 Review
Review

Affiliation:
1Synergo, Institute of Clinical Immunotherapy and Advanced Biological Treatments, 65100 Pescara, Italy
Email: digioacchino@me.com
ORCID: https://orcid.org/0000-0002-9224-5886
Affiliation:
2Respiratory Clinic, Department of Internal Medicine, University of Genoa, 16132 Genoa, Italy
3Respiratory & Allergy Clinic, IRCCS Ospedale Policlinico San Martino, 16132 Genoa, Italy
ORCID: https://orcid.org/0000-0002-3661-5731
Affiliation:
2Respiratory Clinic, Department of Internal Medicine, University of Genoa, 16132 Genoa, Italy
3Respiratory & Allergy Clinic, IRCCS Ospedale Policlinico San Martino, 16132 Genoa, Italy
ORCID: https://orcid.org/0000-0003-2460-4709
Affiliation:
4Department of Biomedical Sciences, Humanitas University, 20089 Pieve Emanuele, Milan, Italy
5Personalized Medicine, Asthma and Allergy, IRCCS Humanitas Research Hospital, 20089 Rozzano, Milan, Italy
ORCID: https://orcid.org/0009-0003-8582-4008
Affiliation:
6Department of Educational Sciences, Faculty of Psychology, University of Catania, 95121 Catania, Italy
7Center of Excellence for the Acceleration of Harm Reduction, University of Catania, 95121 Catania, Italy
ORCID: https://orcid.org/0000-0001-7414-155X
Affiliation:
8Department of Internal Medicine, University of Genoa, 16132 Genoa, Italy
ORCID: https://orcid.org/0009-0002-5246-6892
Affiliation:
9Unit of Respiratory Medicine, Department of Experimental Medicine, University of Rome “Tor Vergata”, 00133 Rome, Italy
ORCID: https://orcid.org/0000-0003-4895-9707
Affiliation:
10Swiss Institute of Allergy and Asthma Research (SIAF), University of Zurich, 7265 Davos, Switzerland
ORCID: https://orcid.org/0000-0001-9951-6688
Affiliation:
4Department of Biomedical Sciences, Humanitas University, 20089 Pieve Emanuele, Milan, Italy
5Personalized Medicine, Asthma and Allergy, IRCCS Humanitas Research Hospital, 20089 Rozzano, Milan, Italy
Affiliation:
11Changzhou Key Laboratory of Respiratory Medical Engineering, Institute of Biomedical Engineering and Health Sciences, School of Medical and Health Engineering, Changzhou University, Changzhou 213164, Jiangsu, China
ORCID: https://orcid.org/0000-0003-1970-8239
Affiliation:
12Department of Pediatrics, Federal University of Paraná, Curitiba 80060-000, Brazil
ORCID: https://orcid.org/0000-0002-8550-8051
Affiliation:
13Allergy and Clinical Immunology Service, Centro Hospitalar Universitário de Santo António, 4099-001 Porto, Portugal
ORCID: https://orcid.org/0000-0001-8956-9145
Affiliation:
14Department of Clinical and Biological Sciences, University of Turin, 10043 Orbassano, Turin, Italy
15Severe Asthma, Rare Lung Disease and Pathophysiology Unit, San Luigi Gonzaga University Hospital, 10043 Orbassano, Turin, Italy
ORCID: https://orcid.org/0000-0003-3876-8587
Affiliation:
16Center of Research Excellence in Allergy & Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand
17Department of Parasitology, Biodesign Innovation Center, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand
ORCID: https://orcid.org/0000-0003-2681-2958
Affiliation:
16Center of Research Excellence in Allergy & Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand
18Department of Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand
ORCID: https://orcid.org/0000-0002-9540-6171
Affiliation:
19Department of Clinical Medicine and Surgery, University of Naples “Federico II”, 80138 Naples, Italy
20Istituti Clinici Scientifici Maugeri IRCCS, Pulmonary Rehabilitation Unit of Telese Terme Institute, 82037 Telese Terme, Italy
ORCID: https://orcid.org/0000-0001-6751-9921
Affiliation:
21Allergy Center, CUF Descobertas Hospital, 1998-018 Lisbon, Portugal
ORCID: https://orcid.org/0000-0003-1837-2980
Affiliation:
22Department of Internal Medicine, University of Genoa and Allergology and Clinical Immunology Unit, Ospedale San Bartolomeo, 19038 Sarzana, Italy
ORCID: https://orcid.org/0000-0002-6403-6905
Affiliation:
23Department of Otorhinolaryngology-Head and Neck Surgery, Seoul National University Bundang Hospital, Seongnam 13620, South Korea
24Department of Otorhinolaryngology-Head and Neck Surgery, Seoul National University College of Medicine, Seoul 13620, South Korea
ORCID: https://orcid.org/0000-0001-5905-057X
Affiliation:
4Department of Biomedical Sciences, Humanitas University, 20089 Pieve Emanuele, Milan, Italy
5Personalized Medicine, Asthma and Allergy, IRCCS Humanitas Research Hospital, 20089 Rozzano, Milan, Italy
ORCID: https://orcid.org/0000-0003-3953-9225
Affiliation:
25Division Allergy and Clinical Immunology, Department of Medicine ASL Salerno, “Santa Maria della Speranza” Hospital, 84091 Battipaglia, Italy
26Postgraduate Programme in Allergy and Clinical Immunology, University of Naples “Federico II”, 80138 Naples, Italy
ORCID: https://orcid.org/0000-0001-5640-6446
Affiliation:
27RISE-Health, MEDCIDS—Department of Community Medicine, Information and Health Decision Sciences, Faculty of Medicine, University of Porto, 4200-450 Porto, Portugal
28Allergy Unit, CUF-Porto Hospital and Institute, 4100-180 Porto, Portugal
ORCID: https://orcid.org/0000-0002-5468-0932
Affiliation:
29Department of Educational Sciences, Faculty of Psychology, University of Catania, 95121 Catania, Italy
ORCID: https://orcid.org/0000-0002-0234-7011
Affiliation:
8Department of Internal Medicine, University of Genoa, 16132 Genoa, Italy
ORCID: https://orcid.org/0000-0002-5393-0071
Affiliation:
23Department of Otorhinolaryngology-Head and Neck Surgery, Seoul National University Bundang Hospital, Seongnam 13620, South Korea
24Department of Otorhinolaryngology-Head and Neck Surgery, Seoul National University College of Medicine, Seoul 13620, South Korea
30Sensory Organ Research Institute, Seoul National University Medical Research Center, Seoul 03080, South Korea
31Institute of Allergy and Clinical Immunology, Seoul National University Medical Research Center, Seoul 03080, South Korea
ORCID: https://orcid.org/0000-0002-1361-8585
Affiliation:
32Allergy and Clinical Immunology, Medicine and Surgery Department, University of Parma, 43121 Parma, Italy
ORCID: https://orcid.org/0000-0003-1439-7902
Affiliation:
33University Clinic of Respiratory and Allergic Diseases Golnik, 4204 Golnik, Slovenia
34Biotechnical Faculty, University of Ljubljana, 1000 Ljubljana, Slovenia
ORCID: https://orcid.org/0000-0002-2596-4952
Affiliation:
351st Respiratory Department, Medical School, Sotiria Chest Hospital, National and Kapodistrian University of Athens, 11527 Athens, Greece
ORCID: https://orcid.org/0000-0003-0138-5582
Affiliation:
36Department of Biological Sciences and Pathobiology, University of Veterinary Medicine Vienna, 1210 Vienna, Austria
ORCID: https://orcid.org/0000-0001-5005-9228
Affiliation:
16Center of Research Excellence in Allergy & Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand
37Rhinology and Allergy Division, Department of Otorhinolaryngology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand
ORCID: https://orcid.org/0000-0003-1995-4798
Affiliation:
38Department of Pulmonology, Celal Bayar University Faculty of Medicine, 45040 Manisa, Türkiye
ORCID: https://orcid.org/0000-0002-4032-0944
Affiliation:
39Institute of Medical Sciences, University of Aberdeen, AB25 2ZD Aberdeen, U.K.
ORCID: https://orcid.org/0000-0002-7138-4078
Affiliation:
40Institute of Allergology, Charité—Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, 12203 Berlin, Germany
41Fraunhofer Institute for Translational Medicine and Pharmacology ITMP, Immunology and Allergology, 12203 Berlin, Germany
ORCID: https://orcid.org/0000-0002-1466-8875
Affiliation:
4Department of Biomedical Sciences, Humanitas University, 20089 Pieve Emanuele, Milan, Italy
5Personalized Medicine, Asthma and Allergy, IRCCS Humanitas Research Hospital, 20089 Rozzano, Milan, Italy
ORCID: https://orcid.org/0000-0001-8467-2557
Explor Asthma Allergy. 2026;4:1009111 DOI: https://doi.org/10.37349/eaa.2026.1009111
Received: December 10, 2025 Accepted: January 28, 2026 Published: March 02, 2026
Academic Editor: Makoto Hoshino, International University of Health and Welfare Atami Hospital, Japan
This review describes the eosinophil journey through the various physiological and pathophysiological phases, from production, maturation, and activation by chemokines and cytokines [especially eotaxin, interleukin (IL)-5, IL-3, and granulocyte-macrophage colony-stimulating factor (GM-CSF)], to interaction with the innate and adaptive immune system and tissue homing. Excessive production and activation of eosinophils lead to the release of granule proteins, such as major basic protein, eosinophil cationic protein, eosinophil peroxidase, and others, resulting in inflammation, cell cytotoxicity, and oxidative stress. The pathogenesis, clinical features, diagnostic processes, and the latest therapeutic approaches to the resulting diseases—which affect the upper and lower airways, gastrointestinal tract, skin, myocardium, and may occur systemically—are discussed.
Eosinophils are a specialized type of granulocyte, characterized by their prominent orange-red cytoplasmic granules visible under an optical microscope after staining with eosin. Generated in the bone marrow from pluripotent hematopoietic stem cells, the maturation of eosinophils is tightly regulated by various cytokines, particularly interleukin (IL)-5 [1]. Once mature, these cells enter the peripheral blood, where they represent approximately 1–4% of circulating leukocytes, before migrating to tissues such as the gastrointestinal tract, lungs, thymus, and spleen. Under physiological conditions, tissue-resident eosinophils contribute to immune homeostasis and maintenance of epithelial barrier integrity [2].
Eosinophils possess a series of cytoplasmic granules containing potent effector molecules, including major basic protein (MBP), eosinophil peroxidase (EPX), eosinophil cationic protein (ECP), and eosinophil-derived neurotoxin (EDN) [3]. These bioactive substances are released upon cellular activation, enabling eosinophils to participate in the destruction of multicellular parasites, particularly helminths, and to modulate inflammatory responses [4]. In addition to their classic role in antiparasitic defence, eosinophils are central mediators in the pathogenesis of allergic diseases such as asthma, atopic dermatitis, and allergic rhinitis (AR), where their accumulation and degranulation contribute to inflammation and tissue remodelling [5].
Eosinophil recruitment and activation are orchestrated by a complex network of signals involving chemokines (such as eotaxins), cytokines [particularly IL-5, IL-3, and granulocyte-macrophage colony-stimulating factor (GM-CSF)], and adhesion molecules [6]. Once activated, eosinophils perform a variety of effector functions: They release granular proteins that are toxic to pathogens but can also damage host tissues; they generate reactive oxygen species (ROS); and they secrete a broad spectrum of immunomodulatory cytokines and growth factors that influence the behaviour of other immune and structural cells. This multifaceted activity underscores their dual nature: instrumental in both protective immunity and destructive inflammation [7].
Dysregulation of eosinophil production, trafficking, or activation underlies a spectrum of eosinophil-associated pathologies, collectively referred to as eosinophilic diseases [8]. These conditions range from organ-limited conditions, such as eosinophilic esophagitis (EoE) and eosinophilic pneumonia (EP), to systemic diseases such as hypereosinophilic syndrome (HES), in which persistent eosinophilia leads to widespread organ damage. Furthermore, emerging evidence suggests that eosinophils play a role in metabolic regulation, tissue repair, and modulation of innate and adaptive immune responses, indicating that their importance extends beyond traditional paradigms [9, 10].
This manuscript will delve into the complex biology of eosinophils, exploring their developmental pathways, the molecular mechanisms of activation, and the diverse clinical implications of their dysregulation. Through this in-depth analysis, we aim to provide readers with a comprehensive understanding of eosinophil pathophysiology and its relevance to human disease.
Eosinophils were first described in the blood of various species, including man, as “coarse granule cells” by Wharton Jones in 1846. In 1879, Schultze described some of the eosinophils’ morphological features, but the term “eosinophile” was coined by Paul Ehrlich based on their strong avidity for acid aniline dyes, most notably eosin. Ehrlich also suggested that eosinophils might arise in the bone marrow and exert their function in the tissues. These early 19th-century observations were followed by a considerable history of research endeavour on eosinophil biology stretching to the present day. Perhaps one of the most fascinating aspects of the eosinophil is how accumulating knowledge has changed the perception of its function from passive bystander, modulator of inflammation, to potent effector cell loaded with histotoxic substances, through to more recent recognition that it can act as both a positive and negative regulator of complex events in both innate and adaptive immunity [1, 11]. Eosinophils are noted for their potent arsenal of diverse pro-inflammatory mediators that include granule-derived basic proteins, lipid mediators, cytokines and chemokines, products that significantly contribute to a wide range of T-helper 2 (Th2)-driven inflammatory conditions, including those that affect the skin, gastrointestinal, and respiratory tract [12]. Multiple studies have further highlighted that eosinophils represent a heterogeneous population of immune cells, whose differentiation and function are critically shaped by transcriptional and epigenetic mechanisms. Distinct subsets, including resting and activated eosinophils, have been identified through high-dimensional cytometry and single-cell transcriptomics, revealing unique phenotypic markers such as Siglec-8 and EMR1 and suggesting functional specialization across homeostatic and pathological contexts [1, 11–15].
Eosinophil granule proteins, cytokines, growth factors, and chemokines are synthesized at early stages of eosinophil maturation in the bone marrow and are packaged into various intracellular organelles, including the eosinophil crystalloid granule prior to secretion in response to receptor stimulation [16, 17]. During activation, eosinophils generate an elaborate tubulovesicular network that is composed of small secretory vesicles and elongated tubules, which appear to carry the contents of the crystalloid granule to the cell surface [18, 19]. The crystalloid granule in eosinophils is comprised of two compartments: A core and a surrounding matrix, both of which are enriched with highly cationic proteins, principally MBP [20]. Electron microscopy images of sectioned eosinophils display the strikingly electron-dense crystalline cores in crystalloid granules, visible in eosinophils from many different mammalian species. In the matrix that envelopes the MBP-rich core, EPX, EDN, and ECP are found in high concentrations, along with many other granule proteins, including cytokines. Eosinophils rarely release granule products during transit through the bloodstream, being relatively benign even as they marginate into tissues, predominantly the gut, in healthy individuals [21]. However, in diseases such as allergy and atopic asthma, eosinophils undergo a high degree of proliferation in the bone marrow and are found degranulating in the nasal and airway mucosa [22]. Degranulation is a general term that describes an activated phenotype ranging from piecemeal degranulation to degradation of cells and cytolysis (necrosis). When eosinophils encounter secretagogues, they will release the contents of their crystalloid granules by mobilizing granules and secretory vesicles to the cell surface, inducing granule-membrane fusion in a regulated manner; hence the term, “regulated exocytosis” [23]. In the case of cytolysis, eosinophils release intact membrane-bound crystalloid granules with their lipid bilayer membranes still surrounding their core and matrix components, and whole granules infiltrating tissues may be readily visible upon appropriate staining of tissue sections [24, 25]. Potential mechanisms that control eosinophil cytolysis include adhesion-induced cytolysis of eosinophils that involves the receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase-like (MLKL) signalling pathway [26]. Beyond classical degranulation, eosinophils also release extracellular vesicles carrying bioactive molecules such as microRNAs and enzymes, which contribute to intercellular communication and systemic immune modulation [27–29].
Eosinophils respond to activation by a wide range of pro-inflammatory mediators through ligation of their cell numerous surface receptors, facilitating their interaction and response with their environmental milieu. However, in vitro induction of eosinophil degranulation by soluble secretagogues is limited, often requiring potent or multiple stimuli to evoke a significant secretory response. As mentioned, blood eosinophils do not readily degranulate but release their granule contents only after their transmigration into tissues and their activation in the inflammatory foci. In some instances, eosinophils appear to undergo degranulation in response to both soluble and immobilized stimuli that activate multiple receptors. These secretagogue-binding receptors include those to complement factors (C5aR), immunoglobulins (FcRI, FcRII), platelet-activating factor (PAF), and fungal extracts (such as Alternaria acting on protease-activated receptor-2, PAR-2) [18, 19].
Receptors are generally classified by their signalling mechanisms, such as G protein-coupled receptors that activate dissociation of and subunits of heterotrimeric G proteins, and immunoglobulin- or cytokine-binding families that activate a cascade of tyrosine kinase phosphorylation. Eosinophils express many of the signalling components necessary for receptor activation of cellular events [30]. Eosinophils do not undergo degranulation in response to the potent eosinophil differentiation and maturation-inducing cytokines, IL-3, IL-5, or GM-CSF if applied individually, all three must be combined together in a “cytokine cocktail” in order to elicit degranulation in human eosinophils [31]. This is likely associated with the relatively quiescent state of eosinophils during their proliferation and maturation in the bone marrow in response to these cytokines.
Following binding and activation of specific receptors, secretagogues induce the mobilization of granules through the cytoplasm of the eosinophil, which may be associated with the formation of large tubulovesicular structures containing small secretory vesicles and elongated tubules that may extend from crystalloid granules [32, 33]. The tubulovesicular structure appears in the cytoplasm in correlation with piecemeal degranulation, where membrane-bound vesicles bud off from crystalloid granules and selectively shuttle specific granule contents to the plasma membrane for release. The tubulovesicular network is responsible for trafficking cytokines and chemokines such as IL-4 and CCL5/RANTES [34, 35]. The movement of granules and vesicles through the cells is controlled by actin cytoskeleton remodelling, regulated in eosinophils by a family of guanosine triphosphatases (GTPases), particularly Rac2 [36]. When vesicles and granules reach the inner leaflet of the lipid bilayer in the plasma membrane, they bind to specific intracellular receptors known as soluble N-ethylmaleimide-sensitive attachment protein receptors (SNAREs), which facilitate their docking and fusion with the cell membrane. This is followed by membrane fusion, in which the internal surface of the granule membrane becomes exposed to the outside of the cell membrane [37–39]. Concurrently, vesicle fusion is mediated by another membrane-bound GTPase, Rab27a, which conveys the secretagogue signal through to SNARE binding and ensures that granule membranes come into close proximity with cell membrane lipid bilayer with granule polarization focused on their leading edges during shape change and degranulation [40], suggesting that they may have the ability to focus their granule contents onto target surfaces, as previously observed in vitro using opsonized helminthic parasites [41]. There is a substantial body of clinical evidence that suggests that eosinophils degranulate upon recruitment and activation at inflammatory foci; these observations largely are the result of the examination of tissue biopsies from different organs and in association with diseases including allergies and asthma [39, 41, 42]. Eosinophil degranulation is thought to be an essential component of the late-phase mucosal tissue response to allergen challenge. There is significant evidence for eosinophil degranulation in tissues in association with AR, cutaneous allergic reactions, and atopic asthma; this observation often correlates with a deteriorating clinical outcome [42]. Furthermore, eosinophils and their major granule proteins have been detected at high levels in tissues and body fluids in response to fungal, parasitic, and viral infections [42–45] and likewise participate in the formation of eosinophil traps that are critical for host defence against bacterial sepsis [46].
In healthy individuals, eosinophils are present in the circulation in low numbers and are rarely found in the lungs, being mostly confined to the tissues surrounding the gut. Eosinophil accumulation during inflammatory events is complex, involving their maturation in and release from the bone marrow, adhesion to and transmigration through the post-capillary endothelium, followed by their chemotaxis to and activation/degranulation at inflammatory foci [47, 48]. The processes controlling eosinophil accumulation are of obvious importance and represent potential therapeutic targets for the antagonism of their accumulation in allergic-based disease. Asthma pathology is characterized by excessive leukocyte infiltration that leads to tissue injury. Cell adhesion molecules, i.e., selectins, integrins, and members of the immunoglobulin superfamily control leukocyte extravasation, migration within the interstitium, cellular activation, and tissue retention. Numerous animal studies have demonstrated essential roles for these cell adhesion molecules in lung inflammation, including L-selectin, P-selectin, and E-selectin, ICAM-1, VCAM-1, together with many of the β1 and β2 integrins. These families of adhesion molecules have therefore been under intense investigation to inform the development of novel therapeutics [49]. In addition, eosinophil accumulation is orchestrated by chemokines such as the eotaxins CCL11, CCL24, CCL26, and their receptor CCR3, as well as local stromal interactions and extracellular matrix (ECM) remodelling, which together determine both physiological tissue surveillance and pathological infiltration in diseases such as asthma and EoE [14, 50–52].
Eosinophil fate is also important; apoptosis and the disposal of apoptotic cells by phagocytic removal (efferocytosis) are vital aspects of inflammation resolution in all multi-cellular organisms. Eosinophils have a limited lifespan in the circulation of 8–18 hours and 3–4 days in the tissues, and up to two weeks in tissue culture conditions that favour their survival. As with neutrophils, they are terminally differentiated cells programmed to undergo apoptosis in the absence of viability-enhancing stimuli [53]. Eosinophil persistence in the tissues is enhanced by the presence of several asthma-relevant cytokines that prolong eosinophil survival by inhibition of apoptosis; these include IL-3, IL-5, IL-9, IL-13, IL-15, and GM-CSF [54]. Furthermore, thymic stromal lymphopoietin (TSLP), IL-25, and IL-33 represent a triad of cytokines released by airway epithelial cells in response to various environmental stimuli or by cellular damage. They act in concert to drive Th2 polarization through overlapping mechanisms, causing remodelling and pathological changes in the airway walls, suggesting pivotal roles in the pathophysiology of asthma. All three have been shown to have multiple effects on eosinophil function, including enhancement of their receptor expression, adhesion, and viability through inhibition of apoptosis [55–57]. Eosinophil interactions with the proteins of the ECM also contribute to their persistence within the tissues. For example, integrin-mediated eosinophil adhesion to fibronectin results in the autocrine production of viability-enhancing cytokines GM-CSF, IL-3, and IL-5 [58]. These interactions between multiple cytokines and ECM components antagonize eosinophil programmed cell death, thereby prolonging their longevity for weeks. Thus, a balance in the tissue microenvironment between pro- and anti-apoptotic signals is likely to greatly influence the load of eosinophils in the tissues [59].
Allergic asthma is a chronic disease characterized by airway inflammation, reversible airway obstruction, and airway hyperresponsiveness. Its pathogenesis is complex and involves interactions among multiple immune cells and inflammatory mediators [60]. Among these, eosinophils play a critical role in the pathological process of allergic asthma. Eosinophils are classic effector cells of the type 2 (T2) immune response, and their activation and infiltration are considered one of the hallmarks of allergic asthma [61]. In patients with allergic asthma, eosinophils directly contribute to the initiation and maintenance of airway inflammation by releasing inflammatory factors [62, 63]. These factors not only induce airway epithelial damage but also further exacerbate the inflammatory response by activating immune cells [64].
Eosinophils in allergic asthma are not only involved in the regulation of inflammation but also participate in the complex modulation of the immune system [65]. Research has elucidated that eosinophils modulate the polarization of Th2 immune responses through interactions with immune cells such as T cells and dendritic cells, thereby influencing disease progression [66–68]. In the early stages of allergic asthma, eosinophilic infiltration is one of its characteristic pathological manifestations [69]. Eosinophils promote airway hyperresponsiveness and mucus hypersecretion by releasing Th2-type cytokines such as IL-5 and IL-13 [70, 71]. As a critical factor for eosinophil survival and activation, IL-5 levels in both blood and tissues are significantly correlated with asthma severity [72]. Furthermore, eosinophils exacerbate the inflammatory response by releasing chemokines such as CCL11 and CCL24, which recruit additional eosinophils and other inflammatory cells to the airways [73, 74]. In the chronic phase of allergic asthma, the role of eosinophils extends beyond simple inflammation. Studies suggest that eosinophils may also influence the establishment of immune tolerance by modulating the function of regulatory T cells [75]. For instance, by modulating T cell secretion of cytokines such as transforming growth factor-β (TGF-β) and IL-10, eosinophils may suppress the overactivation of Th2 immune responses, thereby mitigating disease progression to some extent [76]. However, this regulatory function is often impaired in asthma patients, contributing to the persistence and exacerbation of the inflammatory response.
The involvement of eosinophils in the pathogenesis of allergic asthma also encompasses complex biophysical mechanisms. Eosinophils directly alter the structure and function of airway epithelial cells by releasing EPX and MBP [65]. These proteins disrupt tight junctions between epithelial cells, increase airway permeability, and thereby promote the infiltration of both inflammatory cells and mediators [77]. Furthermore, eosinophils exacerbate tissue damage and inflammation by releasing ROS and the lipid mediator leukotriene C4 (LTC4), which induce bronchoconstriction and increase mucus secretion [78, 79], further exacerbating airway inflammation and remodelling. These biophysical alterations not only lead to airway wall thickening and fibrosis but also heighten airway sensitivity to stimuli, thereby exacerbating asthma symptoms.
As discussed above, the interaction between eosinophils and the ECM plays a pivotal role in their activation and tissue retention. In the airways of asthma patients, provisional ECM components, such as tenascin-C, periostin, and thrombospondin, are enriched and are associated with the tissue activation of eosinophils [80, 81]. ECM proteins not only provide structural support but also act as signalling platforms that modulate eosinophil behaviour, including adhesion and migration [82]. Research indicates that β1-integrin serves as a key receptor for eosinophil interaction with the ECM. Its binding to collagen IV (COL IV) promotes eosinophil adhesion and signal transduction. Activation of β1-integrin not only enhances eosinophil adhesive capacity but also regulates their functions—including cell survival, differentiation, and release of inflammatory mediators—through the downstream FAK/Bag3 phosphorylation signalling pathway [83]. Additionally, eosinophil-derived molecules such as EPO directly modify the ECM, contributing to tissue remodelling and the inflammatory response [80].
The mechanical properties of the tissue microenvironment, including stiffness and physical forces, also modulate eosinophil function. ECM stiffness is transduced intracellularly via integrin-mediated mechanosensing pathways, driving FAK phosphorylation and activation of the RhoA/ROCK signalling axis. This cascade regulates actin cytoskeleton reorganization [84], ultimately impacting eosinophil morphological plasticity and chemotactic migration capacity. Within the stiffened ECM of asthmatic airway remodelling zones, eosinophils exhibit enhanced morphological polarization, accelerated migration velocity, and augmented adhesion strength to the matrix, collectively promoting their recruitment to inflammatory foci [85]. Concurrently, fluid shear stress within blood vessels and tissue tension generated by contractile forces activate mechanosensitive ion channels such as Piezo1, triggering intracellular Ca2+ influx that modulates eosinophil activation states [86].
Eosinophils exhibit significant functional plasticity, enabling dynamic adaptation of their phenotype and functions in response to local signals. In allergic asthma, tissue-resident eosinophils acquire a distinct activation state characterized by upregulated expression of integrins CD11c and CD11b, alongside adhesion molecules, which facilitates their interactions with the ECM and other immune cells [87]. This tissue-activated phenotype mirrors eosinophils engaged in pulmonary morphogenesis, suggesting that evolutionarily conserved morphogenetic programs may drive eosinophil functionality across both physiological and pathological contexts. Furthermore, eosinophils interface with group 2 innate lymphoid cells (ILC2s) and CD4+ Th2 cells to establish a self-perpetuating inflammatory circuit that amplifies T2 immunity through cytokine-mediated feed-forward loops [88]. Eosinophil-derived IL-13 potently recruits and activates ILC2s. In turn, ILC2s amplify T2 inflammation by producing IL-5 and IL-13, which enhance eosinophil survival and activation through autocrine-paracrine signalling loops.
The pathogenic role of eosinophils extends beyond allergic asthma to encompass multiple T2 inflammatory disorders, including EoE, chronic rhinosinusitis (CRS) with nasal polyps (CRSwNP), and atopic dermatitis—all characterized by tissue-specific eosinophil infiltration driving core pathological manifestations. In EoE, eosinophil infiltration into the oesophageal mucosa drives tissue remodelling and fibrosis through the release of pro-fibrotic mediators such as TGF-β, which directly activate subepithelial fibroblasts and stimulate collagen deposition [89]. In CRSwNP, eosinophils serve as pivotal orchestrators of T2 inflammation [90]. Through the release of cytotoxic granule proteins (e.g., MBP and EPO) and pro-inflammatory cytokines (e.g., IL-5, IL-13), they drive polyp formation and sustain chronic inflammation by disrupting epithelial integrity and amplifying immune cell recruitment. Atopic dermatitis is also characteristically defined by eosinophil infiltration into the dermal compartment [91]. These cells potentiate pruritus and inflammation through the release of histamine, proteases (e.g., EDN), and pruritogenic cytokines, directly activating cutaneous sensory neurons and amplifying T2 immune responses.
Therapeutic strategies targeting eosinophils have shown promise in the management of allergic asthma and other eosinophil-associated disorders. The biologics mepolizumab and reslizumab are humanized anti-IL-5 mAb [92, 93], while benralizumab is a monoclonal antibody specific for the a-chain of the human IL-5 receptor [94–96]. These biologics have been demonstrated to be effective in reducing blood and tissue eosinophil counts, glucocorticoid usage, disease exacerbations, together with improved lung function in patients with severe refractory asthma. Additionally, interactions between eosinophils and other immune cells provide novel therapeutic angles for asthma. The crosstalk between eosinophils and ILC2s plays a significant role in allergic asthma pathogenesis. ILC2s promote eosinophil activation and recruitment through IL-5 and IL-13 release, while eosinophils reciprocally activate ILC2s via cytokine and chemokine secretion, establishing a pathogenic positive feedback loop [97]. However, the efficacy of these therapies varies across distinct asthma phenotypes, underscoring the necessity of developing personalized treatment approaches based on the underlying eosinophil biology and disease mechanisms.
While eosinophils clearly respond to signals from other leukocytes, most notably cytokines from Th2 cells such as IL-5, it has become clear that these cells in turn release cytokines and granule proteins that provide signals that promote local immune regulation and have an impact on the function of other leukocyte lineages [98]. Eosinophils have also been implicated in directing the functions of both B and T lymphocytes, including expression of MHC class II, the co-stimulatory molecules CD80 and CD86, together with the observation that eosinophils can process antigen and direct antigen-specific T cell proliferation and cytokine release [99, 100]. Eosinophils can also promote humoral immune responses by promoting the production of antigen-specific IgM [101] and supporting plasma cell growth and development in the bone marrow [102]. Eosinophils also interact directly with innate immune cells and have a role in supporting the viability of alternatively activated macrophages in adipose tissue [103], promote the migration and activation of myeloid dendritic cells [104], participate in extensive bidirectional signalling with tissue resident mast cells [105] and elicit production and release of pro-inflammatory mediators from isolated peripheral blood neutrophils [106].
As discussed above, eosinophils are recruited to the airways and are a prominent feature of the asthmatic inflammatory response, where they are broadly perceived as promoting pathophysiology. Respiratory virus infections can exacerbate this response. Among the recent concepts under exploration is the role of eosinophils in promoting antiviral host defence in this and other settings [107, 108], including the observation that eosinophils can respond to lipopolysaccharide from gram-negative bacteria by releasing mitochondrial DNA complexed with cationic proteins to form distinctive extracellular traps [109, 110]. There is also evidence for a role of eosinophils to provide host defence against bacteria in general and/or bacterial pathogens [111]. This hypothesis is particularly attractive, given the predominance of resident eosinophils in the intestines and the possibility of a more complex role involving eosinophils with commensal bacteria in the gut [112, 113].
Traditionally, eosinophils have been considered destructive agents in allergic responses, primarily due to their characteristic release of cytotoxic granules [114, 115]. However, emerging evidence demonstrates that eosinophils also play essential roles as regulators of homeostasis [116, 117], contributing to the defence against bacterial and viral infections [118], and even facilitating tissue repair [119, 120]. This evolving perspective has shifted the conceptualisation of eosinophils from being viewed solely as cytotoxic effector cells responsible for pathology to being recognized as multifunctional immunomodulators with regulatory capabilities. The ongoing debate focuses on the context-dependent function of eosinophils. Distinct subsets—regulatory and inflammatory—have been identified, and studies in conditions such as cancer [121] or inflammatory bowel disease (IBD) [122, 123] have reported conflicting results regarding their protective versus harmful effects. This challenges the simplistic view of eosinophils as either “friend” or “foe”.
It is now understood that not all eosinophils are identical. There are two main subsets: regulatory eosinophils (rEos) and inflammatory eosinophils (iEos), each distinguished by unique gene expression profiles and anatomical locations [124], challenging the idea of a single eosinophil function. The ongoing debate centres on whether eosinophil function is determined by pre-programmed subsets or by environmental plasticity [125, 126]. Advocates for the “subset” theory argue that eosinophils exist as developmentally distinct lineages, each characterized by specific surface markers and functional identities. For instance, iEos display signs of activation, such as increased granule density or cytoplasmic vacuolation [127]. These cells are typically marked by high expression of Siglec-F and CD101hi, low expression of CD62L, and are highly dependent on IL-5 for survival [128, 129]. In contrast, rEos are typically found in healthy lung parenchyma, express CD62L and low levels of CD101, possess a ring-shaped nucleus, are largely IL-5 independent, and have the capacity to inhibit Th2 responses [130, 131].
Opponents of the fixed subset theory argue that eosinophils are highly sensitive to their microenvironment and “reprogram” their function upon entering a different tissue [132]. In fact, local signals (like IL-33 or TSLP) can induce eosinophils to express markers like CD80 or PD-L1, effectively shifting them from a “basal” state to an “activated” or “regulatory” state in response to local stress [133].
Studies confirm that eosinophils are highly adaptable cells, changing their function (phenotype) and abundance (frequency) to suit the requirements of different tissues like the gut or lungs [134]. In these contexts, they become crucial for tissue maintenance, repair, and immune regulation, influenced by local signals like the aryl hydrocarbon receptor in the gut [135].
Recent 2026 data suggest a residency-time model that bridges both views: Eosinophils continue to mature after entering tissues [136, 137]. In tissues like the small intestine, where they are long-lived (> 15 days), they undergo deep transcriptional reprogramming and diversify into multiple distinct, stable subsets with unique gene and protein signatures that support metabolic regulation and barrier integrity. In contrast, in tissues where they are short-lived, such as the lung (< 5 days), they remain more uniform as there is insufficient time for them to fully specialise. Consequently, in these tissues, they appear more homogeneous and “plastic”, responding quickly to acute inflammatory signals but forming stable, resident subtypes. In this view, this model suggests that eosinophil “plasticity and subsets” are two stages of the same biological process. Upon entering a tissue, all eosinophils initially exhibit plasticity, responding to local environmental cues (like IL-33 or TSLP). If the cell remains in a specific niche long enough, continuous exposure to these cues “locks” the cell into a fixed subset identity [137] (Table 1).
“Molecular toolkit” of surface markers to identify eosinophil subsets through their maturation trajectory.
| Marker | Mature/Resident subset (e.g., small intestine) | Immature/Inflammatory state (e.g., lung) |
|---|---|---|
| CD101 | High expression (stable subset) | Low expression (plastic) |
| CD62L | Low expression | High expression |
| Siglec-F | Intermediate expression | High expression |
| Siglec-8/F | Low expression | High expression |
| Effector genes | Il16, TNF, VEGFA (specialized) | EPX, Prg2 (granular focus) |
Our understanding of the immunological role of the eosinophil is continually evolving, from earlier dogma that emphasised a role in combating helminthic parasitic infections and as a key effector cell in allergic inflammation to more recent discoveries suggesting important roles in immunomodulation. Other emerging roles include functions against numerous pathogens such as respiratory viruses [98, 99], a role in gastrointestinal disease [138], and in interactions with nerves that impact the pathology of many diseases [139]. In summary, eosinophils play multifaceted roles in the pathogenesis of allergic asthma and other allergic diseases, encompassing complex interactions with the immune system, tissue microenvironment, and biophysical processes. Their functional plasticity and adaptability to local signals position them as central players in T2 inflammation and tissue remodelling. Understanding the biophysical aspects of eosinophil function, particularly interactions with the ECM and mechanical forces, may yield novel insights into disease mechanisms and therapeutic strategies. Future research should elucidate the molecular and biophysical determinants of eosinophil function across disease contexts, paving the way for innovative and targeted therapeutic approaches.
Eosinophils are central effector cells in both the physiology and pathology of the upper airways [140]. Normally, they participate in tissue maintenance and repair. Still, in response to inflammatory triggers, such as allergens, pathogens, and environmental irritants, their recruitment and activation initiate a cascade of events that underpin local tissue inflammation, resulting in epithelial injury, remodelling, and sustained immune cell infiltration [141, 142].
Eosinophilic infiltration of the sinonasal mucosa is a pathobiological hallmark across a wide spectrum of upper airway diseases associated with T2 inflammation. This inflammatory milieu is shaped by cytokines such as IL-4, IL-5, and IL-13, as well as epithelial alarmins including TSLP and IL-33, which further support the survival, activation, and migration of eosinophils, mast cells, and ILC2s [143–146].
Disorders such as AR and CRSwNP are among the most common diseases with an eosinophilic component. According to recent European data, CRSwNP affects nearly 11% [143] of the population, with marked geographic variations, while AR impacts approximately 10–23% of individuals in Western countries, ranking among the most prevalent chronic conditions globally [143]. The burden of these diseases is amplified by their frequent coexistence with other eosinophilic disorders, most notably asthma and non-steroidal anti-inflammatory drugs (NSAIDs)–exacerbated respiratory disease (N-ERD). For example, the prevalence of asthma in patients with CRSwNP reaches up to 50%, especially in severe and late-onset eosinophilic phenotypes, while in allergic fungal rhinosinusitis (AFRS), asthma coexistence has been reported in as many as 73% of cases [143]. N-ERD, which encompasses the triad of asthma, CRSwNP, and hypersensitivity to NSAIDs, is a classic example of the clinical and immunological continuum that links the upper and lower airways.
These strong associations have led to the unified airways disease concept, which views the respiratory tract as a single functional and immunological unit [143, 145, 146]. The clinical relevance of this model is underscored by evidence that treating upper airway inflammation can improve asthma outcomes, and, conversely, effective management of asthma may ameliorate sinonasal disease [147].
The degree of eosinophilic infiltration and activation has important clinical implications: It correlates with disease severity, the risk of recurrence after surgical intervention, and an increased likelihood of resistance to corticosteroid therapy. As such, eosinophils occupy a central role at the crossroads of innate and adaptive immunity and are now recognized as crucial therapeutic targets in upper airway diseases driven by T2 inflammation.
The current understanding of eosinophilic inflammation in the upper airways emphasizes the interplay between the epithelial barrier, immune system, and tissue structure. As more in detail described in Pathophysiology of eosinophils (correspondence to Garry Michael Walsh: g.m.walsh@abdn.ac.uk), when the nasal epithelium encounters environmental insults such as allergens or pollutants, it responds by releasing “alarmin” cytokines that serve as early warning signals to the immune system, with activation of dendritic cells and stimulation of ILC2s, setting off a cascade that favors a Th2-skewed immune response [148, 149]. This Th2 polarization is characterized by elevated levels of cytokines like IL-5, which is crucial for the growth and survival of eosinophils, and IL-4 and IL-13, which promote IgE production and further weaken the epithelial barrier [150].
Allergen exposure (e.g., pollen or dust mites) in sensitized subjects induces IgE-mediated mast cell degranulation, leading to the release of mediators that recruit eosinophils and sustain their accumulation. Concurrently, microbial dysbiosis, loss of commensal diversity with overgrowth of pathogenic bacteria (such as Staphylococcus aureus), can impair the epithelial barrier and shift local immunity toward chronic T2 inflammation [150]. S. aureus, for instance, produces exotoxins and superantigens (e.g., staphylococcal enterotoxin B) that can directly stimulate epithelial cells to release TSLP, IL-33, and IL-25, thereby creating a T2 cytokine milieu that favors eosinophil infiltration (Figure 1).
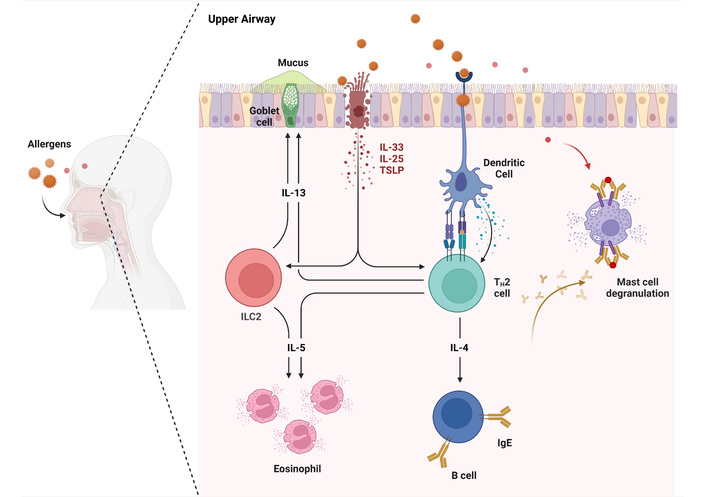
Eosinophilic inflammation pathway in the upper airways. Upon allergen exposure, epithelial cells release TSLP, IL-25, and IL-33, which activate dendritic cells and ILC2s. This promotes TH2 polarization, leading to IL-4, IL-5, and IL-13 secretion. IL-4 induces IgE production and mast cell activation; IL-5 recruits eosinophils; IL-13 enhances mucus production. These pathways contribute to sustained eosinophilic inflammation in allergic airway diseases. IL: interleukin; ILC2: group 2 innate lymphoid cell; TSLP: thymic stromal lymphopoietin.
Beyond driving inflammation, chronically activated eosinophils cause structural remodelling of the nasal mucosa by releasing growth factors, matrix metalloproteinases, and profibrotic mediators. These factors lead to goblet cell hyperplasia, subepithelial fibrosis, basement membrane thickening, and epithelial-mesenchymal transition (EMT), which weakens the epithelial barrier and increases permeability to allergens and microbes. Eosinophil granule proteins disrupt tight junctions, while fibrogenic factors such as TGF-β and vascular endothelial growth factor (VEGF) further promote tissue remodelling and EMT [151]. In eosinophilic chronic rhinosinusitis (ECRS), eosinophils can also form extracellular traps (EETs) that damage the epithelium. Elevated cell-free DNA (cfDNA) in nasal secretions acts as a danger signal recognized by eosinophil Toll-like receptor 9 (TLR9), enhancing EET formation. These mechanisms contribute to persistent tissue injury, chronic inflammation, and remodelling in the upper airways [151, 152].
Eosinophils are involved in several upper airway diseases, contributing to a variety of short-term symptoms and long-term sequelae [153]. CRSwNP, N-ERD, and AR are some of the most representative examples.
CRSwNP is a historical clinical phenotype of CRS, characterized by typical symptoms (nasal congestion or discharge and facial pain or hyposmia) lasting ≥ 12 weeks [154], and the presence of nasal polyps based on nasal endoscopy or computerized tomography (CT) findings [154–156]. It typically presents T2 inflammation and eosinophil infiltration, especially in Western countries [155, 157]. CRSwNP has a high burden, presenting greater morbidity than CRS without nasal polyps (CRSsNP), with higher disease severity, a higher number of surgeries and medication exposure, and an increased risk of comorbid asthma [155, 158].
CRSwNP can be classified based on the histological quantification of eosinophils into eosinophilic and non-eosinophilic CRS [a cut-off of 10 eosinophils/high-power field (eos/hpf) was suggested] [141, 154]. In Europe and the USA, eosinophils are found in up to 90% of the polyps [155, 159, 160]. Eosinophils develop and survive in response to IL-5 and are major effectors of T2 inflammatory response through the release of EPX, cationic proteins, and EDN, which are preformed and stored in cell granules [155, 161]. Degranulation and formation of EETs, extracellular structures containing DNA and granule proteins, is a prominent feature of some patients with CRS [155, 162]. These EETs, together with eosinophil-derived Charcot-Leyden crystals (CLC), contribute to mucus stiffness and further mucin production [155, 162].
Clinically, eosinophilic-CRSwNP is characterized by the presence of bilateral nasal polyps, hyposmia, nasal obstruction, rhinorrhoea, and a close link to asthma (up to 65%) and N-ERD (up to 26%) [162]. The amount of eosinophilic infiltration and the intensity of the inflammatory response are associated with CRS severity and prognosis [159], including recurrence after surgical treatment [163].
It should be noted that, although the presence of nasal polyps predicts high tissue eosinophilia, this feature is not exclusive to CRSwNP: Patients with CRSsNP can also present eosinophilic inflammation [160, 161].
N-ERD is typically an adult-onset triad that includes asthma, CRSwNP, and hypersensitivity to NSAIDs [153, 157, 164]. It has a prevalence of almost 15% in patients with severe asthma that increases to about 30% in those with concurrent asthma and CRSwNP [165]. Its pathophysiology is thought to involve an eosinophilic response in the upper and lower airway mucosa, resulting from leukotriene release and other inflammatory mechanisms driven by dysregulated arachidonic acid metabolism [158, 162]. Tissue eosinophil infiltration (in the lungs and nasal polyps) is higher in NSAID-intolerant compared to NSAID-tolerant individuals [166]. Patients with N-ERD tend to have more severe respiratory disease [167].
AR is an inflammatory disorder of the lining of the nose characterized by nasal symptoms (including rhinorrhoea, sneezing, nasal congestion, and itching) that occurs for more than one hour during at least two consecutive days [31, 168]. It is associated with an IgE-mediated response against allergens [168] and is usually accompanied by local eosinophilia and sometimes by peripheral blood eosinophilia [157]. Eosinophils are involved in the inflammatory response during and after allergen exposure, both in perennial and seasonal AR [169–171]. In patients with pollen allergy, during the pollen season, the number of eosinophils in nasal scrapings has been shown to significantly correlate with clinical symptoms (e.g., total symptom score), inflammatory parameters, nasal flow, spirometry measurements, and bronchial hyperreactivity [169]. Although in patients with indoor allergy, nasal eosinophilia was not a permanent feature [168], the levels of activated and pathogenic eosinophils were found to be higher in patients with moderate-severe house dust mite AR (vs. mild patients and healthy controls) and positively correlated with total nasal symptom score (TNSS) [171].
Activated and degranulated eosinophils were observed in markedly elevated numbers in patients with AR after allergen exposure [172]. A recent study also supported that individuals with chronic rhinitis presenting higher blood eosinophil levels (≥ 0.3 × 109/L) have a higher frequency of asthma [173].
Other rhinitis phenotypes typically present with eosinophilia, such as local allergic rhinitis (LAR) [174] and non-allergic rhinitis with eosinophilia syndrome (NARES). LAR is characterized by a clinical history suggestive of AR, but with negative skin prick tests (SPT) and/or serum specific IgE (sIgE), and a positive response to a nasal allergen challenge [175]. Its pathophysiology involves increased nasal eosinophilic inflammation, with high levels of tryptase and ECP [175, 176]. Eosinophil and ECP levels increase during allergen exposure in LAR patients [176].
NARES is a common condition that is estimated to cause up to one-third of cases of nonallergic rhinitis [153, 177]. It is characterized by the presence of nasal eosinophilia, persistent nasal symptoms, and negative SPT and sIgE [153, 168, 177]. Contrary to AR, anosmia has been described as a prominent feature [39, 153]. It may be a precursor of nasal polyposis and N-ERD [153, 177].
Systemic hypereosinophilic diseases, including eosinophilic granulomatosis with polyangiitis (EGPA), can also be associated with upper airway inflammation [120, 145]. EGPA is a rare small-vessel vasculitis characterized by necrotizing vasculitis and tissue eosinophilia with eosinophil-rich [157, 178]. EGPA can affect multiple organs, but its cardinal feature is respiratory tract involvement [153]. Ear-nose-throat disease is estimated to occur in 60–80% of the patients, beginning in the early disease stages, together with asthma (frequently severe) [141, 178].
Respiratory tract involvement is usually followed by peripheral hypereosinophilia and, finally, progresses to a systemic necrotizing vasculitis [153, 178].
The role of eosinophils is under investigation in several other chronic respiratory conditions, including allergic fungal airway disease and sinonasal eosinophilic angiocentric fibrosis (SEAF) [141, 153]. AFRS is considered a subset of CRSwNP characterized by the presence of eosinophilic mucin together with non-invasive fungal hyphae in the sinus and an IgE-mediated hypersensitivity to fungi [154]. It frequently presents bony erosions and expanded sinus, which are not common in other forms of CRSwNP [179]. SEAF is a rare, non-malignant, obstructive lesion in the upper respiratory tract mucosa, usually involving the sinus and nasal septum [153]. Its etiology is not fully known.
An early identification of these potentially disabling diseases is essential, as they require a multidisciplinary diagnostic and therapeutic approach [141].
Diagnostic procedures, in addition to clinical presentation, are required for the definitive diagnosis of upper airway eosinophilic diseases. Nasal endoscopy and/or CT scan of the paranasal sinus (PNS) are important for diagnosing CRS and are included in the standard diagnostic criteria [154].
For the diagnosis of AR, SPT or sIgE testing is recommended. Nasal endoscopic examination is also important for excluding nasal polyps or CRS as potential causes of nonspecific rhinitis symptoms [180–182]. Additionally, findings such as middle or inferior turbinate oedema, pale/bluish discoloration, or isolated central compartment polypoid changes and/or oedema have been demonstrated to be associated with AR.
For nasal cytology, nasal eosinophils stand out as a biomarker assisting in the identification of AR [45, 46, 182, 183]. Additionally, polysensitized patients demonstrate a higher inflammatory infiltrate than monoallergic patients [163, 184, 185]
However, evidence remains limited, with high heterogeneity in techniques and cut-off values across previous studies. Additionally, the use of nasal cytology in non-allergic rhinitis (NAR) shows low specificity and positive predictive value [183], thus raising questions regarding its role in practical use.
The role of nasal histology in rhinitis is also limited, as nasal tissue biopsy carries a potential risk of bleeding while providing similar information to nasal cytology. In contrast, in CRS, tissue biopsy is a valid and crucial marker for determining endotype and reflects disease severity [154]. The cut-off value per high-power field (HPF) varies across centres, with 10/HPF being the most commonly used threshold for classifying CRS as eosinophilic. Higher eosinophil counts correlate with greater disease severity and recurrence [163, 184, 185].
CT PNS is included in the standard diagnostic criteria for CRS [154]. In diffuse CRS, current practice involves T2 and non-T2 endotyping. The T2 subtype can be further divided into ECRS and central compartment atopic disease (CCAD). Differences between these endotypes have been studied, including clinical presentation, disease onset, sinus involvement, evidence of AR on endoscopy (such as polypoid edema at the central compartment), and, importantly, CT PNS findings [186–188]. In ECRS, CT PNS typically shows diffuse involvement, whereas in CCAD, the disease is centrally located, leading to the term “CCAD” [189].
Peripheral blood eosinophil counts (BECs) are indicative of T2-mediated inflammatory responses and have been used as predictive markers for ECRS [190–192]. They have been demonstrated to correlate with the Lund-Mackay CT and Lund-Kennedy endoscopic scores in patients with nasal polyps [192]. A threshold of 250 cells/μL3 has been suggested as a diagnostic criterion for ECRS [190].
Serum total IgE is associated with increases in sIgE and eosinophilic inflammation and is considered a relevant biomarker for ECRS [193–195]. A total IgE level of 100 kU/L has been linked to poorer clinical outcomes [196].
In AR, despite higher total IgE levels compared to NAR [197–199], an elevated total IgE indicates an atopic condition [200] but is not specific to AR and may also be influenced by other atopic comorbidities, particularly asthma [201]. Therefore, while serum total IgE can be helpful, it serves only as a preliminary or supportive criterion for AR diagnosis.
Beyond conventional biomarkers such as eosinophil counts and total IgE, emerging biomarker candidates have been explored in recent preclinical and clinical studies over the past decades. Among these, serum osteopontin (OPN) and periostin have shown promise. Elevated serum OPN and periostin levels have been found to correlate positively with disease severity, BECs, serum ECP, and T2 cytokines (e.g., IL-4 and IL-5) in patients with AR and CRSwNP [202–206].
As described earlier, systemic markers may not always reliably reflect local inflammation. IgE, for example, can be produced locally in the nasal mucosa, and its levels may be elevated in nasal secretion in AR and CRSwNP, independent of serum IgE levels. This underscores the utility of nasal IgE as a more direct, though practically complicated and less common, diagnostic tool [207]. Indeed, a subset of rhinitis patients who test negative in SPT and lack serum sIgE have shown a positive correlation between positive nasal allergen provocation test, nasal production of sIgE, and increased levels of cellular and soluble T2 inflammatory mediators in nasal tissue, including eosinophils and ECP. These findings support the existence of LAR [175]. The ability to distinguish between T2-biased upper airway conditions, which have high local IgE, and non-T2-biased forms is clinically significant, especially given that the eosinophilic T2 is typically more refractory to treatment and prone to recurrence [207]. Moreover, T2-driven inflammation, marked by elevated IL-5, has been implicated in the pathogenesis of comorbid asthma [191, 208]. In addition, other biomarkers in nasal secretions, such as OPNs, have also been associated with eosinophilic inflammation in patients with ECRS [209], further highlighting the potential of localized biomarkers in refining diagnosis and guiding targeted therapy.
The treatment of upper airway eosinophilic disorders, particularly eosinophilic CRSwNP, has evolved from symptom-based approaches to mechanism-driven strategies targeting T2 inflammation.
In all age groups, intranasal steroids (INS) remain the cornerstone of pharmacological therapy for AR, owing to their potent anti-inflammatory effects on the nasal mucosa. They effectively reduce nasal congestion, rhinorrhea, sneezing, and itching by suppressing the recruitment and activation of eosinophils, mast cells, and other inflammatory cells. INSs are superior to oral antihistamines in relieving nasal obstruction, the most bothersome symptom for many patients, and are recommended by international guidelines as first-line monotherapy, namely in moderate-to-severe AR [210].
In cases of persistent or poorly controlled symptoms, combination therapy of INS with intranasal antihistamines (e.g., azelastine and olopatadine) has demonstrated synergistic effects. This approach provides both rapid symptom relief through H1 receptor blockade and sustained anti-inflammatory action via glucocorticoid receptor activation. Fixed-dose combinations, such as azelastine—fluticasone and mometasone-olopatadine, have shown superior efficacy compared to either agent alone in reducing TNSS, including congestion and ocular symptoms. Such combinations may be particularly beneficial in patients with more severe disease or rapid-onset symptoms. Importantly, the local delivery minimizes systemic side effects, supporting long-term adherence and safety [211].
Overall, INS-alone or combined with intranasal antihistamines—represent a highly effective, targeted treatment option for upper airway eosinophilic inflammation in AR and other related phenotypes, as is the case with LAR and NARES.
Omalizumab, an anti-IgE monoclonal antibody, is not universally approved for the treatment of AR, although its efficacy in this indication is well documented; in Japan, it is officially approved for seasonal AR, particularly due to Japanese cedar pollen, based on robust local clinical trial data, being cost-effective [212, 213]. Similar approvals exist in South Korea and China [214, 215]. In contrast, regulatory agencies, such as the Food and Drug Administration (FDA) and European Medicines Agency (EMA), have not granted formal approval, although off-label use is common in treatment-refractory cases, particularly in severe AR due to pollens; international recommendations acknowledge its role as an add-on therapy in patients with persistent symptoms despite optimized treatment [177, 181].
Conventional pharmacological therapy in CRSwNP is centred on INS, as mentioned in several rhinitis eosinophilic phenotypes/endotypes, which reduce local inflammation and/or polyp volume. Short courses of oral corticosteroids (OCS) remain useful but must be used only for acute control in patients with severe symptoms, while saline irrigation enhances mucosal clearance. Functional endoscopic sinus surgery (FESS) may be required in patients with obstructive polyposis or poor response to medical therapy, although eosinophilic inflammation is associated with frequent post-surgical recurrence and need for long-term control, as other risk factors that include smoking, presence of asthma, N-ERD, or prior FESS [154].
In recent years, biologic therapies targeting IgE and T2 cytokines—notably IL-4, IL-5, and IL-13—have transformed disease management [216, 217]. These agents are particularly effective in patients with comorbid asthma, N-ERD, or recurrent CRSwNP; side effects are infrequent and mostly mild [218–220].
Omalizumab was approved for the treatment of CRSwNP, and benefits were observed regardless of baseline total IgE levels or systemic atopy. In real-world practice, omalizumab may be considered when eosinophilia is less prominent, in patients intolerant to IL-5/IL-4Rα blockers, or when allergic comorbidity is predominant. Its dual effect on both upper and lower airway inflammation reinforces its role within the unified airway model [221].
Anti-IL-5 targeting therapies (mepolizumab and benralizumab) reduce eosinophil survival and activation, thereby lowering polyp size and improving nasal obstruction.
Dupilumab, the first biologic approved for CRSwNP treatment, targeting IL-4Rα and inhibiting both IL-4 and IL-13 signalling, has shown consistent efficacy across randomized trials and real-world studies in improving symptom burden and reducing the need for surgery [222, 223]. Dupilumab is being reported as the most effective treatment in network meta-analysis and studies using RWE data [224–226]. A recent head-to-head clinical trial with omalizumab also showed that dupilumab seems to have better results overall [227].
Biologic selection may be guided by biomarkers such as blood eosinophils, serum IgE, and comorbid T2 traits [e.g., asthma or elevated fractional exhaled nitric oxide (FeNO)]. Studies suggest that patients with prominent eosinophilia and frequent relapses may benefit most from anti-IL-5 therapies, while those with broad T2 endotype or corticosteroid dependence may respond better to dupilumab [228].
Importantly, the success of biologics reinforces the concept of united airways disease, whereby controlling upper airway inflammation may contribute to improved asthma control and overall reduction of T2 inflammation burden. Longitudinal data support the role of these therapies in altering disease trajectory and improving quality of life [229, 230].
Although not yet approved for CRSwNP, anti-TSLP therapy with tezepelumab represents a promising upstream intervention [231]. Early data from asthma trials involving patients with comorbid nasal polyposis suggest potential benefits in reducing sinonasal inflammation, with randomized studies in upper airway diseases currently underway [232, 233].
Eosinophils are key effector cells in the pathogenesis of several upper airway diseases. Despite recent advances, the management of eosinophilic upper airway diseases, including CRSwNP and AR, continues to face significant challenges. Biologic therapies targeting T2 inflammation have improved patient outcomes, but variability in therapeutic response highlights the need for improved disease phenotyping and predictive biomarkers to guide personalized treatment. Better stratification based on molecular and cellular profiles will enable clinicians to select the most effective therapy, minimizing unnecessary exposure and optimizing healthcare resources [141, 217].
Moreover, the long-term safety, cost-effectiveness, and patient access to novel biologics require rigorous, real-world evaluation. There remains an unmet need for effective management in patients unresponsive to conventional and biological therapy, emphasizing the importance of innovative approaches such as upstream cytokine inhibition and combination strategies [217]. Non-invasive, locally reflective biomarkers, such as nasal secretion analyses, should be further explored and validated to refine diagnoses and monitor disease activity with higher specificity [140].
In AR, persistent symptoms and frequent pharmacological polytherapy suggest that current treatment paradigms fall short for a substantial patient subset. Real-world data highlight a gap between guideline recommendations and clinical practice, underscoring the importance of developing region-adapted guidelines and optimizing adherence strategies [234].
Ultimately, future research should focus on multicentre, head-to-head trials, integrating clinical, immunological, and patient-reported outcome measures. Collaborative networks are crucial for advancing precision medicine and closing current gaps, ensuring improved quality of life for individuals affected by eosinophilic upper airway diseases [141, 217].
Eosinophils are granulocytic leukocytes derived from hematopoietic stem cells in the bone marrow. Their development is regulated by cytokines such as IL-5, IL-3, and GM-CSF. Once matured, eosinophils circulate in the blood and can be recruited into tissues, especially in response to allergic and parasitic stimuli, and they are prominent in allergic and inflammatory responses [235].
Under normal conditions, eosinophils are present in low numbers in the circulation and tissues. They are predominantly tissue cells, and their major target organ for homing in the healthy individual is the gastrointestinal tract. However, in diseases such as asthma, they are found in elevated numbers in the blood, sputum, and airway tissues. Eosinophil numbers can remain high in tissues even when peripheral numbers are low, suggesting that their survival is enhanced after extravasation. Once they enter tissues, eosinophils do not return to the blood circulation, although studies in mice suggest that endobronchial eosinophils can travel to regional lymph nodes and act as antigen-presenting cells. In asthma, the bronchial epithelium and submucosa are infiltrated by eosinophils in both large and small airways [236, 237].
Eosinophils contribute significantly to airway inflammation in asthma. They contain cytoplasmic granules rich in toxic, highly charged cationic proteins, including MBP, EPX, ECP, and EDN. Their granules, upon release, not only contribute to tissue damage, airway remodelling, and bronchial hyperreactivity, but also damage epithelial cells, increase vascular permeability, and promote further leukocyte infiltration. In asthmatic airways, eosinophil recruitment is driven by chemokines such as eotaxins (CCL11, CCL24, CCL26) and regulated by adhesion molecules (e.g., VCAM-1). IL-5, predominantly produced by Th2 lymphocytes and ILC2s, plays a crucial role in eosinophil maturation, survival, and activation. Eosinophils can release cytokines (IL-4, IL-5, IL-13) and lipid mediators (e.g., leukotrienes), which amplify Th2-driven inflammation and bronchoconstriction. IL-5 is a key T2 cytokine in promoting eosinophil recruitment into the asthmatic airways [235, 237–239].
Eosinophils can also regulate Th1 and Th2 cytokine secretion [IL-5, IL-13, interferon-γ (IFN-γ)] in response to pathogenic stimulation (staphylococcal enterotoxin B). Thus, the eosinophil is not only an effector cell in the asthmatic airway but also influences Th1 and Th2 evolution of the inflammatory response that may be of relevance to nonallergic asthma [235, 237]. Chronic eosinophilic inflammation leads to structural changes in the airway wall, collectively referred to as airway remodelling. This includes subepithelial fibrosis, smooth muscle hypertrophy, goblet cell hyperplasia, and increased angiogenesis. Eosinophil-derived TGF-β is a key mediator in this process, promoting ECM deposition and fibrosis. Eosinophilic inflammation is associated with markers of airway remodelling, like increased levels of TGF-β expression and thickening of the lamina reticularis underlying the epithelial basement membrane [236, 237].
Remodelling contributes to irreversible airflow limitation in severe asthma and is associated with poor response to conventional therapies such as corticosteroids [237].
Bronchial hyperresponsiveness (BHR) is a hallmark of asthma and is partly mediated by eosinophil-derived mediators. MBP and ECP can damage parasympathetic nerves and epithelial integrity, leading to heightened sensitivity to stimuli. This exaggerated response to allergens or irritants leads to episodes of bronchoconstriction, further exacerbating clinical symptoms. This role of eosinophils is behind the pathophysiology of exercise-induced bronchoconstriction [236, 237].
Asthma is a heterogeneous condition, meaning it can manifest in different ways depending on the underlying biological processes. The presence of eosinophils in the airways helps to categorize different phenotypes of asthma. Eosinophilic asthma is one of the key phenotypes, characterized by elevated eosinophils in the airway and/or blood. It has classically been associated with allergic sensitization and a Th2-dominant inflammatory response [240, 241]. Eosinophil levels can also provide insight into the severity of asthma and predict the risk of exacerbations and long-term outcomes. These patients tend to have more severe asthma and may respond better to targeted therapies, such as biologics that reduce eosinophil levels (anti-IL-5 biologics) [238–247]. Usually, eosinophil-driven severe asthma is an adult-onset phenotype and frequently can be associated with comorbidities such as rhinitis, CRSwNP, but less frequently linked to atopy compared to childhood-onset allergic asthma [191, 228, 248]. Integration of BECs with other biomarkers (e.g., FeNO, IgE) enhances phenotyping and supports personalized treatment strategies.
Eosinophils, in severe asthma, have a triple role, both in pathogenesis and in diagnosis and endotyping, finally they can be used as a predictor of response to targeted biologic therapies too. Their count can be obtained from different samples.
Biomarkers such as BEC, sputum eosinophils, and FeNO are used to identify eosinophilic inflammation in asthma patients [246]. Peripheral BECs are easily obtained and widely available but lack both specificity and sensitivity.
The variability of the eosinophilic count in blood is wide, and it is therefore suggested that it be investigated on several samples, taken on different days, especially in patients with low values, to observe whether the absence of hypereosinophilia is real or only related to physiological fluctuations of this cell in the blood.
The evaluation of eosinophils in sputum is considered the gold standard for identifying eosinophilic airway inflammation. Induced sputum analysis is a more direct method for assessing airway eosinophilia. However, the procedure is technically demanding, time-consuming, and not widely available in routine clinical practice. Therefore, its use is often limited to specialized centres or research settings.
In addition to cell counts, eosinophil-derived products such as EDN and ECP were studied. EDN, in particular, was shown to be stable and correlated with both asthma severity and response to biological treatments, representing a potential additional biomarker for patient stratification [249, 250].
Regarding TSLP, mentioned earlier as a cytokine implicated in T2 inflammation, this cytokine has a slightly different role [251, 252]. Since it is a cytokine produced by the epithelium, it acts more ubiquitously than those precipitating T2 inflammation, also interacting with other cells, capable of producing different cytokines, characteristic of non-T2 inflammation, such as IL-17, IL-6, IL-8, and TH-17 cells. In the specific case of TSLP, the role of eosinophils, as a biomarker, appears to be more “ambivalent”. Although in clinical trials of tezepelumab, the monoclonal antibody directed against this cytokine, it showed greater efficacy in patients with more than 150 eos/mcL on blood, the drug was also effective in reducing disease exacerbations in those without evidence of eosinophilic inflammation, having a count below the parameter.
FeNO measurement is a non-invasive surrogate marker of eosinophilic inflammation. IL-13 stimulates nitric oxide synthase in airway epithelial cells, leading to increased FeNO levels, suggesting eosinophilic airway inflammation and predicting responsiveness to corticosteroids. While not a direct measure of eosinophils, FeNO correlates with sputum eosinophils in many patients and serves as a useful adjunct in asthma diagnosis and management [247].
Sputum cell counts of 1–3% can define eosinophilic asthma and can be used for its diagnosis [243, 244].
Higher eosinophil counts, both in blood and sputum, are associated with an increased risk of asthma exacerbations. These exacerbations are often linked to more intense inflammation in the airways and can lead to hospitalization if not managed properly.
In addition, elevated blood eosinophils (≥ 150 cells/µL), sputum eosinophil proportion ≥ 2%, and/or FeNO values ≥ 20 parts per billion (ppb) in adults suggest a refractory T2 inflammation in patients under high-dose inhaled corticosteroid (ICS) treatment or OCS, helping in diagnosing severe asthma. Monitoring eosinophil levels over time can help track how well asthma is controlled. Persistent eosinophilic inflammation despite treatment may indicate the need for a more aggressive therapeutic approach.
GINA 2025 update has a new appendix with the data on the role of T2 biomarkers (particularly blood eosinophils and FeNO) in the diagnosis, assessment, and management of asthma. This information will also be appreciated while assessing a patient’s eligibility for T2-targeted biologic therapy in clinical practice. There has to be caution when comparing a patient’s biomarker results with absolute thresholds in clinical practice [239].
Despite their usefulness, eosinophil levels can be influenced by corticosteroid therapies, infections, and other comorbidities. In addition, a proportion of patients with severe asthma have non-eosinophilic phenotypes, so eosinophil counts alone are not sufficient for a complete characterization of the disease.
In addition, eosinophil levels can be used as a biomarker to predict how well a patient will respond to certain treatments. For example, patients with elevated eosinophil counts often respond better to ICS or biologic therapies that target eosinophil-related pathways, while those with lower eosinophil counts may not benefit as much from these treatments [253].
In patients who present with asthma-like symptoms (e.g., cough, wheeze, shortness of breath) but whose diagnosis is uncertain, eosinophil levels can act as an additional piece of the diagnostic puzzle. Conditions like chronic obstructive pulmonary disease (COPD), upper airway diseases, or gastroesophageal reflux disease (GERD) can also cause symptoms similar to asthma. Elevated eosinophils in blood or sputum can help to distinguish asthma from these other conditions, especially when symptoms are suggestive, but the classic tests (e.g., spirometry) are inconclusive [254].
Eosinophil levels have a role in eosinophilic asthma diagnosis and phenotyping, correlate with asthma severity, risk of exacerbations, and long-term prognosis, guiding management decisions.
The advent of biologics, especially monoclonal antibodies that specifically target eosinophils, has not only revolutionised asthma treatment, but also transformed our knowledge of the role of eosinophils in asthma [255].
GINA recommends the use of biologic therapy only for patients with severe asthma, with exacerbations and/or poor symptom control despite taking at least high-dose ICS-LABA, and who have allergic or eosinophilic biomarkers or need maintenance OCS, and only after treatment has been optimized.
When choosing between available therapies (anti IgE, anti-IL5/5Rα, anti-IL4Rα or anti-TSLP), one needs to consider local payer eligibility criteria, T2 comorbidities such as atopic dermatitis, nasal polyps, clinical history suggesting allergen-triggered symptoms, predictors of asthma response (see below), cost, dosing frequency, delivery route (i.v. or s.c.; potential for self-administration) and of course patient preference [247].
The regulatory approvals for add-on anti-IL5 or anti-IL5Rα include:
For ages ≥ 12 years: mepolizumab (anti-IL5), 100 mg by SC injection every 4 weeks, or benralizumab (anti-IL5Rα), 30 mg by SC injection every 4 weeks for 3 doses then every 8 weeks.
For ages ≥ 18 years: reslizumab (anti-IL5), 3 mg/kg by IV infusion every 4 weeks.
For ages 6–11 years: mepolizumab (anti-IL5), 40 mg by SC injection every 4 weeks.
Mepolizumab and benralizumab may also be indicated for EGPA, and mepolizumab also for HES and CRSwNP. Self-administration may be an option.
Potential predictors of good asthma response to anti-IL5 or anti-IL5Rα are higher blood eosinophils (which is strongly predictive), a higher number of severe exacerbations in the previous year (strongly predictive), adult-onset asthma, nasal polyps, maintenance OCS at baseline, and low lung function (FEV1 < 65% predicted).
Eligibility criteria (in addition to criteria for severe asthma) for add on anti-IL4Rα for severe eosinophilic/T2 asthma or patients requiring maintenance OCS are more than a specified number of severe exacerbations in the last year, and T2 biomarkers above a specified level (e.g., blood eosinophils ≥ 150/μL and ≤ 1500/μL, or FeNO ≥ 25 ppb) or requirement for maintenance OCS.
Potential predictors of good asthma response to dupilumab are higher blood eosinophils (strongly predictive) including in children, and higher FeNO (strongly predictive) including in children.
Anti-TSLP may also be considered in patients with no elevated T2 markers. Potential predictors of good asthma response to anti-TSLP are higher blood eosinophils (strongly predictive) and higher FeNO levels (strongly predictive).
Regarding treatment, biologics that target the IL-5 pathway are not universally effective in people with severe asthma and elevated eosinophil counts. The efficacy of these biologics appears to be limited to people with an intrinsic, non-allergic phenotype or a mixed phenotype with a dominant intrinsic component. In people with predominantly allergic phenotypes and allergen-driven symptoms (or a phenotype mainly driven by IL-13 rather than IL-5), biologics such as omalizumab, tezepelumab, or dupilumab might be considered as the first treatment choice [255].
Conflicting evidence on eosinophil depletion therapies (like anti-IL-5 biologics) and outcomes stems from eosinophils having dual roles (protective vs. inflammatory), different cell subtypes (tissue vs. blood), inconsistent biomarkers, and varied responses in specific eosinophil-associated diseases (EADs) like asthma or EoE, leading to mixed efficacy signals, especially concerning severe infections, long-term safety, and varying benefits in different patient groups (e.g., pediatric vs. adult). Eosinophils drive inflammation in diseases but also can fight parasites and have immune homeostatic functions, creating uncertainty about depleting them entirely. It is, in fact, recommended to treat helminth infections prior to therapy [256]. Different eosinophil populations (tissue-resident vs. inducible/inflammatory) may have distinct roles, and therapies might not target them equally, affecting outcomes [257]. Moreover, peripheral BECs don’t always perfectly correlate with tissue inflammation or disease activity, complicating trial results [258]. It is also relevant that EADs have varied mechanisms, so a drug effective in one (e.g., asthma), like mepolizumab, might not be effective in another (e.g., EoE) [259, 260]. A concern also regards the long-term safety. In fact, while clinical trials show sustained efficacy, long-term risks (malignancy, infection) remain a focus, though current data lean towards a favourable safety profile [261]. In essence, the conflicting nature arises from the complexity of eosinophil biology, varied disease contexts, and limitations in measuring specific eosinophil functions, highlighting the need for more nuanced therapeutic strategies.
Biologics for asthma have limitations, including significant patient heterogeneity in response and challenges in identifying optimal treatment duration and dosing interval, leading to concerns about long-term efficacy. Around 25% don’t respond at all (primary nonresponders), others lose response over time after 6–12 months of good asthma control (secondary nonresponders—waning efficacy) [262]. In most cases, the waning effect manifests at the end of the dosing interval, requiring time or dosing adjustment [263].
Moreover, while traditional markers like eosinophils and FeNO help identify T2 inflammation, they aren’t specific enough for choosing among biologics targeting IL-4/13, IL-5, or IgE, and non-T2 asthma patients often lack effective options, creating a significant unmet need for predictive tools. Therefore, clinicians are forced to rely on trial-and-error, subjective symptom scores, and a combination of clinical features, leading to costly delays and ineffective treatments for many severe asthmatics [264].
Choosing a biologic drug in the treatment of asthma involves a multi-faceted strategy, focusing on the patient’s biomarker profile (eosinophils, FeNO, IgE) to identify the specific inflammatory pathway (T2), their phenotype/endotype (allergic, eosinophilic, non-T2), and the presence of comorbidities (like nasal polyps or eczema) that might benefit from a specific drug, while also considering patient preference for administration, safety, and cost to guide personalized therapy (Table 2).
Key factors in selecting biologics.
| Biomarker profile (The “endotype”) | ||
|---|---|---|
| Blood eosinophils and FeNO | High levels suggest T2 inflammation | Dupilumab, mepolizumab, benralizumab, reslizumab |
| IgE levels | Elevated IgE points towards allergic asthma | Omalizumab |
| Low eosinophils and IgE | non-T2 asthma | Tezepelumab |
| Clinical phenotype and comorbidities | ||
| Allergic asthma | Omalizumab | |
| Eosinophilic asthma | Mepolizumab, benralizumab, reslizumab, dupilumab | |
| Comorbidities | Biologic that treats both asthma and conditions like atopic dermatitis, nasal polyps, or EGPA | |
EGPA: eosinophilic granulomatosis with polyangiitis; FeNO: fractional exhaled nitric oxide; T2: type 2.
New drugs are at present in clinical trials (itepekimab and astegolimab—anti-IL-33 antibodies), offering the possibility to efficiently treat non-T2 asthma. However, besides new drugs, there is still a need for major research: comparative studies, better biomarkers for predicting response, and the determination of optimal treatment duration. Moreover, one of the most relevant goals will be the discovery of biomarkers identifying non-severe asthmatics who will become severe to anticipate the use of biologics [265].
While traditionally associated with asthma, eosinophilic inflammation has also been recognized in other chronic airway conditions, including COPD and eosinophilic bronchitis (Table 3).
Comparison of eosinophilic COPD and eosinophilic bronchitis.
| Feature | Eosinophilic COPD | Eosinophilic bronchitis |
|---|---|---|
| Definition | COPD phenotype with eosinophilic inflammation in airways and/or blood | Chronic cough with eosinophilic airway inflammation, without airflow obstruction |
| Typical symptoms | Chronic cough, sputum production, exertional dyspnea, wheezing; frequent exacerbations | Isolated chronic non-productive cough |
| Lung function | Persistent airflow limitation | Normal lung function |
| Bronchodilator response | Sometimes shows a high response (FEV1 > 15% and > 400 mL) | No bronchodilator response |
| Airway hyperresponsiveness | May or may not be present | Absent |
| Exacerbation pattern | Frequent, especially non-bacterial | Rare to none |
| Inflammatory profile | T2-high: IL-5, IL-4, IL-13; ILC2s and Th2 cells in airway mucosa | T2-high inflammation localized to the airway without systemic involvement |
| Histopathology | Patchy eosinophil-rich lung areas; GATA3+ cells | Sputum eosinophilia; absence of tissue remodelling |
| Associated biomarkers | Elevated BEC (≥ 150–300 cells/μL or ≥ 2–3% leukocytes), FeNO, T2 cytokines | Sputum eosinophils; no consistent blood eosinophilia |
| Patient demographics | More common in men, ex-smokers, those with higher BMI and ischemic heart disease | Not clearly associated with specific demographics |
| Asthma relationship | Can occur with or without asthma; not necessarily ACO | Not associated with asthma |
| Microbiome | Distinct from neutrophilic COPD; non-Haemophilus dominant | Not well characterized |
ACO: asthma-COPD overlap; BEC: blood eosinophil count; BMI: body mass index; COPD: chronic obstructive pulmonary disease; FeNO: fractional exhaled nitric oxide; IL: interleukin; ILC2s: group 2 innate lymphoid cells; T2: type 2; Th2: T-helper 2.
Eosinophilic COPD refers to a phenotype of COPD characterized by increased eosinophilic inflammation in the airways and/or peripheral blood [233, 234, 266, 267]. This phenotype is typically identified by elevated BECs, often using thresholds such as ≥ 2% of total leukocytes or ≥ 150–300 cells/μL, though there is no universally agreed-upon cutoff. The prevalence of eosinophilic COPD varies based on the diagnostic thresholds used: A BEC of ≥ 3% cells/µL identifies approximately 20% of COPD patients [268], while using a sputum eosinophil threshold of ≥ 3% increases the prevalence to 36% [269]. Some patients may only exhibit this eosinophilic phenotype during disease exacerbations (about 28% of COPD exacerbations) [268]. GOLD notes that BECs are a practical surrogate for airway eosinophilia, but they can fluctuate over time, are influenced by infections, comorbidities, and corticosteroid therapy [270]. Repeated measurements are often necessary to confirm persistent eosinophilic status, as single measurements may not reliably reflect the underlying inflammatory phenotype.
Notably, eosinophilic COPD occurs both in patients with and without a history of asthma [269], suggesting a distinct endotype rather than an overlap syndrome in many cases; therefore, its identification should not be limited to those with asthma-COPD overlap (ACO) [271]. It is present in approximately 20–40% of patients with COPD, independent of a history of asthma, and is associated with a T2 inflammatory profile, including increased IL-5, IL-4, and IL-13 activity, even in the absence of asthma [266, 270]. Molecular studies have shown similar T2 marker expression in eosinophilic COPD and eosinophilic asthma, but the T2 signature is generally more restricted and less robust than in asthma, and the correlation between eosinophilia and T2 markers is weaker in COPD than in severe asthma [272]. The eosinophilic subtype of COPD most commonly affects men, ex-smokers, individuals with higher body mass index (BMI), and those with a history of ischemic heart disease [273].
Why only some patients develop eosinophilic airway inflammation remains unknown. The mechanisms driving this phenotype are multifactorial and incompletely defined. Genetic predisposition, environmental exposures, and local immune microenvironments, such as spatially confined aggregates of T2 cells and eotaxin-producing cells, may contribute to the development of eosinophilic inflammation in only some individuals [270, 274].
The key pathophysiological mechanisms underlying eosinophilic inflammation in COPD involve both ILC2s and Th2 lymphocytes, which drive the production of IL-5 and other T2 cytokines [270, 274]. In eosinophilic COPD, ILC2s and Th2 cells are present in the airway mucosa and contribute to the local T2 immune microenvironment, as evidenced by spatial association with GATA3+ cells and patchy eosinophil-rich foci in lung tissue. IL-5 is central to eosinophilopoiesis, promoting eosinophil maturation, survival, and recruitment to the airways. Overexpression of IL-5, along with chemotactic signalling by eotaxin-1 (CCL11) and CCL24, facilitates eosinophil trafficking into the lung parenchyma. The patchy and focal nature of eosinophil-rich microenvironments in the lung further underscores this heterogeneity [274].
The GOLD Science Committee also noted that the airway microbiome may play a role, as eosinophilic inflammation is associated with distinct, non-Haemophilus-dominant microbiome patterns, in contrast to neutrophilic COPD [270]. It has been suggested that TSLP is upregulated in the airways of some patients with COPD and may contribute to eosinophilic airway inflammation by promoting T2 immune responses [275, 276]. However, the association between TSLP and eosinophilic inflammation in COPD is less robust than in asthma. While TSLP and its receptor are present and upregulated in COPD airways, the correlation between TSLP levels and BECs is not as strong as in atopic asthma, and some studies suggest that TSLP may be more closely linked to airway remodelling and non-eosinophilic inflammation in COPD [277].
The typical symptoms of eosinophilic COPD are chronic cough, exertional dyspnoea, and sputum production, often with persistent and progressive airflow limitation [267, 270]. A highly positive bronchodilator response (increase in FEV1 > 15% and > 400 mL) in a patient with eosinophilic COPD is clinically significant because it suggests a phenotype with overlapping features of asthma and COPD, but not necessarily true ACO [278]. This degree of reversibility is uncommon in typical COPD and is more characteristic of asthma or ACO [271]; however, in eosinophilic COPD, it identifies a subgroup with heightened T2 inflammation and corticosteroid responsiveness [270]. Indeed, such patients are more likely to experience better responses to ICS therapy [279] with greater improvements in lung function and symptom control, and a marked reduction in exacerbation risk when ICS is continued. Conversely, withdrawal of ICS in these patients is associated with an increased risk of exacerbations, particularly in those with a history of frequent exacerbations and elevated BECs [270, 280].
Patients may also experience wheezing and are at increased risk for episodes of worsening dyspnoea, cough, and sputum, sometimes with increased sputum purulence. These symptoms are similar to those of non-eosinophilic COPD, but the eosinophilic phenotype is distinguished by a higher frequency of exacerbations, particularly those not associated with bacterial infection [268, 270, 281]. This is supported by evidence that higher BECs are associated with increased exacerbation risk, especially in those with a history of frequent exacerbations, and that these exacerbations are less likely to be driven by bacterial pathogens.
In contrast, eosinophilic bronchitis is a distinct cause of chronic cough characterized by sputum eosinophilia (> 3%) in the absence of variable airflow obstruction, airway hyperresponsiveness, or abnormal spirometry, features that differentiate it from asthma. According to the GOLD Science Committee, eosinophilic COPD represents a T2-high, corticosteroid-responsive phenotype with airflow limitation, whereas eosinophilic bronchitis is defined by isolated cough and sputum eosinophilia in the absence of physiological compromise [270]. Patients typically present with a chronic, non-productive cough, normal lung function, and no evidence of airway hyperreactivity on methacholine challenge testing [282, 283]. There is no dyspnea, wheezing, or airflow limitation, and patients do not experience the acute exacerbations characteristic of COPD [284]. While both eosinophilic bronchitis and eosinophilic COPD involve elevated eosinophilic inflammation, they differ significantly in pathophysiology. In eosinophilic bronchitis, the eosinophilic infiltration is limited to the bronchial mucosa, without the structural or functional changes seen in asthma or COPD [285].
The treatment approach for eosinophilic COPD differs from that for eosinophilic bronchitis in several key ways (Table 2). For eosinophilic COPD, GOLD recommends ICSs in combination with long-acting bronchodilators (LABA and/or LAMA) for patients with frequent exacerbations and elevated BECs, as these patients are more likely to benefit from ICS-containing regimens [237]. In select cases with persistent exacerbations despite optimal inhaled therapy and elevated eosinophils, biologic agents targeting T2 inflammation (such as anti-IL-5, such as mepolizumab and reslizumab, or anti-IL-5R therapies, such as benralizumab) may be considered, though their benefit is limited and not universal [270]. Indeed, these therapies do not significantly improve lung function or quality of life beyond what is achieved with standard inhaled therapy, and their use should be restricted to highly selected patients with persistent exacerbations and evidence of eosinophilic inflammation [286–288]. Dupilumab inhibits IL-4 and IL-13 signalling, which is central to T2 inflammation that is not fully addressed by anti-IL-5 therapies [286]. It has demonstrated a clinically meaningful role as an add-on therapy for patients with COPD who have evidence of T2 inflammation, specifically those with elevated BECs (≥ 300 cells/μL) and a high risk of exacerbations despite receiving maximum inhaled triple therapy [289, 290]. The effect is most pronounced in patients with higher eosinophil counts and/or elevated FeNO. The GOLD Science Committee specifically supports a phenotype-driven approach to ICS and biologic use in COPD [270]. However, mepolizumab is not approved for eosinophilic COPD by either the United States FDA or EMA, while dupilumab is approved for this indication by the FDA but not by the EMA (Table 4).
Comparison of treatment strategies for eosinophilic COPD and eosinophilic bronchitis.
| Feature | Eosinophilic COPD | Eosinophilic bronchitis |
|---|---|---|
| Primary goal of treatment | Reduce exacerbation frequency, improve airflow, control inflammation | Suppress eosinophilic airway inflammation and resolve chronic cough |
| Role of ICS | Indicated in patients with blood eosinophil count ≥ 300 cells/μL or frequent exacerbations | First-line therapy due to corticosteroid responsiveness |
| Bronchodilators (LABA/LAMA) | Commonly used in combination with ICS for symptom control and airflow limitation | Not typically indicated, as airflow obstruction is absent |
| Systemic corticosteroids | Reserved for acute exacerbations | Occasionally used for short-term control if symptoms persist despite ICS |
| Biologic therapy | May be considered in select patients with high eosinophil counts and frequent exacerbations | Not routinely used; insufficient evidence for benefit |
| Antibiotics | Used during bacterial exacerbations | Not part of routine management |
| Methacholine challenge test | May help exclude the asthma component | Used to confirm the absence of airway hyperresponsiveness |
| Monitoring strategy | Regular lung function tests, symptom tracking, and exacerbation history | Monitor cough resolution and eosinophil counts in sputum |
| Treatment response time | Gradual, varies with baseline lung function and eosinophil burden | Typically rapid (within weeks) with corticosteroid therapy |
COPD: chronic obstructive pulmonary disease; ICS: inhaled corticosteroids.
In contrast, the mainstay of treatment of eosinophilic bronchitis is ICSs alone, which are highly effective in reducing cough and airway eosinophilia, but the optimal dose and duration are not clearly defined in the literature [282, 283, 291]. Long-acting bronchodilators are not indicated because this condition is defined by the absence of variable airflow obstruction and airway hyperresponsiveness and does not feature the bronchoconstriction or exacerbation risk that these therapies are designed to address [291]. Systemic corticosteroids may be considered in exceptional cases where symptoms are severe and refractory to high-dose ICSs, but this is uncommon [291, 292]. Long-term systemic corticosteroid use is avoided due to well-established risks [293]. Biologic therapies should not be considered part of routine management for eosinophilic bronchitis, and their use should be limited to select, refractory cases after careful consideration of risks, benefits, and costs [294, 295].
Leukotriene receptor antagonists (e.g., montelukast) may be considered in select cases, but their efficacy in eosinophilic bronchitis is less well established, and they are not first-line [296]. Patients with eosinophilic bronchitis who are most likely to benefit from leukotriene receptor antagonists, such as montelukast, as alternative or adjunct therapy are those with coexisting atopic features (e.g., AR), aspirin-exacerbated respiratory disease, or exercise-induced symptoms, and those who are unable to tolerate or are poorly adherent to ICSs [296]. However, in patients with severe, corticosteroid-dependent disease and persistent airway eosinophilia, the addition of montelukast has not demonstrated significant further reduction in eosinophilic inflammation [297].
EGPA and allergic bronchopulmonary aspergillosis (ABPA) are considered clinical and pathological entities within the spectrum of eosinophilic-driven diseases. From a historical perspective, the description of these diseases has followed very different trajectories according to their natural history and the systems of classification.
EGPA is classified as a medium and small vessel anti-neutrophil cytoplasmic antibodies (ANCA) associated vasculitis, grouped with granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA). The recent 2022 American College of Rheumatology (ACR) classification criteria allow EGPA to be distinguished from MPA and GPA when a diagnosis of small- or medium-vessel vasculitis has been made in the presence of a BEC of at least 1 × 109/L, obstructive airway disease, nasal polyps, extravascular eosinophilic-predominant inflammation, and mononeuritis multiplex [298]. The hallmark of EGPA is the essential presence of a history of asthma, often associated (> 90%) with CRSwNP. Regardless of the duration or severity of asthma or CRSwNP (in the prodromal or allergic phase), the eosinophilic and vasculitic phases can develop sequentially. This is associated with rising peripheral eosinophils and damage to organs infiltrated by eosinophils, primarily the lungs, gastrointestinal tract, and heart. This is followed by endothelial cell adhesion and leukocyte activation, leading to vasculitis that primarily affects the kidneys, peripheral nerves, and skin [299]. Nevertheless, the diagnosis of EGPA remains a clinical challenge [300], as the disease has heterogeneous clinical presentations and the three phases may overlap. In addition, obtaining a histological demonstration of pauci-immune necrotising vasculitis or eosinophil-rich granulomatous inflammation from damaged tissue is not always easy for each patient. Perinuclear ANCA (p-ANCA) directed against myeloperoxidase (MPO) are potential biomarkers useful to predict different clinical phenotypes of EGPA. In ANCA-positive patients, peripheral nervous system, renal, and skin involvement, and histopathological evidence of vasculitis are more common, albeit not exclusive. Conversely, cardiac involvement and gastroenteritis are more prevalent in ANCA-negative patients.
Immunological dysregulation in EGPA is thought to result from T-lymphocyte antigen-driven activation. Th2-screwed cells are detected in biopsies and fluid from active EGPA, leading to the recruitment of eosinophils via eotaxin3 and IL5. Eosinophils exert cytotoxic functions in infiltrating organs through the degranulation of enzymes such as MBP, ECP, EPO, and EDN. They also contribute to tissue fibrosis, thrombosis, allergic inflammation, and neural damage through the release of active molecules. In addition, Th2 cells guide the IL4-mediated B-cell response, resulting in the production of IgG4, IgE, and ANCA [301]. The autoreactive CD4+ T-cell population responds to the MPO autoantigen by secreting IFN-γ and IL-17, while the CD8+ cells in EGPA are clonally expanded and have an effector memory phenotype, expressing cytotoxic markers upon stimulation [302]. These data suggest a role for both Th1 CD4+ and Tc CD8+ cells in granuloma formation and vasculitic damage. Genetic analysis revealed differences in pathology based on ANCA status, with an HLA-DQ association in MPO-ANCA-positive cases suggesting an antigen-driven disease. GPA33 and IL5 gene expression was found in ANCA-negative EGPA cases, which are involved in mucosal barrier function and eosinophil pathways [303].
Historically, the treatment options for EGPA have been borrowed from those for ANCA-associated vasculitis. Systemic corticosteroids and immunosuppressants are the mainstay of treatment and have demonstrated the ability to induce disease remission in many patients. However, they often leave behind a permissive dose of chronic corticosteroids, which are not free of long-term adverse effects. Furthermore, the risk of relapse and disabling outcomes remains. It is crucial to stratify patients into risk categories according to organ or life-threatening manifestations in order to achieve timely and effective disease remission. Evidence-based guidelines for the management of EGPA [304] recommend remission induction in patients with new-onset, active severe EGPA with rituximab or cyclophosphamide, followed by remission maintenance through rituximab or traditional DMARDs [305].
Treatment with monoclonal antibodies targeting IL-5 or the IL-5 receptor has been recognised as an effective therapy for relapsing EGPA, and more recently for the induction of non-severe EGPA. This therapy has a documented steroid-sparing effect, as demonstrated in clinical trials and in real-life experience.
Many unmet clinical needs are currently under investigation. Disease-specific biomarkers, tools for assessing therapeutic response, and patient-reported outcomes need to be identified and validated. Additionally, the role of IL-5-targeting agents in severe organ or vasculitic manifestations, as well as the differential therapeutic approach in EGPA subsets, requires further investigation.
ABPA is a clinical condition caused by hypersensitivity reactions to inhaled and colonising Aspergillus fumigatus (A. fumigatus). The airways of patients with asthma or cystic fibrosis (CF) develop bronchitis, bronchiectasis, and/or mucus plugs due to impaired epithelial barrier dysfunction, local T2 inflammation, and eosinophilia [306]. Although considered an organ-limited condition, ABPA is often compared to other eosinophil-driven diseases due to its unique pathogenic mechanisms.
The development of ABPA is driven by a complex interplay between A. fumigatus and the host immune response. In patients with asthma or CF, defects in innate or adaptive immune responses, combined with impaired clearance of A. fumigatus and abnormal mucus production, induce a chronic state of airway inflammation in which repair mechanisms are impaired [307]. A hypersensitivity reaction to A. fumigatus, as indicated by A. fumigatus-sIgE and IgG responses, is a necessary and unavoidable condition for the development of the disease. A T2 allergic reaction to A. fumigatus activates eosinophil-driven inflammation and hyper IgE synthesis, enhancing mast cell activation. In patients with chronic asthma, A. fumigatus triggers and sustains T2 inflammation via different mechanisms. Activation of the protease induces IL-33 production, which is an alarmin that can recruit ILC2s and stimulate IL-13 and IL-5 eosinophil production. The process of Eosinophil ETosis (EETosis) amplifies oxidative stress, perpetuates tissue damage and the cycle of mucus [308]. On the other hand, β-glucans and carbohydrate constituents of fungal cell walls activate receptors on innate immune cells, leading to the synthesis of inflammatory cytokines by epithelial and macrophage cells, the activation of Th17-type lymphocytes, and the chemotaxis of neutrophils. The elastase activity of Aspergillus serine proteases cleaves epithelial tight and adherens junctions, leading to epithelial damage. This chronic process disrupts the integrity of the airways, leading to the development of bronchiectasis and mucus plugs, which can result in nodularity and consolidation. Clinically, the disease presents as remitting-relapsing episodes of wheezing, coughing, and phlegm production, which are sometimes accompanied by systemic symptoms or severe local complications, such as haemoptysis and a decline in lung function. The final stages of the disease are characterised by steroid dependence and fibrosis or fibrocavitary findings [309].
The diagnostic algorithms for ABPA have evolved from the initial Rosenberg-Patterson criteria to the 2013 ISHAM diagnostic criteria, which were defined in the context of patients with asthma or CF. According to these criteria, a positive type I skin reaction to A. fumigatus or sIgE levels of > 0.35 kUA/L and serum total IgE levels of > 1,000 IU/mL are mandatory, while eosinophilia of > 500 cells/mL, radiographic abnormalities, and an immunological IgG response to A. fumigatus can be variably present [310]. Good performance of diagnostic criteria is important in terms of prognosis and cost management. Hasano highlighted the limitations of traditional diagnostic criteria in the context of physician-diagnosed ABPA, given that fungi other than A. fumigatus can trigger a similar pathological picture (ABPM). The presence of fungal hyphae in allergic mucin, as well as radiological evidence of mucus plugs in the bronchi, may serve as a surrogate for pathological or immunological findings, thereby increasing the sensitivity of physician-diagnosed ABPA [311]. Molecular allergology is becoming a new tool for identifying the endotype of A. fumigatus sensitisation, and for improving the specificity of ABPA screening and diagnosis [307].
The goals of ABPA treatment are to control symptoms, prevent or treat pulmonary exacerbations, and halt the progression of the disease to its final stage. While minimising exposure to fungi by identifying personal risk factors is desirable, corticosteroids remain the mainstay of treatment once the inflammatory mechanisms are primed the management of ABPA. Antifungals play an important yet supplementary role in the management of ABPA [312]. The aim of antifungals is to reduce prolonged high-dose systemic corticosteroids and decrease the burden of fungal colonisation [309]. More recently, biological agents have been explored as a treatment option for underlying steroid-dependent severe asthma in ABPA. Omalizumab, which targets IgE, acts on the central core of the disease. In real-life studies, it has shown the ability to improve exacerbations, lung function, and decrease steroid use. In addition, IL-4/IL-13R and IL-5 target molecules appeared to specifically modify the radiological impact of the disease [313].
Eosinophilic lung diseases include: 1) acute and chronic eosinophilic pneumonias (CEP) characterized by increased eosinophils in peripheral blood and bronchoalveolar lavage (BAL) fluid and/or by eosinophilic infiltration of lung parenchyma; 2) other lung diseases characterized by eosinophilic infiltration, like Lӧffler syndrome, ABPA, drug-induced EP, and EGPA—Churg-Strauss syndrome [314].
Eosinophilic lung disease may be acute or chronic, idiopathic or due to known causes, and is usually severe and does not resolve without treatment. High-resolution computed tomography (HRCT) is the gold standard imaging procedure for eosinophilic lung diseases diagnosis. Lung biopsy may be useful to characterize the histopathology of pulmonary eosinophilic infiltrates and define the diagnosis.
Acute eosinophilic pneumonia (AEP) is a rare disease of unknown cause mainly affecting men of 20–40 years of age, smokers, and without a history of atopy [285, 315]. AEP may be triggered by a hypersensitivity reaction to an inhaled antigen, including tobacco and environmental contaminants. AEP may also be associated with several drugs and parasitic, fungal, and viral infections [316–319].
AEP is characterized by an acute onset with fever, bilateral diffuse infiltrates, PaO2 ≤ 60 mmHg or oxygen saturation ≤ 90% on room air, BAL eosinophilia (≥ 25%) or EP at lung biopsy [320]. HRCT reveals ground glass opacities, interlobular septal thickening, pleural effusion, thickening of broncho-vascular bundles, air space consolidations, and centro-lobular nodules [321, 322]. Blood eosinophilia (> 500/mm3) may develop later in the course of the disease [315]. Pulmonary function tests may reveal restriction and a reduced diffusing capacity for carbon monoxide (DLCO), which resolves after therapy [323]. Usually, a lung biopsy is not necessary for diagnosis, but when performed, it reveals interstitial edema and eosinophilic infiltration of bronchial walls, interstitium, and/or alveolar spaces.
AEP typically responds to corticosteroids within 48 hours and resolves within one month. High intravenous doses (500 mg/day methylprednisolone) are required for severe respiratory failure, which are switched to oral prednisone (40–60 mg/day) and continued for 2–4 weeks, followed by tapering [315, 324]. The recurrence of AEP is uncommon.
CEP is a rare disease of uncertain aetiology accounting for 1–3% of interstitial lung diseases characterized by marked tissue and peripheral blood eosinophilia [320, 325]. CEP is more frequent in women of 30–45 years of age, non-smokers, and with a history of adult-onset asthma and/or AR. CEP patients develop progressive respiratory symptoms over 2–4 weeks, low-grade fever, night sweats, malaise, and unintentional weight loss.
HRCT findings reveal dense and patchy areas of consolidation and ground glass affecting two-thirds of the mid to upper lung fields bilaterally. Less common manifestations include reverse halo (atoll) sign, septal thickening, nodular infiltrates, mediastinal adenopathy, bronchial wall thickening, and pleural effusion [326]. Peripheral blood eosinophilia (> 1,000/mm3) and BAL eosinophilia (> 40%) occur in most patients. Erythrocyte sedimentation rate and C-reactive protein are elevated. Serum IgE levels may be elevated in about half of the patients [327]. Pulmonary function testing may reveal obstruction, restriction, or normal physiology. Symptoms, imaging, laboratory findings, and response to steroid treatment are often sufficient for the diagnosis, avoiding lung biopsy [320, 328]. When a biopsy is performed, histopathological findings show large numbers of eosinophils infiltrating alveolar spaces and interalveolar septa, accompanied by macrophages and lymphocytes. Clusters of eosinophils can form eosinophilic micro-abscesses.
Corticosteroids are the choice therapy. Patients respond dramatically; however, long-term low-dose oral steroid therapy (0.5 mg/kg/day for about 4–6 weeks) is required to prevent relapse and to obtain full remission [329, 330]. Recently, biological agents, such as the anti-IgE antibody (omalizumab), the anti-IL-5 antibody (mepolizumab), and the anti-IL-5 receptor antibody (benralizumab), could be alternative CEP steroid-sparing treatments [331].
Lӧffler syndrome is characterized by transient pulmonary infiltrates, absent or mild respiratory symptoms, and peripheral blood eosinophilia. HRCT shows migratory, non-segmental unilateral or bilateral air space opacifications with peripheral distribution. Pleural effusions and lymphadenopathy are not present. Löffler-like syndrome may occur after helminthic infections with Ascaris lumbricoides and Anchilostoma duodenale. Löffler syndrome often spontaneously recovers within one month [332, 333].
Drug-induced EP represents an important subset of eosinophilic pulmonary diseases with infiltrates causing severe respiratory symptoms. Numerous drugs are associated with EP, including antibiotics, anti-inflammatory non-steroidal drugs, and cardiac anti-arrhythmic drugs such as amiodarone. Radiotherapy is also implicated [334–339]. Diagnosis is suspected if there is a temporal relationship between drug intake and symptom onset. Generally, patients demonstrate a good response to steroid treatment and drug cessation.
HESs are defined as a wide spectrum of diseases characterised by blood and/or tissue eosinophilia. These syndromes can present with a variety of clinical manifestations and cause hypereosinophilia (HE)-associated organ damage. Over the last few decades, the most difficult aspect of HES classification has been distinguishing between known causes of HE, such as parasitic, allergic, drug-induced, or malignant forms, and idiopathic forms. The latter are characterised by confirmed blood eosinophil levels of > 1,500/mm3 that have been sustained or persistent for a period of 2–4 weeks and are accompanied by eosinophil-mediated organ damage or dysfunction. However, the discovery of haematological variants of HESs characterised by identifiable molecular clonal mechanisms, as well as the description of organ-specific eosinophilic disorders such as CEP and eosinophilic gastrointestinal disorders, which are not necessarily accompanied by high BEC levels, has prompted a change in the practical approach to patients with hypereosinophilia [254].
Following an evaluation of the secondary causes of eosinophilia (reactive HESR), the World Health Organization (WHO) and the International Consensus Classification recommend a diagnostic work-up for primary eosinophilias (HESN). Myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusion (MLN-eo-TK) are characterised by rearrangement of the PDGFRA, PDGFRB or other genes (e.g., FGFR1 or PCM1-JAK2). Diagnosis is confirmed by fluorescence in situ hybridization (FISH) or reverse transcription polymerase chain reaction (RT-PCR). The underlying myeloid or stem cell neoplasm, including chronic eosinophilic leukaemia (CEL), can be identified using the morphological, immunological, and histomorphological criteria provided by the WHO and the International Cooperative Working Group (ICOG-EO) [340]. The lymphocytic variant of HES (L-HES) is now included among the HESR, which are characterised by T-cells that produce IL-5 and have an abnormal immunophenotype and clonal T-cell receptor gene rearrangement. A diagnosis of idiopathic HES (HES-i) can be made once all primary and secondary causes of HES have been excluded [341].
HES is a rare disease that may present with non-specific constitutional symptoms and signs of eosinophil-induced organ damage. Bone marrow infiltration may be indicated by neutrophilia, basophilia, or myeloid immaturity with varying degrees of dysplasia or anaemia. Sustained eosinophilia may affect all organ systems. The skin, lungs, and gastrointestinal tract are the organs most commonly affected by HE infiltration. Cardiac involvement may cause progressive heart failure and be complicated by endocardial damage, mural thrombotic thrombi evolving to the fibrotic stage, and restrictive cardiomyopathy. Neurological manifestations of the central nervous system or peripheral neuropathy, as well as ocular damage, are not uncommon. When the BECs are below 1,500 cells/μL but eosinophils are the convincing primary mechanism of tissue or organ damage, the terms “tissue-restricted HES” or “organ-restricted HES” have been proposed. In HES, HE-induced organ dysfunction/damage derives from a dysimmunity process in which the Th2 inflammatory cascade overlaps with eosinophil-mediated impairment of cytotoxic and coagulation homeostasis. Demonstrating tissue infiltration by massive eosinophils or the extracellular deposition of eosinophil granule proteins histologically is not always feasible, with different cutoffs applicable depending on the organ under investigation [342].
The therapy for HES depends on the organ involvement and the underlying disease [341, 343], which impacts the risk stratification. Concurrent myeloproliferative diseases such as CEL and cardiac disease are predictive of a worse outcome, as are vascular, infective, or thrombo-embolic complications. In HESN, therapy is dictated by molecular markers. For PDGFRA/B-rearranged eosinophilia-positive myeloid neoplasms, imatinib is the first-line treatment, leading to complete haematological remission, lasting in a minority of patients after discontinuation. For other MLN-EO-TK forms of HES at risk of an aggressive course, the combination of tyrosine kinase or JAK inhibitors with hematopoietic stem cell transplantation is recommended. For idiopathic adult lymphocytic variants of HES, the treatment approach is still to use steroids as the first-line option at a high dose in cases of severe, life-threatening organ involvement, with immunosuppressive agents reserved as second-line options. The recent availability of anti-IL-5 targeted treatment has paved the way for a customised treatment for eosinophilic disorders. Mepolizumab was approved in 2020 for HES-i after showing a significant steroid-sparing effect and a significant reduction in flare-onset in clinical trials [344].
Eosinophilic myocarditis (EM) is an inflammatory heart disease whose clinical presentation can be variable and complex, with a significant mortality rate [345]. Eosinophilic inflammatory infiltration is the characteristic histological feature and the target of treatment. Inflammatory myocardial cell damage is secondary to eosinophilic infiltration, often image of peripheral eosinophilia. Intramyocardial eosinophilic infiltration can range from mild to severe. Notably, the most common causes of EM are allergic reactions/hypersensitivity (34% of patients), EGPA (about 13%), early giant cell myocarditis, idiopathic HES (about 8%). It should also be emphasized that EM can also manifest itself during myeloproliferative disorders, infections also including parasitosis and malignancy [346, 347]. Moreover, it should be remembered that chemotherapy is not free from organ complications. Indeed, chemotherapy of acute myeloid leukaemia (AML) can also induce the onset of severe eosinophilia and consequently of EM among the possible cardiac complications [346]. Lipof et al. [348] described a case of AML with a pathogenic mutation involving plant homeodomain finger 6 (PHF6) and eosinophilia who developed EM. Furthermore, EM may be part of the visceral manifestations of chronic immune-mediated inflammatory diseases, including systemic lupus erythematosus (SLE), antiphospholipid syndrome (APS), systemic sclerosis (SSc), sarcoidosis, dermatopolymyositis (DPM), chronic IBD, EGPA, and other vasculitis [349]. It should be noted that giant cell myocarditis and necrotizing EM represent an autoimmune process. On the other hand, the borderline between EM and autoimmune myocarditis is extremely thin since it should never be forgotten that in addition to the intramyocardial eosinophilic infiltrate, it is now established that many heterogeneous factors play a role in the onset and maintenance of the inflammatory process. Among these, viral infections, HLA, gender, exposure to cryptic antigens, mimicry, and a deficit in thymic training/induction of Treg cells can certainly play a triggering role. Once the inflammatory process has been triggered, various elements of the immune response maintain the intramyocardial inflammation with specific immunopathogenic characteristics. Th17 cells favour the chronicization of the inflammatory process, as in any chronic immune-mediated inflammatory diseases [318, 350] that can clinically evolve into dilated cardiomyopathy. Chronicity is supported by fibroblasts, which in turn maintain inflammation through specific cytokines and consequently intramyocardial fibrosis [351–353]. Furthermore, eosinophils also support microvascular damage, resulting in activation of the coagulation process, and promote and maintain autoimmune activation [354, 355]. Notably, Barin et al. [356] demonstrated the important role of IFN-γ and IL-17A in the inflammation of EM, so much so that IFNγ−/−IL17A−/− mouse models developed rapidly fatal EM. There seems to be a genetic predisposition in some forms of EM. In particular, the HLA-DRB1*07 and HLA-DRB4 gene alleles have been shown to represent genetic risk factors for EGPA [357, 358]. In support of this, Wojnicz et al. [359] demonstrated a strong diffuse expression of HLA-A, -B, -C antigens localized exclusively on microvascular endothelium, other interstitial cells, and induced HLA-A, -B, -C antigens on cardiac myocytes in proximity to areas of inflammatory infiltration. Increased expression of HLA class II antigens found on endothelial cells and other interstitial cells. However, making a more realistic estimate of the incidence of EM does not appear simple, even if it would seem to be approximately 20% in hearts removed for transplantation and in 0.5% of unselected autopsies [360, 361]. However, the clinical and technical limitations of endomyocardial biopsy (EMB) and the fact that peripheral eosinophilia is not present in approximately 25% of patients still make EM difficult and underestimated. In confirmation of this, myocarditis still represents 6% of the causes of sudden cardiac death as reported in a Danish study [362]. There are no significant differences in the onset of EM between males and females, with an age at diagnosis of around 41 years if accompanied by histological examination and 46 years if only clinical and without histological examination [347].
However, EM, like any form of myocarditis, can manifest itself clinically in a paucisymptomatic form, or with severe manifestations including potentially lethal arrhythmias, cardiogenic shock, and even sudden death [363, 364]. It should be emphasized that approximately 40% of cases of acute myocarditis have a favourable evolution with spontaneous remission [365], while in the remaining patients, there is a chronicity supported by the anomalous immune response, with development of dilated cardiomyopathy and clinical progression towards heart failure [366, 367]. Patients usually complain of acute chest pain or tightness and dyspnoea, sometimes asthenia, nausea, and myalgia that can confuse the clinical picture. Laboratory tests show elevated levels of creatine kinase MB and troponin. In patients with EM during EGPA, in addition to bronchial asthma attacks, chronic rhinosinusitis, and nasal polyposis, the patient may develop myocarditis, pericarditis, coronary vasculitis, heart failure, and often peripheral neuropathy [368]. The prognosis is clear and will depend on the multiorgan involvement and the consequent damage. In suspected EGPA, ANCA should be tested, although these are generally present in ANCA-related nephritis, but often absent in cardiac involvement [369, 370]. In cases where there is an absolute eosinophil count greater than 1.5 × 109/L for more than six months, the possibility of being faced with an HES must be taken into consideration. Therefore, it will be necessary to investigate the involvement of other viscera such as the bone marrow and the nervous system in addition to the heart. It should be remembered that HES can be idiopathic, i.e., without a well-defined cause, or secondary in most cases associated with myelo- or lymphoproliferative haematological diseases [354]. In patients with HES, in many cases, EM is paucisymptomatic/asymptomatic, while in the forms of chronic myocarditis, it evolves towards cardiac failure with dyspnoea and oedema of the legs. It is hoped that in the face of severe peripheral hypereosinophilia, the presence of skin lesions/rash and itching is sought and leads to the suspicion of HES [371, 372]. It should be noted that patients who develop EM do not always have peripheral eosinophilia and that it may appear later than the onset of EM [353]. There are no elements suggestive of myocarditis in the standard 12-lead electrocardiogram (ECG), while echocardiography can help exclude other possible causes of acute heart failure, including cardiomyopathies, genetic heart disease, and valvular heart disease, and the presence of associated pericardial effusion [373]. Cardiovascular magnetic resonance imaging (CMR) represents the gold standard diagnostic imaging for the confirmation of myocarditis. The Lake criteria, defined in 2009, allow to confirm the diagnosis of myocarditis with CMR. In particular, hyperemia, intracellular and interstitial oedema, necrosis, and fibrosis are considered specific markers, and are identified with T1 and T2 images, and with early and late acquisitions after infusion of gadolinium [374]. Notably, the acute phase is characterized by oedema that is hyperintense on T2 sequences. The chronic phase evolves into fibrosis or necrosis, with a patchy subendocardial distribution, highlighted on sequences with late gadolinium enhancement [375]. This allows us to differentiate chronic myocarditis fibrosis from post-ischemic fibrosis, which, on the contrary, has a transmural distribution along the territory of one or more coronary arteries [375]. In patients with EGPA-associated myocarditis, CMR, in addition to the typical features of acute/chronic inflammation, allows to confirm abnormalities of the coronary macro- and microcirculation and/or small vessel vasculitis, which occur during systemic vasculitis and also in subclinical cases [376]. Cardiac involvement in EGPA is extremely variable, so much so that three variants are recognized on the basis of cardiac enzymes, CMR, and EMB, namely EGPA-EM, chronic inflammatory myocarditis/cardiomyopathy, and EGPA-control [377]. However, EMB is the invasive diagnostic gold standard that allows confirmation of the presence of myocardial inflammatory infiltrates, non-ischemic degeneration/necrosis, and thus, the confirmation of myocarditis. The Dallas criteria include the histological elements [378]. It should be noted and underlined that the absolute contraindications for performing EMB are represented by acute myocardial infarction, left ventricular thrombosis, and ventricular aneurysm. Furthermore, the importance of EBM is also evident from the fact that it allows to define myocytolysis, which is characterized by severe interstitial and perivascular eosinophilia. The severity of myocytolysis depends on the degranulation of eosinophils and the action of the specific enzymes released, up to the most serious picture of necrotizing EM [379]. EM therapy involves both a pharmacological and a non-pharmacological approach. Physical activity should be limited in acute EM and for 6 months thereafter. The pharmacological approach primarily involves the administration of drugs that reduce myocardial remodelling such as low-dose beta-blockers, angiotensin converting enzyme inhibitors/angiotensin receptor blockers, and aldosterone receptor antagonists. The treatment of haemorrhagic EM is based on the administration of immunosuppressive agents, including high-dose corticosteroids, in order to limit the progression of cardiac damage, towards thrombotic necrosis and fibrosis. Corticosteroids have a potent anti-inflammatory action. The use of corticosteroids is the first-line therapy in EM associated with EGPA, HES, and EM hypersensitivity [353, 375]. There are still no definitive indications on the initial dosage of corticosteroids. However, good clinical practice suggests adjusting the initial dosage and duration of treatment to the severity of EM, and, therefore, to the presence and severity of left ventricular dysfunction, to troponin I levels, to whether EM was associated with EGPA, HES and, finally, to the evolution of inflammation, given their ability to induce rapid improvement also in cardiac kinetics. Useful in this regard during the follow-up, the outcome of any control biopsy and/or CMR [380]. The use of DMARDs, in particular cyclophosphamide, methotrexate (especially EM associated with EGPA), azathioprine, hydroxyurea, or interferon-α (especially in cases of steroid-refractory HES), in association with corticosteroids, represents the most indicated therapeutic armamentarium in the advanced stages of EM [381]. Among biological drugs, mepolizumab, a monoclonal antibody that acts by blocking IL-5, is gaining increasing consensus and represents an effective choice in EM associated with EGPA and HES, even initially in association with corticosteroid therapy, with the aim of reducing corticosteroid doses and preventing side effects [375]. Benralizumab, a humanized antibody against the IL-5 receptor, is proving effective in reducing eosinophilia in peripheral blood and tissues [381]. The administration of anticoagulant drugs should also be considered as prophylaxis in the acute phase of EM to prevent the formation of mural and intravascular thrombi, in subjects with persistent eosinophils that favor thrombotic and fibrotic evolution, determining the evolution in Loeffler’s endomyocarditis [382]. Finally, albendazole is indicated in the therapy of EM associated with helminthic infections [383].
The main mechanisms of pathogenesis and myocardial damage in EM are reported in Figure 2.
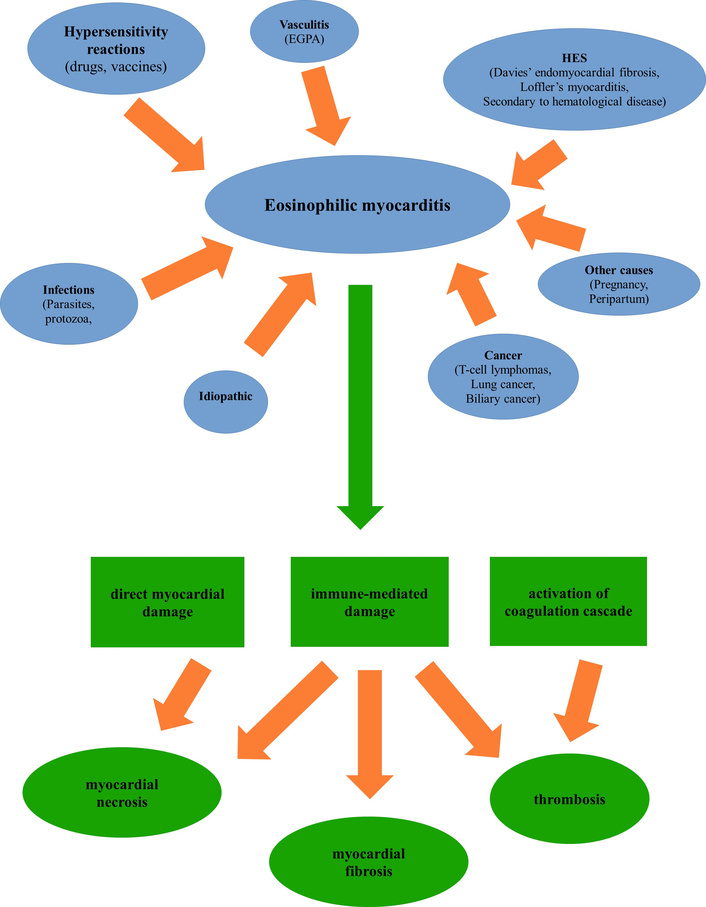
The main mechanisms of pathogenesis and myocardial damage in EM. EGPA: eosinophilic granulomatosis with polyangiitis; EM: eosinophilic myocarditis; HES: hypereosinophilic syndrome.
EM is a rare, potentially life-threatening inflammatory disorder of the myocardium characterized by dense infiltration of eosinophils into the cardiac tissue. Clinical manifestations are variable, ranging from mild dyspnea and chest discomfort to fulminant presentations with acute heart failure, severe arrhythmias, or sudden cardiac death. Peripheral eosinophilia is frequently observed, though not universally present, and the clinical course may vary from self-limiting to rapidly progressive disease. Reported aetiologies are heterogeneous and include drug-induced hypersensitivity reactions, EGPA, HES, infections, and idiopathic cases [383–385].
Pathophysiological models proposed by the American Heart Association describe EM as a progressive disease: an initial necrotic phase due to eosinophil-mediated tissue injury, a thrombotic phase related to mural thrombosis, and ultimately a fibrotic phase characterized by extensive scarring and remodelling, potentially leading to restrictive cardiomyopathy and chronic heart failure. Eosinophil-mediated damage is largely attributable to the release of cytotoxic proteins and inflammatory mediators, including MBP, ECP, and IL-4. IL-4 has been identified as a driver of the transition from acute myocarditis to inflammatory dilated cardiomyopathy, establishing a pathogenic bridge between acute inflammation and chronic structural disease [386, 387].
Diagnosis remains challenging. EMB continues to be the gold standard for definitive diagnosis, but it is invasive and not always feasible. Increasingly, CMR is used for non-invasive evaluation, offering diagnostic and prognostic value, particularly in the detection of myocardial oedema, fibrosis, and thrombus formation [384, 385]. Therapeutic approaches are primarily based on high-dose glucocorticoids, often leading to rapid improvement in both symptoms and imaging findings. In refractory or fulminant cases, additional immunosuppressive agents may be employed, as endorsed by the American Heart Association guidelines [388].
EM may coexist with or evolve in the context of systemic eosinophilic disorders, and pulmonary involvement is frequently observed. EP represents a related but distinct entity, characterized by pulmonary eosinophilic infiltration and variable systemic manifestations. While EM and EP share overlapping immune-mediated mechanisms, they represent separate clinical syndromes with differing prognostic implications [331, 387, 389].
The psychological and psychopathological burden of EM and EP remains underexplored. To date, disease-specific literature has primarily concentrated on cardiac, pulmonary, and systemic features, with only limited attention paid to psychiatric comorbidities. The psychiatric literature mentions depression and anxiety as common comorbidities in chronic and life-threatening cardiopulmonary diseases, but specific data on EM and EP cohorts are lacking [331, 384, 385, 389]. Consequently, psychiatric symptoms are often discussed only within the broader context of chronic illness, immune dysregulation, and psychosomatic vulnerability, rather than as direct manifestations of eosinophilic disease.
Insights from psychosomatic research in heart failure may be particularly relevant. Altamura et al. [390] reported that psychosomatic syndromes—particularly persistent somatization and demoralization—were present in more than half of ischemic heart failure patients. Notably, elevated levels of IL-6 were significantly associated with psychosomatic psychopathology, even after controlling for disease severity, lifestyle, and demographic confounders. Interestingly, IL-6 correlated with psychosomatic syndromes but not with depressive symptom scores, suggesting a psychoneuroimmune pathway distinct from classical depression. This aligns with evidence that IL-6 and other pro-inflammatory cytokines can activate the indoleamine-2,3-dioxygenase (IDO) pathway, leading to tryptophan depletion, serotonergic dysregulation, and accumulation of neurotoxic tryptophan catabolites (TRYCATs). These processes have been implicated in somatization, fatigue, irritability, and related psychosomatic phenomena.
Large-scale genetic studies further support a link between immune dysregulation and psychopathology. Genome-wide analyses have shown shared liability across immune-mediated disorders and psychiatric conditions, particularly depression and anxiety, underscoring the role of immune-inflammatory pathways as common biological substrates [391]. Such findings suggest that in EM and EP, where immune dysregulation is central to pathogenesis, psychiatric comorbidities may extend beyond depression and anxiety to include a broader spectrum of psychosomatic syndromes.
In conclusion, no studies in the current medical literature specifically characterize the prevalence or nature of psychiatric comorbidities in EM or EP cohorts. The psychiatric burden in these patients is likely like that seen in other chronic or severe cardiac and pulmonary diseases, with depression and anxiety being the most frequently observed comorbidities [391]. However, there is no evidence for disease-specific psychopathological syndromes directly attributable to EM or EP. The emerging evidence from heart failure and other immune-mediated cardiovascular conditions suggests that psychosomatic syndromes—such as somatization, demoralization, and vital exhaustion—may be clinically relevant but underrecognized in this population.
This highlights an important knowledge gap. Systematic studies are urgently needed to:
Define the prevalence and profile of psychiatric and psychosomatic syndromes in EM and EP.
Clarify the biological mechanisms linking immune dysregulation to psychopathology, with particular focus on cytokine-mediated pathways.
Evaluate the prognostic impact of psychiatric comorbidity on clinical outcomes such as treatment adherence, quality of life, and survival.
Addressing these questions would not only expand our understanding of EM and EP as systemic and multidimensional disorders but also improve holistic patient care. Integrating psychiatric and psychosomatic assessment into the management of eosinophilic cardiopulmonary diseases may represent a crucial step toward optimizing outcomes in these rare but severe conditions.
Eosinophilic gastrointestinal diseases (EGIDs) comprise a spectrum of chronic, immune-mediated disorders of the gastrointestinal tract that are defined by dense tissue eosinophilia in the absence of secondary causes. The spectrum includes EoE and the less common non-esophageal forms such as eosinophilic gastritis (EoG), enteritis, and colitis. While these conditions share key pathogenic mechanisms such as Th2-driven inflammation and epithelial barrier disruption, they differ in their anatomical location, clinical manifestations, histologic thresholds, and treatment strategies. In addition, eosinophilia is a hallmark of many parasitic infections, particularly those involving tissue-invasive helminths, where it may reflect both protective immunity and tissue-damaging inflammation. This chapter reviews the current understanding of EoE, non-esophageal EGIDs, and eosinophil-associated parasitic diseases, with emphasis on epidemiology, pathogenesis, diagnosis, and treatment, and highlights both shared mechanisms and distinct features across these conditions.
EoE is a chronic, immune-mediated inflammatory disease of the esophagus characterized by symptoms of esophageal dysfunction and dense eosinophilic infiltration [392]. Once considered rare, EoE is now a leading cause of dysphagia and food impaction in children and adults in developed regions, with a prevalence of 1–5 per 1,000 individuals [393]. It is more common in males and often coexists with allergic diseases such as asthma, eczema, AR, and food allergy [393].
EoE pathogenesis is driven by a Th2-skewed immune response triggered by food or environmental allergens [394]. Damaged epithelial cells release IL-33, TSLP, and IL-25, which activate dendritic cells and ILC2s, promoting IL-5 and IL-13 production by Th2 cells. IL-5 drives eosinophil recruitment, while IL-13 impairs epithelial barrier function and induces eotaxin-3, enhancing eosinophil chemotaxis [395]. Recent findings indicate that TSLP and IL-33 contribute independently to EoE pathogenesis, with TSLP promoting IL-13 secretion via mast cell activation and regulating genes involved in epithelial barrier function and remodelling, while IL-33 primarily drives eosinophilic inflammation without significantly affecting tissue remodelling [396].
Barrier dysfunction is a hallmark of EoE. Reduced desmoglein-1 expression, altered tight junction proteins, and impaired epithelial differentiation weaken barrier integrity [397]. Environmental exposures such as detergents can directly impair epithelial barrier function, induce IL-33 production, and promote eosinophilic inflammation [398]. Eosinophils themselves may initially exert barrier-protective roles, but persistent T2 inflammation drives their profibrotic activities [399]. In addition, B-cell-derived mediators and other immune-epithelial interactions contribute to barrier disruption [400]. Transcriptomic and proteomic analyses show that periostin upregulation and broad changes in epithelial differentiation gene expression further compromise the barrier, enabling allergen penetration and perpetuating inflammation [397, 401]. Mast cells and basophils, often increased in EoE mucosa, release mediators such as IL-9 and tryptase that contribute to smooth muscle dysfunction and fibrotic remodelling [402, 403]. Disruption of the epithelial barrier not only facilitates allergen exposure but also alters epithelial immune cell crosstalk, shaping the chronic inflammatory niche.
Beyond Th2 inflammation, cow’s milk-specific CD4+ T cells with a T follicular helper (TFH)-like phenotype, expressing CXCR5, IL21, and IL10, are enriched in active disease and may influence B cell-driven responses [404]. B cells, particularly the CD49b+CD73+ subset enriched among IgG4-switched B cells, are elevated in EoE tissue and blood, and secrete angiogenic mediators such as VEGFA, FGF2, and CYR61, potentially driving tissue remodelling.
Unlike food allergy, EoE does not appear to be strongly dependent on allergen-sIgE, as illustrated by the poor predictive value of SPT and no clinical improvement in response to anti-IgE therapy [405–407]. Elevated levels of systemic and local food-specific IgG4 antibodies have been reported in EoE [408–410], and the presence of immune complexes consisting of IgG4 and food allergens in the esophagus of active EoE suggests a potential involvement of these antibodies in EoE pathogenesis [411]. However, circulating food-specific antibodies of other IgG subclasses and isotypes beyond IgG4 are also increased in EoE, suggesting a broad humoral food-specific response in EoE [412].
Recent findings show that circulating food allergen-specific antibody profiles in EoE extend beyond IgG4, with elevated IgG and IgA subclasses to cow’s milk, wheat, and egg allergens, and differences between active and inactive disease [412]. These patterns suggest a complex, localized humoral response, with antibody production likely restricted to the inflamed esophagus.
Genetic risk factors, including variants in TSLP, CAPN14, and FLG, contribute to epithelial barrier defects and disease susceptibility [413, 414]. Histologically, EoE is characterized by an eosinophil count of at least 15 per HPF, basal cell hyperplasia, expansion of intercellular spaces, and, in chronic stages, fibrosis within the lamina propria [394]. If left untreated, persistent inflammation can result in stiffening of the esophagus and the development of strictures [394].
Clinical presentation varies by age: Children often have vomiting, feeding difficulties, and poor growth, whereas adolescents and adults most commonly present with dysphagia and food impaction [415]. Current diagnostic criteria, established through international consensus during the AGREE conference and endorsed by major societies, consist of three components: (1) symptoms of esophageal dysfunction; (2) a peak eosinophil count of ≥ 15 eos/hpf on esophageal biopsy; and (3) exclusion of other conditions that could account for esophageal eosinophilia [1, 24, 25, 392, 416, 417]. A high index of suspicion is warranted in patients with coexisting atopic conditions such as asthma, eczema, AR, or food allergies, or a family history of EoE. While routine screening is not currently recommended, emerging clinical prediction tools and severity indices like the Index of Severity in EoE (I-SEE) may support future diagnostic and disease monitoring strategies [418].
EoE exhibits heterogeneity in both clinical and histological presentation. The inflammatory phenotype is more prevalent in younger patients and early-stage disease, while chronic cases often develop fibrostenotic features such as narrowing and scarring of the esophagus. Some individuals display characteristics of both types. Recognizing these variations is important for determining appropriate treatment and predicting outcomes. Recent transcriptomic analyses have proposed the existence of molecular endotypes of EoE, each associated with distinct immunological and therapeutic profiles [419–421]. Variants of esophagitis that mimic EoE, such as EoE-like, nonspecific, lymphocytic, and mast cell-predominant esophagitis, share molecular features with EoE but do not always exhibit prominent eosinophil infiltration [422, 423]. These forms appear to exist along a spectrum with classical EoE, with potential transitions over time and progression to the fully developed eosinophil-rich phenotype.
Management of EoE focuses on reducing inflammation, alleviating symptoms, preventing complications like strictures, and achieving long-term remission. Treatment approaches typically include dietary modifications, medication, and, in some cases, endoscopic procedures. Dietary therapy is often effective and includes elemental diets composed of amino acid-based formulas, empiric elimination diets, and elimination based on allergy testing. Although elemental diets produce the highest rates of histologic remission, they are difficult to maintain due to poor taste and social restrictions. Empiric strategies like the six-food elimination diet (removing milk, wheat, egg, soy, nuts, and seafood) have proven successful in many patients [424]. Simplified versions like the four-food or step-up elimination diets have gained popularity for their improved adherence rates [425].
Topical corticosteroids remain the cornerstone of pharmacological treatment. Agents like swallowed fluticasone and viscous budesonide effectively reduce eosinophilic inflammation and improve symptoms in both pediatric and adult populations [426]. These medications are generally preferred over systemic corticosteroids because they minimize systemic side effects. Histologic improvement is achieved in roughly 50 to 80 percent of cases, though maintenance therapy is often necessary due to the likelihood of relapse following discontinuation [426]. Newer biological therapies have also emerged as promising options. For instance, dupilumab, an IL-4 receptor alpha antagonist, has demonstrated significant clinical efficacy and has received approval for treating EoE. Ongoing studies are exploring monoclonal antibodies targeting IL-13, TSLP, and Siglec-8 [406].
In patients with significant fibrotic changes or esophageal narrowing, endoscopic dilation may be required to relieve symptoms. While this intervention does not modify the inflammatory process, it can improve swallowing and enhance quality of life. When performed by experienced clinicians, the procedure is generally safe, though temporary chest discomfort and rare instances of perforation have been documented [427].
Long-term management involves periodic monitoring of clinical symptoms, histologic inflammation, and structural changes in the esophagus. Endoscopy with biopsy remains the gold standard for assessing disease activity and treatment response, though alternative, less invasive methods such as the esophageal string test and cytosponge are being studied [428, 429].
Despite advancements in understanding and managing EoE, the condition remains chronic and prone to relapse. Delayed diagnosis can lead to irreversible esophageal damage, underscoring the importance of early detection and timely intervention. The future of EoE care may lie in personalized medicine strategies that leverage molecular profiling to guide therapy. Effective management typically involves a multidisciplinary approach, bringing together gastroenterologists, allergists, dietitians, and pathologists for comprehensive care.
Non-esophageal EGIDs are chronic, inflammatory disorders of the gastrointestinal tract characterized by tissue eosinophilia in the absence of identifiable secondary causes [430]. According to the 2022 international consensus, this category includes EoG, eosinophilic enteritis (EoN), and eosinophilic colitis (EoC) [431]. These conditions may rarely occur in association with EoE [432]. Although epidemiological data remain limited, non-esophageal EGIDs appear less common than EoE in Western countries. For instance, in Israel, the estimated prevalence is approximately 39.54 per 100,000 persons for EoE compared with 11.89 per 100,000 for non-EoE EGIDs [433].
Non-esophageal EGIDs can be classified according to the depth of eosinophilic infiltration, which is closely related to clinical presentation. The mucosal subtype is associated with abdominal pain, vomiting, diarrhea, and gastrointestinal bleeding, whereas the muscular subtype is characterized by obstructive symptoms such as nausea and vomiting. The serosal subtype may manifest as eosinophilic abdominal ascites [434]. Muscular and serosal forms are often accompanied by mucosal eosinophilic infiltration, suggesting a centrifugal disease progression from the mucosa toward the deeper layers of the gastrointestinal wall [435].
The pathogenesis of non-EoE EGIDs remains incompletely understood but appears to involve a Th2-driven immune response. In EoG, studies have demonstrated overproduction of Th2 cytokines, such as IL-13, and chemokines including CCL26 (eotaxin-3), eotaxin-1, and IL-5, which collectively promote eosinophil recruitment and activation [436]. In infants with eosinophilic gastroenteritis, a simultaneous increase in TSLP and IL-33 has also been observed [437]. Genomic studies have revealed distinct molecular profiles for each subtype. In EoG, differential expression of CCL26, CLC, IL13RA2, BMP3, IL5, CDH26, CCL18, NPY, HPGDS, and SST has been reported compared with controls [438]. In EoC, 987 differentially expressed genes, including CCL11 and CLC, have been identified, with transcriptomic signatures distinct from those of IBD and other EGIDs, supporting EoC as a separate disease entity [439]. Similar transcriptomic alterations have been described in eosinophilic duodenitis (EoD), including increased transcription of genes involved in IL-4/IL-13 signalling, mast cell activation, and myeloid progenitor cell pathways [440].
Eosinophilic gastritis
EoG typically presents with symptoms of gastric dysfunction, including abdominal pain or cramping, bloating, vomiting, anorexia, weight loss, early satiety, hematemesis, heartburn, and dyspepsia [441]. Its prevalence in the United States has been estimated at approximately 6.3 cases per 100,000 persons [442]. Endoscopic findings most often involve erythema, raised lesions, erosions or ulcers, and mucosal granularity, particularly in the gastric antrum. Notably, up to 8% of patients with active histopathology may have normal endoscopic appearances [443]. Diagnostic histologic thresholds include ≥ 30 eos/hpf along with features such as eosinophilic glandulitis, eosinophils in the muscularis mucosa or submucosa, and lamina propria fibrosis or fibroplasia (Figure 3) [420, 443, 444].
Eosinophilic infiltration of the gastric mucosa (magnification for A and B was 100× and for C and D was 400×).
Eosinophilic enteritis
EoN occurs more frequently in individuals younger than 20 years [442] and is challenging to diagnose due to its non-specific symptoms, such as diarrhoea, abdominal pain, nausea, and vomiting. Endoscopic appearances include erythema, ulceration, nodularity, and mucosal friability [445]. Histologic evaluation may reveal villous atrophy, crypt hyperplasia, and epithelial degenerative or regenerative changes [403]. The proposed diagnostic thresholds are > 52 eos/hpf in the duodenum and > 56 eos/hpf in the ileum [446].
Eosinophilic colitis
EoC is the rarest of the non-esophageal EGIDs, with an estimated prevalence of 3.3 per 100,000 persons in the United States [442]. Clinical manifestations include diarrhoea, haematochezia, and tenesmus [441]. Endoscopic findings may consist of erythema, friability, congestion, and ulceration [420]. Histologic thresholds are ≥ 100 eos/hpf in the cecum and ascending colon, ≥ 84 eos/hpf in the transverse and descending colon, and ≥ 64 eos/hpf in the rectosigmoid colon, often accompanied by lymphoid aggregates and eosinophilic cryptitis [446].
There are currently no approved therapies for non-esophageal EGIDs. Glucocorticoids remain the most frequently used drugs, but the supporting evidence is limited, and standardized dosing and formulations are lacking [447–449]. To reduce systemic steroid exposure, topical budesonide may be used with some success [450]. Dietary interventions, including elemental and elimination diets, have been evaluated in EoG and EoN with encouraging results [447]. Biologic therapies targeting eosinophil-associated pathways are under investigation. In a clinical trial of benralizumab for EoG, histologic remission occurred in 77% of treated patients compared with 8% in the placebo group, although symptom improvement was not significant [451]. Lirentelimab (anti-Siglec-8) demonstrated an average 86% reduction in gastrointestinal eosinophil counts compared with a 9% increase with placebo in a phase 2 trial [452], and a subsequent phase 3 study (NCT04322604) confirmed greater histologic improvement in the active arm, but again without significant symptom benefit [453]. Dupilumab is currently being evaluated in a phase 2 trial for EoG in subjects aged 12–70 years (NCT03678545), and an additional trial is actively recruiting (NCT05831176).
Non-esophageal EGIDs represent a heterogeneous group of rare but increasingly recognized gastrointestinal disorders with distinct clinical, histologic, and molecular features. Although their prevalence is lower than that of EoE, they contribute substantially to gastrointestinal morbidity. Advances in molecular profiling have clarified key immunologic and transcriptomic pathways, highlighting differences between disease subtypes and distinguishing them from other inflammatory gastrointestinal conditions. These insights are refining diagnostic thresholds and opening the door to targeted treatments. Despite progress in understanding disease mechanisms, management remains challenging. Current therapies rely largely on glucocorticoids and dietary interventions, which may be effective but are often associated with limitations, including incomplete symptom control and the need for long-term maintenance strategies. Emerging biologic agents show promise in achieving histologic remission, but the persistent gap between histologic and symptomatic improvement underscores the complexity of these disorders. Future research should focus on identifying biomarkers that better predict treatment response, optimizing therapeutic regimens, and exploring combination approaches that address both inflammation and symptom burden. Through these advances, the goal will be to provide more effective, durable, and individualized care for patients with non-esophageal EGIDs.
Eosinophils are closely associated with helminth infections, participating in both host defence and disease pathogenesis. Their accumulation at sites of infection has been documented in many host species, with the magnitude of eosinophilia varying according to host factors, parasite species, and stage of infection [454, 455]. While often beneficial for parasite clearance, eosinophilia can also contribute to tissue injury and inflammatory disease.
Helminths with life cycles that include tissue migration such as Trichinella spiralis, Ascaris lumbricoides, filarial worms, and Schistosoma species, typically induce sustained eosinophilia in blood and tissues [455]. In contrast, parasites confined to the gut lumen, such as tapeworms and Trichuris trichiura, or in cystic stages, such as Echinococcus species, generally do not provoke persistent eosinophilia [456]. The variability in response reflects differences in parasite biology, host immune status, and the degree of tissue invasion.
Helminths are complex, multicellular organisms with lifecycles that expose the host to multiple antigenically distinct stages. Eosinophilia reflects both quantitative expansion and qualitative activation of eosinophils, enhancing their antiparasitic capacity [457]. In allergic diseases such as asthma, eosinophils are similarly activated within a Th2 inflammatory response [458].
Tissue-invasive parasites cause epithelial damage, releasing alarmin cytokines including IL-33, IL-25, and TSLP. These molecules initiate Th2 inflammation by activating dendritic cells, ILC2s, and Th2 lymphocytes, which secrete IL-5 and IL-13 [459, 460]. IL-5 drives eosinophil proliferation and recruitment, while IL-13 contributes to barrier remodelling. IL-33, in particular, is essential for worm expulsion but may also promote fibrosis in chronic disease [461].
Eosinophils release a range of cytotoxic granule proteins, including MBP-1 and MBP-2, EPX, ECP, and EDN, that damage parasites but can also injure host tissues [3, 17]. Their activity extends beyond antiparasitic effects to antimicrobial defence against bacteria, viruses, and protozoa. Glucocorticoids reduce eosinophilia partly by suppressing these toxic mediators. Eosinophils are now recognized as multifunctional cells with roles in tissue homeostasis, repair, and immune regulation [462]. Eosinophil-deficient mouse models have revealed their involvement in modulating both innate and adaptive immunity during parasitic and allergic diseases [463, 464].
CLCs, composed of galectin-10, are a hallmark of eosinophil death and can persist in tissues for months [465, 466]. Galectin-10 contributes to eosinophil differentiation and piecemeal degranulation, enabling extracellular effector functions without damaging the cell itself [3, 467]. Genetic variants in the CLC gene have been linked to AR risk [468, 469], and mixed eosinophil-mast cell infiltrates rich in CLCs are associated with worse prognosis and higher relapse rates in rhinitis [470].
IL-5 remains the central driver of eosinophil production, bone marrow release, and activation. In helminth infections, both IL-4-dependent and IL-4-independent pathways contribute to IL-5 production [470]. IL-10, produced by multiple immune and epithelial cells, serves as a potent anti-inflammatory regulator, particularly in chronic T2 inflammation [471]. These cytokine pathways have therapeutic significance. Biologics targeting IL-5 or its receptor may inadvertently alter immune control of helminths in endemic regions [472, 473].
Eosinophils not only participate in parasite clearance but also modulate autoimmune and inflammatory disorders, highlighting their broad immunoregulatory potential. They maintain tissue and metabolic homeostasis under physiological conditions and contribute to damage, repair, and fibrosis in disease [473].
The disruption of host-parasite equilibrium, such as after anthelmintic therapy, can lead to abrupt antigen release and heightened eosinophilic inflammation. In one case, a patient with chronic urticaria and ascariasis developed severe urticaria and angioedema shortly after treatment, requiring epinephrine [474]. Corticosteroid use in patients with unrecognized Strongyloides stercoralis infection can precipitate hyperinfection syndrome, with larvae disseminating to multiple organs and frequently leading to Gram-negative sepsis [475, 476]. This can occur even after short steroid courses for asthma or COPD.
Eosinophilia is typically most marked early in infection or during tissue invasion [395]. In institutionalized children with generally low parasite burdens, ascariasis did not cause significant eosinophilia or IgE elevation, possibly due to chronic low-level infection. Following treatment, eosinophil counts normalized within about seven weeks [477]. Experimental high-dose infection with Ascaris suum has produced pulmonary infiltrates, elevated total IgE, marked eosinophilia, and asthma. Notably, individuals mounting the strongest immune responses had the lowest worm burdens, suggesting a protective effect [478]. Trichuris trichiura infection, confined to the colon and lacking a tissue-invasive stage, primarily causes pathology in heavy infections. Infected children showed higher eosinophil counts than controls, but there was no correlation between parasite load, IgE levels, and eosinophilia [479].
Helminth infections remain highly prevalent in tropical and subtropical areas. Both allergen- and helminth-driven immune responses involve eosinophilia, elevated IgE, and Th2 inflammation [480]. Chronic helminth infection can induce regulatory networks that suppress allergic inflammation [481, 482]. Some epidemiological studies suggest helminths may protect against allergic diseases, while others report increased risk depending on the parasite. Ascaris lumbricoides, Strongyloides stercoralis, and Toxocara species have been linked to asthma or atopy [483–487] and Clonorchis sinensis to food allergy [488], whereas hookworm infection may confer protection [489, 490]. Variability in findings likely reflects differences in endemicity, infection intensity, lifecycle, diagnostic accuracy, and allergy definitions. Importantly, eosinophilia and high IgE are not invariably present in either condition.
Biologics targeting IL-5 are effective in eosinophilic asthma, but other pathways, including IL-13, TSLP, IL-33, and the IL-3/5/GM-CSF axis, also support eosinophilia [491]. Siglec-8-targeted antibodies such as lirentelimab, induce apoptosis of cytokine-primed eosinophils, and blockade of TSLP or IL-33 reduces eosinophilia in asthma [491, 492]. While biologics could theoretically increase susceptibility to helminths, evidence is limited. FDA adverse event data from 2004 to 2021 show only 79 parasitic infection reports among 175,888 cases involving omalizumab, mepolizumab, reslizumab, dupilumab, and benralizumab, although the association was statistically disproportionate [492]. This observation warrants further investigation but does not confirm causality.
Helminth-associated eosinophilia is a complex biological process in which the same immune mechanisms can contribute both to protection against infection and to the development of pathology. Sustained eosinophilia is most often seen in tissue-invasive infections, whereas luminal or cystic parasites rarely provoke a prolonged response. The elimination of parasites, particularly through anthelmintic therapy, may in some cases, trigger allergic exacerbations as a result of sudden antigen release. The relationship between helminth infection and allergic disease is influenced by many variables, including parasite species, infection burden, host immune regulation, and environmental exposures. Although biologic therapies that target T2 inflammatory pathways are generally safe, their use in areas where helminthiasis is common should be approached with caution. Ongoing research is essential to deepen our understanding of eosinophil biology, refine therapeutic strategies, and clarify the interplay between parasitic infection, allergy, and immune regulation.
EoE, non-esophageal EGIDs, and eosinophil-associated parasitic infections illustrate the diverse clinical contexts in which tissue eosinophilia can occur. Despite differences in presentation and disease course, these conditions share important immunopathogenic processes, including Th2 cytokine signalling, eosinophil recruitment via chemokines, and epithelial barrier impairment, all of which contribute to inflammation and tissue remodelling. Molecular and transcriptomic advances are refining diagnostic criteria and guiding the development of targeted therapies. In EoE, biologic treatment targeting IL-4Rα with dupilumab is now approved and offers a significant step forward in disease management. Other biologics directed against IL-5, IL-13, TSLP, and Siglec-8 are in various stages of clinical development for EGIDs. Sustained symptom control, however, remains a challenge, and in helminth-endemic settings, eosinophil-targeted therapies require careful consideration to avoid compromising host defence against parasites. A comprehensive understanding of the similarities and differences among these disorders is essential for developing tailored management strategies that address both disease activity and patient quality of life.
Chronic spontaneous urticaria (CSU) is traditionally characterized as a mast cell-driven disorder marked by the recurrent appearance of transient wheals, angioedema, or both, persisting for six weeks or more [493]. The clinical manifestations of CSU are generally attributed to inappropriate activation of skin mast cells and subsequent release of histamine and other vasoactive mediators. While mast cells remain central to disease pathogenesis, growing evidence indicates that additional immune cell subsets, including basophils, neutrophils, and particularly eosinophils, are also key contributors to the complex immunopathology of CSU [494]. The upcoming 2025/2026 international urticaria guideline places greater emphasis on these additional players, reflecting a shift toward a broader view of disease mechanisms. In particular, eosinophils are increasingly recognized not as passive bystanders but as active participants in disease activity, autoimmunity, and therapeutic responsiveness. This section explores their diverse roles, from tissue infiltration and mediator release to immune crosstalk and neuroimmune interactions, and highlights their potential as biomarkers and therapeutic targets.
While some patients with CSU exhibit an increase of blood eosinophils as well as an extravasation of eosinophils into wheals, a number of CSU patients exhibit eosinopenia, a reduced absolute eosinophil count in the blood, most likely reflecting recruitment from circulation to cutaneous lesions during active disease [494]. In both cases, apparently most important factor is the presence of eosinophils in the tissue. Histologic analyses confirm eosinophil infiltration and activation within wheals, evidenced by extracellular granules and localized elevation of EDN, correlating with disease severity [494].
Eosinopenia in CSU has strong clinical relevance: Studies show associations with high disease activity, type IIb autoimmune features, and poor response to antihistamines or omalizumab [495]. About 10% of CSU patients exhibit eosinopenia, which correlates with female sex, elevated C-reactive protein, positive autoantibody markers, and reduced therapeutic responsiveness [495]. Combining eosinopenia with basopenia, characterized by low basophil counts, improves the prediction of poor antihistamine response (odds ratio 9.5 vs. 4.8 for eosinopenia alone) [495]. This suggests that eosinophil count could be both a practical and cost-effective biomarker in clinical assessments.
Eosinophils, as part of the innate immune response, appear to be—at least partly—regulated by iron status [496–498]. Iron deficiency may itself act as a danger signal priming eosinophil activity. From an evolutionary perspective, a host’s iron restriction is a central mechanism of nutritional immunity, depriving pathogens—particularly parasites—of a critical resource [499]. In this context, iron deficiency could serve as a cue for heightened eosinophil readiness, anticipating helminth infection where iron loss and nutrient competition are common. Experimental models support this view, showing that eosinophils are promoted under iron-deficient conditions and suppressed when iron is sufficient [500]. Clinically, serum iron in asthmatic patients is inversely correlated with eosinophil counts [501], and inadequate fetal iron has been proposed as a risk factor for infant eosinophilia [502]. Iron deficiency has been associated with a range of skin manifestations—including dermatitis, rashes, urticaria, and pruritus—particularly in contexts where eosinophil-driven and allergic-type responses are involved [496–498, 503, 504]. Lower iron status has repeatedly been associated with chronic idiopathic urticaria, with one clinical trial showing that two months of oral iron supplementation resolved symptoms in all 81 patients with mild sideropenia [505]. Beyond urticaria, iron deficiency contributes to chronic generalized pruritus and uremic pruritus in chronic kidney disease [506]. Together, these observations highlight the immune-modulatory role of iron deficiency in promoting eosinophilia, mast cell activation, and skin inflammation, thereby aggravating allergic and pruritic diseases [507].
Eosinophils contribute mechanistically to urticarial pathology in several ways. In chronic urticaria lesions, eosinophils express tissue factor, initiating the extrinsic coagulation cascade and fibrinolysis, which may promote wheal formation through microvascular changes and localized oedema [508]. They are also a significant source of VEGF in CSU lesions, driving increased vascular permeability and vasodilation that lead to wheal formation [509]. In addition, eosinophilic granule proteins, especially MBP, may directly induce mast cell degranulation, amplifying histamine release and wheal intensity [510]. These pathways underscore how eosinophils interact with other key effector cells, mast cells, and basophils, contributing to both initiation and perpetuation of urticarial activity.
Given the emerging significance of eosinophils, targeting their activity has surfaced as a potential therapeutic strategy. Biologics such as mepolizumab, reslizumab, and benralizumab, designed to reduce eosinophil maturation, survival, or recruitment, have shown promise in reducing CSU symptoms [511]. The prostaglandin D2 (PGD2)-CRTH2 axis mediates eosinophil and basophil chemotaxis to skin lesions, and an oral CRTH2 antagonist (AZD1981) improved pruritus in antihistamine-refractory CSU, suggesting that eosinophil recruitment via this pathway plays a pathogenic role [512]. However, not all eosinophil-directed therapies yield consistent benefits. For example, benralizumab did not demonstrate therapeutic efficacy in a more recent CSU cohort, highlighting heterogeneity in patient response and disease endotypes [513]. The recent phase IIb ARROYO trial evaluating benralizumab in antihistamine-refractory CSU did not achieve its primary endpoint, despite complete and sustained depletion of peripheral eosinophils [513]. This paradox highlights the heterogeneity of CSU. While eosinophils contribute to disease activity in some patients, they are not universal drivers across all endotypes. In most individuals, mast cells remain the dominant effector cells, and redundant inflammatory cascades may compensate for eosinophil loss. Rather than ruling out eosinophils as therapeutic targets, the benralizumab experience underscores the need for precision medicine approaches that can identify and treat eosinophil-driven CSU more selectively.
In addition to biologics, nutritional strategies may complement CSU management. Iron deficiency has been linked to eosinophil activation and skin inflammation, with anemia common in CSU patients [514, 515]. The study reporting oral iron supplementation resolving chronic idiopathic urticaria in all patients with iron-deficiency [505] suggests that correcting iron deficiency could offer a simple, cost-effective adjunct in selected patients, fitting into precision approaches that match treatment to disease endotypes.
Beyond classical immune interactions, eosinophils may also influence neuroimmune pathways. Activated eosinophils are thought to enhance sensory nerve expression of substance P (SP) in CSU, thereby heightening pruritus, which forms part of a broader narrative linking immune cell infiltration to itch modulation [516]. While this remains an emerging line of investigation, it suggests that eosinophils may not only drive wheal development but also contribute to symptom severity through neuroimmune crosstalk.
Collectively, these findings indicate that eosinophils play a multifaceted role in urticaria pathogenesis, ranging from vascular and coagulative mechanisms to mast cell activation, immune crosstalk, and itch modulation. Peripheral eosinopenia emerges as a potentially valuable biomarker, particularly for identifying patients with active, autoimmune-predominant CSU who may require escalated management or non-IgE-directed biologics. Although therapies targeting eosinophils have shown variable results, they nonetheless represent a promising avenue for future personalized treatment strategies. With ongoing clarification of eosinophil pathways, particularly their crosstalk with mast cells and their involvement in neuroimmune signalling, more effective, targeted modalities may eventually emerge.
Importantly, iron deficiency appears to modulate eosinophil biology and has been implicated in chronic idiopathic urticaria, where iron repletion led to marked clinical improvement; integrating iron status into endotype definitions may therefore help refine eosinophil-predominant profiles and expand therapeutic options.
Anaphylaxis and drug reaction with eosinophilia and systemic symptoms (DRESS) are both severe hypersensitivity reactions, but they differ in onset and immune mechanisms. Anaphylaxis is a life-threatening, systemic hypersensitivity reaction characterised by its rapid onset and potential for fatal outcomes. It is mostly triggered by food, drugs, or insect stings. While the activation of mast cells and basophils via IgE-mediated pathways is widely regarded as the central mechanism driving anaphylaxis, increasing evidence indicates that other immune pathways, effector cells, and mediators may also contribute significantly to its pathophysiology [517, 518]. Among these, eosinophils play a secondary but potentially significant role in anaphylaxis by amplifying inflammation [518]. In contrast, DRESS is a delayed T-cell-mediated multisystem life-threatening drug hypersensitivity reaction mostly associated with anticonvulsants, antibiotics, and allopurinol, characterised by marked eosinophilia [519]. In DRESS, eosinophils are central to disease pathology, promoting tissue damage and systemic symptoms through the release of cytotoxic granules and proinflammatory mediators [519].
Eosinophils express receptors for IgE (FcεRI), IL-5, eotaxins, and other chemokines on their surface; their engagement induces their recruitment and activation and the release of a broad range of mediators including cationic proteins (EPX, MBP, ECP, EDN), lipid mediators (LTC4, PAF), and a variety of cytokines (IL-1, IL-3, IL-4, IL-5, IL-13, GM-CSF, TGF-a/b, TNF-a), chemokines (CCL3, CCL5, CCL11), and neuromodulators (SP, vasoactive intestinal peptide) [518]—which could sustain or augment the immune response.
Changes in eosinophils and eosinophil-derived proteins have been observed in both blood and tissues during or following anaphylactic reactions. Increased expression of eosinophil-related genes has been reported in patients presenting to emergency departments with severe anaphylaxis, as well as in murine models of anaphylaxis [520]. Additional evidence shows that eosinophil counts tend to decline during anaphylactic episodes, suggesting active migration from the bloodstream to target tissues [521]. This migration is associated with PAF, which not only recruits eosinophils but is also secreted by them—establishing a feedback loop that may intensify tissue inflammation and allergic symptoms [521]. Postmortem studies of fatal anaphylaxis cases reveal eosinophilic infiltration in bronchial smooth muscle [522] and elevated serum levels of ECP [523]. Similarly, patients with food-dependent exercise-induced anaphylaxis exhibit elevated serum levels of ECP and EDN, indicating eosinophil activation during anaphylactic episodes [524]. Recent findings suggest that while eosinophils are not essential for the acute phase of oral food allergen-induced anaphylaxis, they play a key role in early life by regulating mast cell proliferation and production of allergen-sIgE during sensitisation [525].
Eosinophilia associated with the use of drugs is not uncommon. In a Spanish study, using a Pharmacovigilance Program for Laboratory Signals based on abnormal laboratory values, 274 cases of drug-induced eosinophilia were identified among 164,379 admissions (incidence of 16.67 per 10,000 admissions). Of the 274 cases, 154 (56.2%) were asymptomatic hypereosinophilia, but there were 64 cases of potential DRESS, which is the main drug reaction associated with eosinophilic inflammation [519]. The diagnosis of DRESS is based on characteristic clinical features, laboratory findings, and the use of several scoring systems: the Bocquet criteria, the RegiSCAR criteria (the most frequently used in Europe), and the Japanese J-SCAR criteria; all of them refer to eosinophilia (> 1.5 × 109/L) as a diagnostic feature [526]. While eosinophilia is present in over 90% of reported cases, DRESS can occasionally occur with normal eosinophil levels [527]. Hematological abnormalities, including atypical lymphocytes and elevated eosinophil counts, are defining features of DRESS.
The pathophysiology of DRESS is multifactorial and associated with aberrant drug metabolism, specific human leukocyte antigens, and viral reactivation of latent infectious, especially of the Herpesviridae family. Viral reactivation seems to play an important role in promoting and sustaining abnormal T-cell and eosinophil responses to drugs and other offenders, leading to organ damage. The main proposed mechanisms of drug-induced T cell activation in DRESS are the hapten/prohapten, the pharmacological interaction (p-i concept), and the altered self-peptide model [528]. Drug-specific T-cells infiltrate the dermis and visceral organs, releasing cytotoxic proteins like granulysin, granzymes, and perforin. The expansion of regulatory T cells (T-regs) and T helper cells (Th1 and Th2) occurs especially in a later stage of DRESS [529]. Eosinophils may be stimulated by IL5 from innate-like lymphoid cells after alarmin release from infected and damaged cells, promoting a shift of CD4+ T cells to a Th2 profile with an increase of IL4, IL5, and IL13. Besides primary eosinophil activation following cell damage, systemic eosinophilic responses may be sustained by T-cell activation. IL-5 in synergy with other Th2-associated chemokines (TARC and macrophage-derived chemokine) promotes further eosinophil attraction, activation, proliferation, and infiltration in tissues, leading to eosinophilic inflammation and tissue damage due to the release of its toxic mediators from granules (MBP, EPO, EDN), enzymes, and an extensive panel of cytokines.
The most common histopathological findings in DRESS are dyskeratosis, spongiotic changes, vacuolisation, perivascular lymphocytic infiltration, and eosinophilic infiltration. More severe DRESS is associated with increased keratinocyte necrosis [530]. CD4+ and CD8+ T lymphocytes are identified in biopsies of the skin and of the internal organs. IL-5 produced by the Th2 cells and innate lymphoid cells induces differentiation, activation, and migration of eosinophils to the peripheral blood and to tissues, which explains the eosinophilia.
Persistently elevated eosinophil counts are thought to correlate with the organ damage and eosinophil infiltration of tissues such as the skin, the liver, the heart, and the nervous system. Although tissue damage is often driven by drug-specific T lymphocytes, eosinophils may directly contribute to complications such as EM. In fact, the myocardium is a preferential site for eosinophilic infiltration, and myocarditis is one of the main prognostic factors in DRESS patients.
A recent retrospective study showed a correlation between cardiac involvement and the degree of eosinophilia. Patients exhibited high eosinophil counts and elevated levels of ECP, which correlated with higher RegiSCAR scores, supporting the role of eosinophils in DRESS pathogenesis. The study proposed including ECP measurements in routine evaluations for DRESS [531].
Another indirect evidence of the implication of IL5 and eosinophils in the pathophysiology of DRESS is the rising data pointing to the potential benefits of the use of biological agents targeting the IL-5 axis (IL-5 antagonist mepolizumab and reslizumab or the IL-5 receptor blocker benralizumab) in the treatment of complicated or steroid-resistant DRESS, drugs that are already FDA and EMA approved for other eosinophilic disorders [532].
In summary, eosinophils play a multifaceted role in the pathophysiology of anaphylaxis and DRESS, though their involvement differs in extent and mechanism. While they are less central than mast cells in the immediate allergic response in anaphylaxis, their ability to modulate immune activity and promote prolonged inflammation positions them as potential therapeutic targets. Despite these findings, eosinophils appear to play a less dominant role in anaphylaxis compared to asthma. Further research is needed to fully elucidate their mechanisms and to explore new treatment strategies. In contrast, eosinophilic inflammation is a hallmark of DRESS, where eosinophils are key effectors contributing directly to tissue damage and organ dysfunction. Persistent eosinophilia, elevated eosinophil-derived proteins, and eosinophilic infiltration correlate with disease severity. Given their pathogenic role, particularly in DRESS, targeting the IL-5/eosinophil axis offers a promising therapeutic approach for severe or steroid-refractory cases in both conditions.
A. fumigatus: Aspergillus fumigatus
ABPA: allergic bronchopulmonary aspergillosis
ACO: asthma-chronic obstructive pulmonary disease overlap
AEP: acute eosinophilic pneumonia
AFRS: allergic fungal rhinosinusitis
AML: acute myeloid leukaemia
ANCA: anti-neutrophil cytoplasmic antibodies
AR: allergic rhinitis
BAL: bronchoalveolar lavage
BECs: blood eosinophil counts
CCAD: central compartment atopic disease
CEL: chronic eosinophilic leukaemia
CEP: chronic eosinophilic pneumonia
CF: cystic fibrosis
CLC: Charcot-Leyden crystal
CMR: cardiovascular magnetic resonance imaging
COPD: chronic obstructive pulmonary disease
CRS: chronic rhinosinusitis
CRSwNP: chronic rhinosinusitis with nasal polyps
CSU: chronic spontaneous urticaria
CT: computerized tomography
DRESS: drug reaction with eosinophilia and systemic symptoms
EADs: eosinophil-associated diseases
ECM: extracellular matrix
ECP: eosinophil cationic protein
ECRS: eosinophilic chronic rhinosinusitis
EDN: eosinophil-derived neurotoxin
EETs: eosinophil extracellular traps
EGIDs: Eosinophilic gastrointestinal diseases
EGPA: eosinophilic granulomatosis with polyangiitis
EM: eosinophilic myocarditis
EMA: European Medicines Agency
EMB: endomyocardial biopsy
EMT: epithelial-mesenchymal transition
EoC: eosinophilic colitis
EoE: eosinophilic esophagitis
EoG: eosinophilic gastritis
EoN: eosinophilic enteritis
eos/hpf: eosinophils per high-power field
EP: eosinophilic pneumonia
EPX: eosinophil peroxidase
FDA: Food and Drug Administration
FeNO: fractional exhaled nitric oxide
FESS: functional endoscopic sinus surgery
GM-CSF: granulocyte-macrophage colony-stimulating factor
GPA: granulomatosis with polyangiitis
GTPases: guanosine triphosphatases
HE: hypereosinophilia
HES: hypereosinophilic syndrome
HPF: high-power field
HRCT: high-resolution computed tomography
IBD: inflammatory bowel disease
ICS: inhaled corticosteroid
iEos: inflammatory eosinophils
IFN-γ: interferon-γ
IL: interleukin
ILC2s: group 2 innate lymphoid cells
INS: intranasal steroids
LAR: local allergic rhinitis
LTC4: leukotriene C4
MBP: major basic protein
MPA: microscopic polyangiitis
MPO: myeloperoxidase
NAR: non-allergic rhinitis
NARES: non-allergic rhinitis with eosinophilia syndrome
N-ERD: non-steroidal anti-inflammatory drugs (NSAIDs)–exacerbated respiratory disease
NSAIDs: non-steroidal anti-inflammatory drugs
OCS: oral corticosteroids
OPN: osteopontin
PAF: platelet-activating factor
PNS: paranasal sinus
ppb: parts per billion
rEos: regulatory eosinophils
ROS: reactive oxygen species
SEAF: sinonasal eosinophilic angiocentric fibrosis
sIgE: specific IgE
SNAREs: soluble N-ethylmaleimide-sensitive attachment protein receptors
SP: substance P
SPT: skin prick tests
T2: type 2
TGF-β: transforming growth factor-β
Th2: T-helper 2
TNSS: total nasal symptom score
TSLP: thymic stromal lymphopoietin
VEGF: vascular endothelial growth factor
WHO: World Health Organization
MDG, GWC: Conceptualization, Supervision, Writing—review & editing. GMW, LD, VP: Conceptualization, Writing—original draft (wrote section Pathophysiology of eosinophils (correspondence to Garry Michael Walsh: g.m.walsh@abdn.ac.uk)). GP, AMP, CDV, PT, DK, F Buta, NL, JO, MMA, CSR: Conceptualization, Writing—original draft (wrote section Eosinophilic inflammation across the upper airways: mechanisms, biomarkers, and therapeutic advances (correspondence to Giovanni Paoletti: giovanni.paoletti@hunimed.eu)). AY, F Braido, NR, DB: Conceptualization, Writing—original draft (wrote section Eosinophils and asthma (correspondence to Arzu Yorgancıoğlu: arzuyo@hotmail.com)). MC, MM: Conceptualization, Writing—original draft (wrote section Eosinophilic COPD (correspondence to Mario Cazzola: mario.cazzola@uniroma2.it)). GG: Conceptualization, Writing—original draft (wrote section Eosinophilic granulomatosis with polyangiitis and allergic bronchopulmonary aspergillosis (correspondence to Giuseppe Guida: giuseppe.guida@gmail.com) and Hypereosinophilic syndromes (correspondence to Giuseppe Guida: giuseppe.guida@gmail.com)). FP, RGC: Conceptualization, Writing—original draft (wrote section Other eosinophilic lung diseases (correspondence to Francesco Puppo: puppof@unige.it)). GM: Conceptualization, Writing—original draft (wrote section Eosinophilic myocarditis (correspondence to Giuseppe Murdaca: giuseppe.murdaca@unige.it)). PC, GCP: Conceptualization, Writing—original draft (wrote section Psychological and psychopathological components of eosinophilic myocarditis and pneumonia, a disease (correspondence to Pasquale Caponnetto: pasquale.caponnetto@unict.it)). WvdV, ER, NRF: Conceptualization, Writing—original draft (wrote section Eosinophils in gastrointestinal diseases (correspondence to Willem van de Veen: willem.vandeveen@siaf.uzh.ch)). TZ FRW: Conceptualization, Writing—original draft (wrote section Eosinophils and urticaria (correspondence to Torsten Zuberbier: torsten.zuberbier@charite.de)). MR, ERG: Conceptualization, Writing—original draft (wrote section Eosinophils in anaphylaxis and DRESS (correspondence to Matija Rijavec: Matija.Rijavec@klinika-golnik.si)). All authors read and approved the submitted version.
Giorgio Walter Canonica is the Editor-in-Chief of Exploration of Asthma & Allergy; Mario Di Gioacchino is the Co Editor-in-Chief of Exploration of Asthma & Allergy; Diego Bagnasco, Pasquale Caponnetto, Willem Van de Veen, Linhong Deng, Nelson Rosario Filho, Eva Rebelo Gomes, Giuseppe Murdaca, Vincenzo Patella, Ana Margarida Pereira, Francesco Puppo, Erminia Ridolo, Matija Rijavec, Nikoletta Rovina, Franziska Roth-Walter, Pongsakorn Tantilipikorn, Arzu Yorgancıoğlu are Editorial Board Members of Exploration of Asthma & Allergy; Fulvio Braido, Mario Cazzola, Giuseppe Guida, Mauro Maniscalco, Mário A. Morais-Almeida, Giovanni Paoletti, Chae-Seo Rhee, Garry M. Walsh, Torsten Zuberbier are Associate Editors of Exploration of Asthma & Allergy. They all had no involvement in the decision-making or the review process of this manuscript. The other authors declare no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
All datasets generated for this study are included in the manuscript.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 11682
Download: 268
Times Cited: 0
