inhibiting individual mediators downstream of the mast-cell effector function. Pharmacological inhibition of FAK, suppressing the allergic inflammatory responses and clinical signs of allergic disease in an AC mouse model, suggests that FAK plays a role in FcεRI-linked mast-cell degranulation signaling. This directionally supports further testing of FAK inhibition (and related kinase nodes) as a method of reducing both mast-cell outputs that depend upon histamine and those that do not require histamine without utilizing corticosteroids [46, 57].
Topical steroids remain effective; however, their utility is limited due to systemic exposure associated with repeated use. Selective glucocorticoid receptor agonists (SEGRAs) continue to be considered as a viable option to provide the anti-inflammatory efficacy of steroids with a reduction in the systemic elevated pressures/cataracts, typically associated with long-term steroid use [58, 66]. Mapracorat (SEGRA) was found to induce apoptosis of eosinophils in the conjunctiva during late-phase ocular allergy models, and thus its effect was directly aligned to the later-phase infiltrate that causes chronicity [58]. There is an active clinical trial record (NCT01289431) for mapracorat ophthalmic formulation in AC, supporting that this is not simply speculative and has progressed to human trials.
The most significant challenge to treating AC is heterogeneity, seasonal vs. perennial disease, mild SAC/PAC vs. severe VKC/AKC. The increasing number of review articles is advocating for biomarker-based stratification to avoid a one-size-fits-all, all-escalating treatment approach [16, 17]. Tear biomarkers have been proposed and reviewed for allergic conjunctival diseases, including chemokines that correlate with severity and biomarkers that can potentially identify phenotypes or monitor response, providing a basis for a personalized medicine approach [34, 67]. Periostin continues to be referenced as a type-2 inflammation-linked biomarker in multiple allergic disease contexts, including ocular allergic disease contexts, providing support for its possible application in ocular phenotypes characterized by high levels of type-2 inflammation [68].
Topical treatments may fail even if the mechanisms are correct due to short residence times, poor penetration into the eye, preservative intolerances, and compliance issues; therefore, drug delivery is becoming a primary component of the therapeutic target [69, 70]. Ocular delivery via nanotechnology has been developed to enhance the retention and penetration of the active ingredient(s) in addition to providing long-lasting (sustained) release. This strategy is important for anti-inflammatory agents requiring chronic/long-duration exposure to downregulate late-phase biology [70, 71]. An example of advancements in delivery/formulations of topical treatments for ocular allergies is the development of preservative-free, once-daily topical antihistamines. Bilastine 0.6% preservative-free formulations were found to demonstrate rapid onset and prolonged duration in randomized clinical trials, thereby demonstrating how advances in formulation can provide improved real-world control for patients using existing drug classes [72, 73]. A forward-thinking approach is that “mechanism-forward” drugs (i.e., aldehyde modulators, kinase inhibitors, CRTH2 antagonists) will most likely require parallel advancements in delivery (preservative-free multidose, sustained-release delivery systems, contact lenses, and/or in-situ gelling platforms) to obtain consistent therapeutic exposure at the ocular surface [69, 70].
Clinical trials are essential for developing new therapies by rigorously testing potential treatments (drugs, devices, vaccines) in humans, proving their safety, effectiveness (efficacy), and optimal dosage, transforming lab discoveries into approved, life-saving medicines, and providing evidence to regulatory bodies like the FDA for patient access, all through structured phases (I–IV) that assess risks vs. benefits for better care. Table 1 summarizes the ongoing clinical trials for AC.
Ongoing clinical trials for allergic conjunctivitis (AC).
| Trial ID | Phase/Patients number | Aim | Strategy | Status/Results |
|---|---|---|---|---|
| NCT07273747 | N/A72 | To test whether oral Kelulut honey is safe and more effective than a honey-flavored placebo at improving AC symptoms. | One-month, parallel-group, placebo-controlled supplementation trial where adults with AC are assigned to take oral Kelulut honey or a honey-flavoured placebo, and outcomes are compared from baseline to 1 month. | Recruiting patients. |
| NCT01743027 | Phase III902 | To determine whether olopatidine (0.77%, 0.2%, 0.1%) is safe and provides relief of ocular itching in AC with rapid onset and 24-hour duration. | Multicenter, randomized, double-masked, phase 3 conjunctival allergen challenge (CAC) model where adults receive a single dose in both eyes and post-CAC itching scores at minutes 3/5/7, and 24 hours are measured. | Completed.Olopatadine 0.77% showed a rapid onset and sustained superiority in reducing ocular itching vs. vehicle and lower-dose olopatadine, with a favorable safety profile. |
| NCT07220408 | Phase II60 | To evaluate the safety/efficacy of TL-925 ophthalmic emulsion 0.1% vs. vehicle for treating seasonal AC in the CAC model. | Single-center, randomized, double-masked, vehicle-controlled phase 2 study assigning participants to either bilateral TL-925 vs. vehicle eye drops, then comparing post-challenge outcomes. | Recruiting patients. |
| NCT03186755 | Phase IV42 | To compare whether Hylo-Dual (hyaluronic acid 0.05% + ectoine 2.0%) is as effective as olopatadine 0.1% at controlling seasonal AC in children. | Phase 4, randomized, parallel, double-masked trial assigning 8 weeks of bilateral drops (Hylo-Dual TID vs. Patanol BID), then comparing clinical outcomes. | Recruiting patients. |
| NCT04207736 | Phase III95 | To test whether reproxalap ophthalmic solution 0.25% is effective and safe vs. vehicle for seasonal AC under controlled allergen exposure. | Single-center, randomized, double-masked, crossover, vehicle-controlled phase 3 study using an environmental exposure chamber (EEC) comparing outcomes after subjects received reproxalap vs. vehicle. | Completed.Reproxalap was statistically superior to vehicle across AC symptoms in the chamber model, with favorable tolerability/safety reported. |
The clinical challenge posed by AC is best understood from an immunological standpoint as an active inflammatory process due to a combination of innate and adaptive immune processes. Early symptoms are due to rapid mediator-related release from activated mast cells; however, persistent symptoms reflect late-phase responses supported by continued production of lipid mediators and cytokines/chemokines. Epithelial-derived signals such as TSLP, IL-33, and IL-25 can activate and sustain type-2 inflammatory responses via ILC2. Finally, neuroimmune signaling also explains why ocular itch can continue even when histamine release is blocked. The above mechanistic rationale supports the view that antihistamines alone are often insufficient for the treatment of many patients with AC and supports the use of next-generation therapies directed at interrupting the upstream amplification of the response. Further advances in this area are likely to occur when combining mechanism-based therapeutic targets with biomarker-based stratification of patients and improved ocular delivery systems to allow for more durable efficacy with reduced toxicity and increased compliance.
AC: allergic conjunctivitis
AKC: allergic keratoconjunctivitis
AR: allergic rhinitis
C3a: complement component 3a
CALT: conjunctiva-associated lymphoid tissue
CCL11: C-C motif chemokine ligand 11
ECP: eosinophil cationic protein
FAK: focal adhesion kinase
GPC: giant papillary conjunctivitis
ICAM1: intracellular adhesion molecule 1
IL: interleukin
ILC2: group 2 innate lymphoid cell
MCP: monocyte chemoattractant protein
PAC: perennial allergic conjunctivitis
PGD2: prostaglandin D2
RANTES: Regulated upon Activation, Normal T cell Expressed and presumably Secreted
SAC: seasonal allergic conjunctivitis
SEGRAs: selective glucocorticoid receptor agonists
TLRs: toll-like receptors
TNF-α: tumor necrosis factor α
TSLP: thymic stromal lymphopoietin
VCAM-1: vascular cell adhesion molecule 1
VKC: vernal keratoconjunctivitis
VLA-4: very late antigen-4
TS: Conceptualization, Writing—original draft. AJ, HM, and JJ: Writing—original draft. VR: Conceptualization, Writing—review & editing, Validation, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The authors received no specific funding for this study.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 7916
Download: 76
Times Cited: 0
Darrell O. Ricke
