 Review
Review

Affiliation:
1Graduate Program in Pharmacology, Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto 14049-900, São Paulo, Brazil
2Department of Biomolecular Science, School of Pharmaceutical Sciences of Ribeirão Preto, University of São Paulo, Ribeirão Preto 14049-900, São Paulo, Brazil
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0003-3300-8327
Affiliation:
1Graduate Program in Pharmacology, Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto 14049-900, São Paulo, Brazil
2Department of Biomolecular Science, School of Pharmaceutical Sciences of Ribeirão Preto, University of São Paulo, Ribeirão Preto 14049-900, São Paulo, Brazil
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0001-6247-4138
Affiliation:
1Graduate Program in Pharmacology, Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto 14049-900, São Paulo, Brazil
2Department of Biomolecular Science, School of Pharmaceutical Sciences of Ribeirão Preto, University of São Paulo, Ribeirão Preto 14049-900, São Paulo, Brazil
Email: sabrinalisboa@usp.br
ORCID: https://orcid.org/0000-0002-2069-3524
Explor Neuroprot Ther. 2022;2:182–209 DOI: https://doi.org/10.37349/ent.2022.00028
Received: April 06, 2022 Accepted: August 22, 2022 Published: October 31, 2022
Academic Editor: Raymond Chuen-Chung Chang, The University of Hong Kong, China
The article belongs to the special issue Intervention of Neuroimmune Responses
Different stressors can elicit neuroinflammatory responses modulated by innate immunity receptors, such as the family of Toll-like receptors (TLRs). The TLR4, a pattern recognition receptor (PRR), is involved in many diseases, such as inflammatory and central nervous system (CNS) diseases. Stress exposure can regulate the expression of PRRs, including TLR4, in the brain of animals, especially in the hippocampus and prefrontal cortex. Moreover, TLR4 modulates behavior and neuroinflammatory responses in the brain. In addition, to TLR4, the endocannabinoid (eCB) system plays a role in stress response and immunity, acting as a regulatory, stress-buffer system. This system is involved in many TLRs-mediated immune responses, such as microglia activation. Therefore, pharmacological approaches targeting the eCB system could modulate neuroinflammatory responses to stress by interfering with the TLR4 pathway. Although the connection between TLR4, stress, and neuroinflammation is well documented, almost no pre-clinical studies investigate the possible direct relationship between TLR4, behavior, stress, and the eCB system. Studies exploring the relationship between stress, neuroinflammation, TLR4, and the eCB system were searched using Pubmed, Web of Science, and Embase databases. Based on this search, this review is focused on the involvement of TLR4 receptors and signaling in neuroinflammation and the behavioral consequences of stress exposure. Moreover, evidence of the eCB system modulating TLR4-mediated responses was brought to the attention, pointing out a possible regulatory role of these responses by eCBs in behavior changes related to mood disorders.
Stress is a physiological response of the organism to any external or internal challenge, called stressor. The physiological alterations induced by stress exposure include behavioral and cognitive changes, and the inability to overcome stress is related to the development of pathologies, including those associated with the central nervous system (CNS) [1, 2]. Acute or chronic exposure to several psychosocial stressors in lab animals, for example, can promote morphological and neuroplastic alterations in the brain, especially in limbic areas [3, 4].
Different neuronal networks are engaged by different types of stressors, although they overlap at some points. Physical stressors, such as infections and hemorrhage, induce the activation of brain regions such as the paraventricular nucleus of the hypothalamus (PVN), the nucleus of the solitary tract (NTS), and locus coeruleus (LC) [5]. Psychological stressors, such as exposure to aversive stimuli and predator-related cues, engage components of the limbic system, including the prefrontal cortex (PFC), amygdala, hippocampus, ventral tegmental area (VTA), and nucleus accumbens (NAc). Limbic-PVN connections are relayed specially by gamma-aminobutyric acidergic (GABAergic) neurons. Chronic stress affects this circuitry resulting in enhanced PVN excitability, and considering corticotropin-releasing hormone (CRH) neurons are expressed in the PVN, this could result in hypothalamus-pituitary-adrenal (HPA) axis activation [5, 6].
Several neurotransmitter systems, including the noradrenergic, glutamatergic, serotonergic, nitrergic, and cannabinoid systems, are involved in neuroplasticity processes after stress. Moreover, alterations in the cytoarchitecture of the amygdala, hippocampus, and PFC [7–11] are also involved. Among these brain regions, the medial PFC is extremely sensitive to stress and suffers significant changes in its morphology and function after chronic stress [7–11].
Besides the neural alterations, stress activates microglial cells, the resident macrophages of CNS, a phenomenon proposed to contribute to and shape the responses of the organism to threats [12]. Microglia cells are vulnerable to both infectious and sterile stimuli, such as psychological stress, so their actions range from maintaining homeostasis to induced neuroinflammation, depending on the type, intensity, and duration of the stimulus [13, 14]. The collection of microglial receptors allows these cells to detect and respond to signals of stress deflagrated by the neuroendocrine, immunologic and nervous systems [12, 15].
Therefore, microglial cells act as sensors of the environment and are highly responsive to local disturbances, which could lead to different reactive states [16]. Microglial activation has a complex classification but is generally designated as microglial M1, or proinflammatory, and microglial M2, or anti-inflammatory [13, 16–18]. Several studies demonstrated that exposure to acute or chronic stress induces behavioral changes and also induces the expansion of microglial processes in many cerebral areas, including the hippocampus and PFC [13, 19, 20]. Moreover, stress exposure also induces other alterations in molecules associated with the immune response in the brain, particularly in the PFC, such as increased expression of the enzymes inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), the transcription factor nuclear factor kappa B (NFκB), and Toll-like receptors (TLRs) such as the TLR4 [2, 21, 22].
The TLR4 is a pattern recognition receptor (PRR) expressed in the membrane, mostly by innate immune cells, such as in microglia in the brain [23]. As a PRR, TLR4 can detect pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), and damage-associated molecular patterns (DAMPs), such as heat shock proteins (HSPs) and high mobility group box 1 (HMGB1) [24].
Activation of TLR4 depends on the myeloid differentiation factor 2 (MD-2) co-receptor and the recruitment of adaptor proteins, such as myeloid differentiation factor 88 (MyD88). This process triggers an intracellular signaling cascade that culminates in phosphorylation and consequent degradation of inhibitor of NFκB (IkB) kinase via the proteasome; IkB kinase is an inhibitor of the NFκB transcription factor in the cytoplasm. The activated NFκB then translocates to the nucleus, where it binds to gene promoter regions, initiating the transcription of several pro-inflammatory genes which originate proteins such as COX-2, iNOS, interleukin 1β (IL-1β), IL-6, and tumor necrosis factor α (TNF-α) [25]. Pharmacological inhibition of the TLR4 pathway by systemic administration of TAK-242 (resatorvid) reduce neuroinflammation in the PFC of rats exposed to acute restraint stress [26]. However, whether TLR4 receptors in the PFC directly participate in the behavioral consequences of stress remains to be investigated. Studies evaluating TLR4 in stress response will be discussed further.
The endocannabinoid (eCB) system is another important system activated by stress response and that modulates this response is the eCB system [27, 28]. Stress increases glutamate release, which through N-methyl-D-aspartate (NMDA) receptors increases neuronal activity. Activation of NMDA and metabotropic glutamate receptor 5 (mGluR5) receptors by glutamate results in intracellular calcium influx in the postsynaptic terminal, culminating in activation of eCB synthesis enzymes, leading to their production and release by the postsynaptic terminal in the synaptic cleft [29]. Cannabinoid type 1 (CB1) and CB2 receptors (CB2Rs), the eCBs anandamide (AEA, also known as N-arachidonoylethanolamine), and 2-arachidonoylglycerol (2-AG) are the most studied components of the eCB system [30–34].
The eCB system can also modulate the neuroimmune response, including in stressful conditions. For example, repeated stress-induced neuroinflammation in the PFC of mice was attenuated by CB1 and CB2 agonists [35, 36]. Furthermore, a non-selective CB1/CB2 agonist administered for six days during social defeat stress decreased neuroinflammation and the anxiogenic response, and prevented the later sensitized conditioned fear response [37].
In addition, several works, including from our research group, demonstrate that eCB signaling in the medial PFC has an important role in responses related to stress and anxiety, including controlling the HPA axis [29]. The presence of CB1Rs in corticolimbic circuits that regulate the HPA axis, the anti-stress properties of cannabis use, and several other pieces of evidence, including from animal models, support the eCB signaling involvement in the inhibition of stress response [37, 38]. Moreover, the presence of the eCB system, mainly CB2Rs, in immune cells, especially in microglia cells, and the involvement of this system in neuroimmune modulation [39, 40] strengthen the idea that eCB effects in modulating behavioral responses could involve the modulation of neuroimmune mechanisms.
Neuroinflammation has been discussed to play a central role in the neurobiology of neuropsychiatric disorders [41]. Several targets are involved in neuroinflammatory responses, including some induced by stress exposure, such as TLR4 and eCB signaling. However, fewer studies have evaluated the interaction between these systems in regulating neuroinflammation and behavior changes after stress [42, 43]. Therefore, it was hypothesized that the TLR4 signaling involvement in stress response, including behavioral changes related to psychiatric disorders, could be modulated by the eCB system. Before summarizing the findings of the TLR4 receptors in stress response and evidence of relationship with the eCB system, it is noteworthy to give a brief overview of how stress exposure can impact the immune system and neuroinflammation. Also, it is important to briefly address the TLR4 pathway to understand how the impact of stress on this signaling could result in behavioral changes. The studies mentioned in this review were obtained in PubMed, Embase, or Web of Science. Only full-text articles in English were considered.
The sympathetic nervous system and the HPA axis, activated during a stress response, are the main drivers of the physiological systems, including the immune system [44]. In turn, the immune system is affected by acute and chronic stressors, resulting in various cellular changes and humoral responses both in the periphery and CNS [45]. The understanding of these mechanisms is essential to comprehend the consequences of stress [46].
The immune system in the brain, for example, develops several responses to stressful situations in the brain, including morphological and functional changes [47]. There is bidirectional communication between the brain and the immune system, which involves efferent and afferent pathways through which the brain and the periphery exchange information about the body’s homeostatic state. This process is an essential element of the response to environmental, physiological, and psychological factors that affect homeostasis [45].
Exposure to acute or chronic stress can have several immune consequences, such as increased cortisol levels, increased circulating pro-inflammatory cytokines/chemokines (IL-6 and TNF-α), and other molecules such as DAMPs and prostaglandins [24]. Some of these mediators can be found in the brain, where they could mediate neuroinflammation and be involved in several behavioral changes [45, 48]. The neurochemical alterations in the brain arising from inflammation include activation of the kynurenine pathway, which affects tryptophan metabolism and serotonin levels, reduction of brain-derived neurotrophic factor (BDNF) production, among others [49, 50]. These changes are mostly associated with mood disorders such as anxiety and depression [51], but not all patients with mood disorders will present signs of immune activation [52]. Therefore, stressful experiences can induce activation of many aspects of peripheral immunity and central neuroimmune processes, contributing to various forms of host defense, stress recovery, and, ultimately, disease susceptibility [37, 45]. There are, therefore, a variety of neuroimmune signaling pathways that can be activated in response to stressful experiences. In animal studies, these effects often depend on specific individual characteristics of the subjects [46]. It is beyond the scope of the present review to address all these pathways; there are excellent reviews about this topic (for example, [53]).
In addition to the observed changes in the expression of cytokines and other inflammatory signaling molecules, exposure to stress is often accompanied by cellular changes manifestations of neuroimmune activation, such as dynamic changes in the state of microglial activation. These cells are the primary brain source of immune mediators [54]. Several human studies suggest that microglial changes could be related to mood disorders [55–59]. Moreover, inhibition of microglia with minocycline, a tetracycline antibiotic that inhibits microglial activation at low doses [60], was benefic to depressive patients [60–62] and demonstrated to improve antidepressant response in treatment-resistant patients [62]. However, the exact role of microglia cells in mood disorders remains uncertain. More recent data, almost exclusively from lab animal studies, suggest that PRRs in microglia, namely the nucleotide oligomerization domain-like receptor protein 3 (NLRP3) and the TLR4, are involved in the behavioral consequences of stress exposure [63–70]. This review will focus on evidence pointing out the involvement of TLR4, therefore is essential to give a brief overview of this pathway.
As described in the Introduction, the activation of TLR4 by PAMPs, such as LPS, or DAMPs, such as HMGB1, HSPs, and fibrinogen, can activate two pathways, MyD88-dependent pathway and MyD88-independent pathway [71].
The MyD88-dependent pathway leads to the recruitment and activation of IL-1 receptor-associated kinase (IRAK) and TNF receptor-associated factor 6 (TRAF6) proteins, which activates transforming growth factor β-activated kinase 1 [TAK1, also known as mitogen-activated protein kinase (MAPK) kinase kinase 7 (MKK7)]. This molecule leads to the activation of MAPK pathways, promoting the nuclear translocation of activator protein 1 (AP-1) [71]. TAK1 can also activate IkB-kinase (IKK) complex [formed by NFκB essential modulator (NEMO), IKKα, and IKKβ], which phosphorylates the inhibitor protein of NFκB complex (subunits p65 and p50) and the IkBα. Phosphorylated IkBα (p-IkBα) is degraded by the proteasome and releases NFκB to translocate to the nucleus, where it will promote the transcription of several proinflammatory genes, including those necessary for the NLRP3 inflammasome [71, 72] (see Figure 1).
The activation of the MyD88-independent pathway promotes the endocytosis of TLR4 dimer, which through Toll/IL-1 receptor-domain-containing adapter-inducing interferon-β (TRIF) and TRAF3 proteins leads to the activation of the transcription factor interferon regulatory factor 3 (IRF3), which favors the expression of type I interferons [e.g., interferon α (IFNα) and IFNβ] [71, 72].
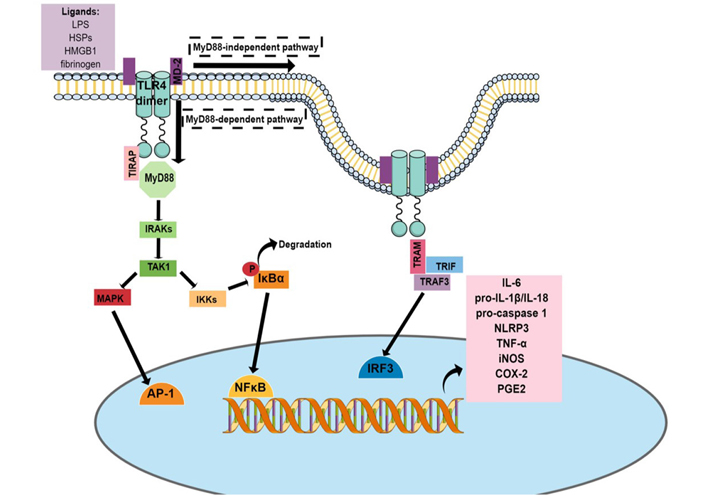
TLR4 pathway. The activation of the TLR4 pathway results in the translocation of transcription factors related to inflammation to the nucleus, such as AP-1, NFκB, and IRF3. These transcription factors bind to specific regulatory regions in the DNA, leading to the transcription of several inflammatory genes, resulting in the synthesis of messenger RNA (mRNA) of proinflammatory mediators, including IL-6, pro-IL-1β, pro-caspase-1, NLRP3, and iNOS. P: phosphorylation site; TIRAP: Toll/IL-1 receptor domain-containing adapter protein; TRAM: TRIF-related adaptor molecule; PGE2: prostaglandin E2
Genetic, chemical, and pharmacological approaches are used to study the influence involvement of TLR4 in stress response and behavior. Some of these strategies will be briefly discussed below.
LPS from Gram-negative bacteria, a TLR4 activator, is extensively used to evaluate sickness behavior, cognitive deficits, and depressive-like behaviors in animals [73–76]. Overall, LPS-induced behavioral changes and neuroinflammation are attenuated by antidepressants from different classes [selective noradrenaline reuptake inhibitors, selective serotonin reuptake inhibitors (SSRIs) and serotonin-noradrenaline reuptake inhibitors (SNRIs), and tricyclic antidepressants] [75, 77–79]. Therefore, this model is helpful for investigating mechanisms involved in the consequences of LPS exposure, as demonstrated for the indoleamine 2,3-dioxygenase (IDO) enzyme [80–82] and the NLRP3 inflammasome [83]. Moreover, the LPS model is used to evaluate the potential effect of drugs in attenuating depressive-like behavior in the context of immune system activation. Ketamine [84–86], agonists of CB2Rs [86], and the phytocannabinoid cannabidiol [87], for example, attenuated the behavioral consequences of LPS administration in lab rodents. Therefore, these data are relevant to the study of depression associated with immune changes because in this case, the condition can be resistant to conventional treatments [88–90].
Some drugs were designed as inhibitors of the TLR4 pathway, including E-5531, eritoran, and TAK-242, mainly for the treatment of sepsis [91–93]. However, they fail to present therapeutic effects in this condition [42, 94]. These compounds aim to block the activation of the TLR4 pathway by DAMPs and PAMPs, blocking the induction of chronic and sterile inflammation, which can be involved in neuropsychiatric diseases [70, 95]; therefore, they are essential experimental tools.
TAK-242 is a cell-permeable compound that selectively binds to the cysteine residue of TLR4, disrupting its interaction with the adaptor molecules TIRAP and TRAM. TAK-242 attenuated neuroinflammation and behavioral changes induced by LPS challenge [96] and acute restraint stress [26]. Recently, Shirayama, et al. [70] showed an antidepressant effect of TAK-242 in a learned helplessness model after intracerebroventricular administration. Interestingly, this effect was attenuated by local administration of a glutamate AMPA receptors antagonist (NBQX) or an inhibitor of BDNF-tropomyosin- related kinase receptor B (TrkB) signaling (ANA-12), highlighting the involvement of these receptors in the effects of TAK-242.
Eritoran, a synthetic analog of the lipid A portion of LPS, competes with LPS for binding to the MD-2 portion of the TLR4 receptor complex [97]. This drug attenuated depressive-like behaviors and neurochemical changes induced by chronic restraint stress (CRS) in a dose-dependent manner [98].
Genetic models are also widely used, such as the TLR4 knockout (KO) and C3H/HeJ mice; in the latter, TLR4 has a defective response to LPS [2]. Moreover, it is also possible to use interfering peptides that disrupt the beginning of the TLR4 pathway [99]. Several studies show that TLR4 KO is resistant to the behavioral effect of stress and has lower levels of neuroinflammation [100, 101]. However, contradictory data show no effect [102] or even an opposite effect, an anxiogenic behavior [103]. These data will be discussed in the next session.
New genetic models have emerged since microglia has gained importance in many neurological and psychiatric disorders [104, 105]. Conditional KO mice, such as C-X3-C motif chemokine receptor 1 (CX3CR1, CX3CR1+/−CreER), when crossed with mice with a floxed gene, can promote the deletion of specific gene targets in CX3CR1-positive cells, including microglia (CX3CR1+ cells). After some weeks following the treatment with the estrogen receptor agonist tamoxifen, the deletion is obtained. These animals were not yet used to study the brain microglia’s TLR4 in behavior but are helpful to study neurodegenerative diseases [106, 107]. Below we will discuss studies using these different approaches to study TLR4 in a stress context.
Several pieces of evidence show that exposure to stressors in lab animals alters TLR4 pathway molecules expression in brain areas related to neuropsychiatric disorders, supporting an essential role for TLR4 in mood disorders. Most importantly, pharmacological or genetic manipulation of the TLR4 pathway modifies animals’ behavior and neuroinflammation after stress exposure. These studies are summarized in Table 1 and will be discussed below. Most of these studies use heterotypic stressors, such as exposure to chronic unpredictable mild stress (CUMS) (see Table 1, Figure 2).
Involvement of the TLR4 pathway and related mediators in stress response, neuroinflammation, and behavioral effects in animal models
| Animal (strain, sex, size/age) | Stress model | Behavioral assessment | Major findings | Modulation of TLR4 pathway | Reference |
|---|---|---|---|---|---|
| C3H/HeN mice, maleadult | Immobilization stress (1 eCB/7 days) | None | Stress induces | C3H/HeJ mice | Caso et al., 2008 [126] |
| C57BL/6N mice, male6–12 weeks old | Single or repeated social defeat stress (4 or 10 days) | Social interaction test, EPM | Repeated stress induces microglia activation, | TLR2/4 KO mice | Nie et al., 2018 [129] |
| C57Bl/6 mice, male8–12 weeks old | Footshock stress (one or two sessions of 180 inescapable footshocks, 0.3 mA duration of 6 s) | Learned helplessness; number of failures to scape footshocks | Stress in WT animals but not TLR4 KO promotes | TLR4 KO mice | Cheng et al., 2016 [100] |
| ICR mice, maleWeighing 18–22 g | CUMS (8 weeks) LPS (5 days, 0,83 mg/kg, i.p./day) | SPT, OFT, TST, FST | Stress: | TAK-242 (3 mg/kg, i.p.) and baicalin (60 or 30 mg/kg, i.g.) | Guo et al., 2019 [96] |
| C57Bl/6 mice, male8–10 weeks old | CUMS (6 weeks) | FST, TST, OFT, SPT | Stress | TLR4 KO mice and arctigenin (25, 50, or 100 mg/kg, i.p.) | Xu et al., 2020 [101] |
| C57Bl/6J mice, male8 weeks old | LPS (1 mg/kg, i.p.) | TST, SPT, FST | LPS | None | He et al., 2020 [74] |
| ICR mice, maleweighing 18–22 g | CUMS (6 weeks) | SPT, TST, OFT, FST | Stress | TAK-242 (referred as Cli-095 in this paper) (3 mg/kg, i.g.) | Fu et al., 2019 [110] |
| BALB/c mice, male8 weeks old | CUMS (4 weeks); i.c.v. administration of fr-HMGB1 or non-oxid HMGB | SPT, TST, OFT | Stress | TAK-242 (3 mg/kg, i.p.) | Lian et al., 2017 [117] |
| C57Bl/6 mice and ob/ob mice, male7–8 weeks old | CUMS (3 weeks) | SPT, OFT, Morris water maze | Stress induced depressive-like behavior and alterations in target quadrant in Morris water mazeStress | TAK-242 (3 mg/kg, i.p.) | Wang et al., 2018 [43] |
| ICR mice, male6–8 weeks old | LPS (0,83 mg/kg, i.p.) | OFT, TST, FST, SPT | LPS induced | Saikosaponin-d (1 mg/kg, i.g.) | Su et al., 2020 [112] |
| ICR mice, male8–10 weeks old | LPS (1 mg/kg, i.p.) | FST, SPT, NSFT | LPS induced | Molecular docking simulation indicates possible interaction between TLR4 and fast green FCF (100 mg/kg, i.p.) | Yang et al., 2019 [73] |
| NMRI mice, maleAdultWeighing20–25 g | LPS (0,83 mg/kg, i.p.) | OFT, FST | LPS induced | Modulation of TLR4 pathway with GM- CSF (30 μg/kg, i.p.) | Hemmati et al., 2019 [111] |
| Wistar rats, maleWeighing 150–180 g | CRS (6 eCB/day, 28 days) | SPT, OFT, FST, social interaction test | CRS induced | TLR4 antagonist, eritoran (5 mg/kg, i.p.) | Aboul-Fotouh et al., 2018 [98] |
| Wistar rats, maleadultWeighing 250–300g | LPS (20 μg or 80 μg, i.c.v.) | OFT, EPM, FST, Morris water maze | LPS induced | None | Na et al., 2021 [131] |
| Wistar Hannover rats, maleWeighing 200–225g | CMS (21 days) | FST, SPT, splash test, EPM | CMS induces bacterial translocation, | None | Martín-Hernández et al., 2016 [130] |
| Offspring of C57BL/6 mice, male and female | MIA (single dose of LPS on embryonic day 12, 50 μg/kg, i.p.) | None | Pro-inflammatory profile of cytokines and | None | O’Loughlin et al., 2017 [114] |
↑: increase; ↓: decrease; EPM: elevated plus-maze; WT: wild-type; SPT: sucrose preference test; OFT: open field test; TST: tail suspension test; FST: forced swim test; Iba-1: ionized calcium binding adaptor molecule 1; TNFR1: tumor necrosis factor receptor 1; GM-CSF: granulocyte-macrophage colony-stimulating factor; NO: nitric oxide; 5-HT: 5-hydroxytriptamine; p-NFκB: phosphorylated NFκB; RANTES: regulated on activation, normal T cell expressed and secreted; MCP-1: monocyte chemoattractant protein-1; NE: norepinephrine; p-p38: phosphorylated p38; SOD: superoxide dismutase; GPx: glutathione peroxidase; MDA: malondialdehyde; MBP: myelin basic protein; NSFT: novelty suppressed feeding test; GFAP: Glial fibrillary acidic protein; GABA: γ-aminobutyric acid; GAD: glutamate decarboxylase; CMS: chronic mild stress; MIA: maternal immune activation
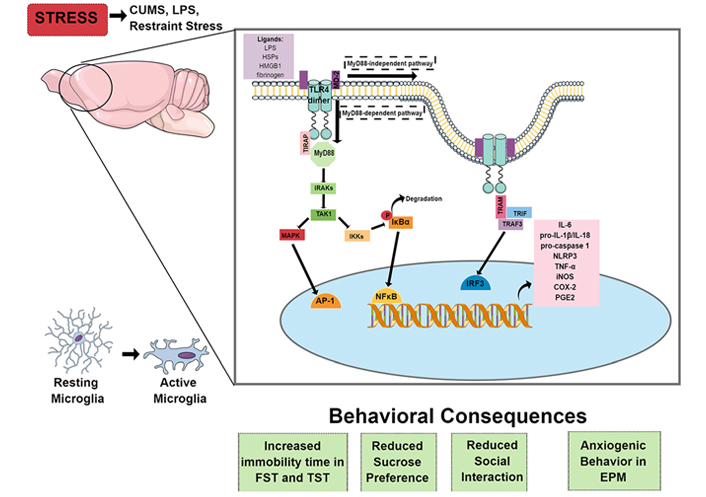
Behavioral and neuroimmune consequences of stress. In rodents, psychological and immunological stressors cause microglial activation, especially in limbic areas, such as the hippocampus and PFC. Microglia can be activated through the activation of the TLR4 pathway by PAMPs, like LPS, or DAMPs, like HSPs, HMGB1, and fibrinogen, leading to an increase in proinflammatory cytokines, such as IL-1β and TNF-α, enzymes that mediate immune/inflammatory responses, such as iNOS, COX-2, and PGE2, and transcription of components of the NLRP3 inflammasome pathway (pro-IL-1β, pro-IL-18, pro-caspase-1, and NLRP3). Stressors’ exposure also induces depressive-like and anxiety-like behaviors, which could be related to the downstream activation of TLR4. Therefore, pharmacological or genetic inhibition of this pathway, by administering drugs that antagonize TLR4 or prevent its activation, such as TAK-242, or using TLR4 KO or transgenic mice, could prevent the development of behavioral consequences of stress exposure
However, fewer articles evaluate the effect of homotypic stress, such as footshock or restraint stress exposure. Several reports show differences in immune system activation after exposure to homotypic or heterotypic stressors, including microglial activation. This effect is related to habituation of the HPA activation in the first but not in the second condition [4, 108, 109].
As already briefly discussed, stress exposure can increase pro-inflammatory cytokines and enzymes in the brain, especially in the hippocampus and PFC, and decrease neurotrophic factors and monoamines in the same brain regions [13, 19, 100, 101, 110, 111]. Also, there is an alteration in microglial markers, suggesting the involvement of these cells in stress response [74, 101, 112] (see Table 1).
Acute or repeated homotypic stressors increase the expression of TLR4 pathway components in the brain, including TLR4, MyD88, and NFκB [2, 73, 102, 113–116]. One of the molecules responsible for triggering the TLR4 response is HMGB1 [100, 101, 112, 117]. Several studies showed that severe stress increases HMGB1 levels in the brain and induces neuroinflammation [116, 118, 119]. Considering that HMGB1 binds to TLR4 and CD14, resulting in the release of several cytokines [120], and considering that behavioral changes after stress involve HMGB1 release [121, 122], this DAMP could be responsible for the behavioral consequences of TLR4 activation after stress exposure.
Moreover, several studies show that stress increases the expression of components of the microglial NLRP3 inflammasome in the brain [63, 64, 118]. This inflammasome is primed by activation of TLR4 (Figures 1 and 2). When primed, other stimuli can activate it, such as activating purinergic P2X7 receptors by ATP. NLRP3 inflammasome activation results in caspase-1 activation and conversion of pro-IL-1β/pro-IL-18 in IL-1β/IL-18 [73, 100, 101, 112] (see Table 1). Therefore, activation of this pathway could be one of the readouts of TLR4 activation.
The benefits of modulating the TLR4 pathway are described in several conditions such as Parkinson’s and Alzheimer’s diseases [123, 124] and traumatic brain injury [125]. Moreover, stress-induced inflammatory changes can be modulated by interfering with the TLR4 pathway. These data are summarized in Table 1 and discussed below.
TLR4 KO mice are resistant to depressive-like behavior in the learned helplessness paradigm and also have a blunted cytokine response to stress, with lower hippocampal levels of TNF-α, IL-6, and IL-1β than wild-type mice [100]. These mice also present a protective phenotype after exposure to repeated homotypic restraint stress [126] or CUMS [101].
However, the data of TLR4 KO mice in models predictive of anxiolytic drugs are controversial, with reports of no effect [102, 127, 128] or anxiogenic effect [103, 116]. For example, the anxiogenic effect in TLR4 KO mice has been previously reported in different animal models, both in males and females [103]. However, one study [102] did not report this anxiogenic effect. These mice also showed deficits in the contextual conditioned fear paradigm, among other cognitive changes [102].
A recent work evaluated social interaction, but not anxiety behavior, in TLR4 KO and double TLR2/TLR4 KO mice exposed to repeated social defeat stress [129]. Stress-induced social interaction reduction depends on TLR2/TLR4 receptors specifically expressed in medial PFC (mPFC) microglia. Furthermore, reduced neuronal activity, microglial activation, and dendritic atrophy in the mPFC after stress also depend on these receptors. Although TLR4 KO animals did not present changes related to anxiety behavior in this study, these animals’ cellular responses to stress were not evaluated [129]. Furthermore, whether the deletion of TLR4 only in microglial cells participates in behavioral and cellular responses promoted by stress exposure is still an open question.
The pharmacological antagonism of TLR4 with TAK-242 restores sucrose preference and ameliorates depressive-like behavior after CUMS [110] and reduces helplessness behavior and expression of TNF-α in the hippocampus induced by chronic social defeat stress (CSDS) [116]. TAK-242 also reversed behavioral alterations and hippocampal increase of TNF-α after intracerebroventricular injection of reduced forms of HMGB [disulfide HMGB1 (ds-HMGB1) and fully reduced HMGB1 (fr-HMGB1)] [117]. In addition, TAK-242 reversed the CUMS-induced depressive-like behavior both in C57bl/6 and in ob/ob (mutant mouse for leptin gene) mice, reversing the increase of TNF-α, IL-6, and IL-1β in the hippocampus and frontal cortex [43]. Overall, these results suggest that blockade of the TLR4 pathway can be beneficial to coping after stress exposure, similar to several data obtained with TLR4 KO mice (Table 1).
Concerning anxiety behavior, to our knowledge, few studies have evaluated if pharmacological inhibition of TLR4 can modify this behavior after stress. Intracerebroventricular administration of a TLR4 antagonist, the inhibitory LPS from Rhodobacter sphaeroides (R. sphaeroides), in naive animals induced an anxiolytic-like effect [102], contrasting with some reports in TLR4 KO mice indicating an anxiogenic effect (Table 1).
Therefore, pharmacological and genetic tools used to study the role of TLR4 in behavior, particularly related to anxiety, can render contradictory effects. The exact role of TLR4 in behavioral responses to stress still needs to be further elucidated. The resultant effect observed with TLR4 KO mice, for example, could involve the absence of these receptors during brain development [128, 129].
Interestingly, CMS promotes intestinal translocation and depressive like-behavior [130], and intestinal decontamination prevents the increase of TLR4, COX-2, and iNOS expression in the rat frontal cortex after stress exposure which indicates an essential role of bacterial translocation in activating the TLR4 pathway after stress. These effects of intestinal decontamination can also be observed with other types of stressors, such as CMS [113] and repeated restraint/acoustic stress [2]. However, in these studies, the behavior was not evaluated. Therefore, it is still not completely clear how bacterial translocation impacts behavior, and this discussion is beyond the scope of this review.
Altogether, these experimental data support the involvement of the TLR4 pathway in the neuroinflammatory and behavioral responses triggered by stress. Therefore, these receptors could be a potential target for therapeutic intervention in conditions of overactivation of the immune system. It is crucial to evaluate if similar alterations are observed in humans and if drugs currently used to treat stress-related disorders, such as antidepressants, or drugs potentially used to treat these disorders, could change the TLR4 pathway [96].
Recent data indicate that major depressive disorder (MDD) patients present changes in TLRs expression, or components of their signaling pathway, in blood cells [131–137]. TLR4 expression, for example, is increased in several MDD patients [132, 134, 136–138]. Interestingly, postmortem evaluation of TLRs in the PFC and dorsolateral PFC (DLPFC) of suicide and non-suicide depressive patients found higher levels of TLR4 mRNA and other TLRs [26, 139, 140]. However, no changes were found in the DLPFC of MDD patients [50]. Some reports suggest that TLR4 levels could predict the severity of depressive symptoms in MDD [132, 137]. Patients with severe symptoms, for example, presented significantly lower levels of methylation (an epigenetic process related to repression of gene transcription) in the tlr4 gene, specifically in the cytosine-phosphate-guanine (CpG) site cg05429895, when compared to patients with mild symptoms; these findings were in line with the increased plasmatic levels of TLR4 mRNA [137]. However, there are also data showing no association between TLR4 levels and symptom severity [69]. In addition, single nucleotide polymorphisms (SNPs) of the tlr4 gene could predict some traits, such as anxiety and psychomotor retardation, observed during the first episode of depression in MDD patients [68]. Also, in MDD patients, peripheral levels of TLR4 expression could predict anxiety traits and weight loss [69]. Accordingly, TLR4 levels could be a potential biomarker in MDD.
A recent review summarized animal and human studies investigating if SSRI and SNRI attenuate neuroinflammation by modulating immune pathways [141]. In fact, human studies evidence anti-inflammatory effects of antidepressants [132–137, 139, 140]. Part of these effects could be mediated by TLRs, because chronic treatment with antidepressants from different classes attenuated the expression levels of several TLRs [133–135], including increased TLR4 levels [134, 135], in blood cells from MDD patients.
In vitro and animal studies with antidepressants, such as amitriptyline, escitalopram, and fluoxetine, support that their effects can result from TLR4 expression modulation [142–144]. For instance, several studies showed that fluoxetine decreases TLR4 levels in various brain regions [145, 146], a mechanism that could be related to subsequent inhibition of the NFκB pathway and the NLRP3 inflammasome [49, 147, 148].
Furthermore, not only antidepressants, but other compounds can modulate the TLR4 pathway. Compounds from plants used in the traditional Chinese and Indian cultures can modulate the TLR4 pathway [95, 149]. For example, asperosaponin VI (ASA VI), isolated from the Radix Dipsaci, used in traditional Chinese medicine, improved LPS-induced depressive-like behavior in mice. ASA VI also suppressed microglia-mediated neuroinflammatory response by inhibiting the TLR4/NFκB signaling pathway [149]. Furthermore, arctiin, isolated from the plant Fructus arctii, induced a dose- dependent antidepressant effect in mice. This compound also reduced excessive microglia activation, decreased the release of HMGB1, and attenuated the expression of TLR4 in the PFC of mice exposed to CUMS; it also attenuated the inflammatory profile of primary microglia stimulated with HMGB1 and TNF-α [101]. Baicalin, a flavonoid compound isolated from Scutellaria baicalensis, has anti-inflammatory and antioxidant properties [150]. This flavonoid attenuated CUMS-induced depressive-like behaviors and attenuated the increase in HMGB1/TLR4/NFκB expression [151]. Other flavonoids, such as flavones, apigenin, and hesperidin, demonstrated antidepressant effects by inhibiting TLR4 signaling in animal models [70, 152, 153]. Finally, curcumin, the yellow pigment in Indian saffron, isolated from the rhizome of Curcuma longa, has been investigated as a neuroprotective agent in several pathological conditions [154]. In a traumatic brain injury model and in an in vitro model, curcumin attenuated microglial activation and the expression of the TLR4/MyD88/NFκB pathway, and reduced neuronal apoptosis [155]. Moreover, it also attenuated neuroinflammation and long-term cognitive deficits induced by a high dose of LPS [156].
Therefore, not only drugs already used in the clinic, such as antidepressants, but also other compounds primarily used in popular medicine could exert at least part of their effects by modulating neuroinflammation via inhibition of the TLR4 pathway. Considering all experimental data showing anti-inflammatory and anti-stress effects of drugs that interfere with the TLR4 pathway and evidence of changes in TLR4 in MDD patients, we suggest that drugs interfering with this pathway could be used as adjuvant treatment in stress-related disorders. Also, they could be an alternative treatment in treatment-resistant patients, especially when there are signs of immune alteration. Finally, considering evidence that some antidepressants modulate the TLR4 pathway, those antidepressants with an anti-inflammatory profile would be a better choice for patients with altered immune parameters.
The eCB system modulates many functions in the CNS, such as neuroplasticity, the release of cytokines by microglia, cell homeostasis, and behavior [29, 157]. This system comprises endogenous lipidic messengers/neurotransmitters (the eCBs), receptors, and anabolic and catabolic enzymes [158]. The most well understood eCBs are AEA and 2-AG. The N-acylphosphatidylethanolamine-specific phospholipase D (NAPE-PLD) and the fatty acid amide hydrolase (FAAH) are responsible for the biosynthesis and hydrolysis of AEA, respectively. The diacylglycerol lipase α (DAGLα) and DAGLβ, monoacylglycerol lipase (MAGL), and α/β-hydrolase domain 6 (ABHD6) are responsible for the synthesis and degradation of 2-AG, respectively. eCBs interact with CB1Rs and CB2Rs; but they can interact with other targets, such as transient receptor potential vanilloid type 1 (TRPV1) and peroxisome proliferator-activated receptor α (PPARα) and PPARγ receptors [157]. Other eCBs exist, such as the N-palmitoylethanolamide (PEA) [159], which has several anti-inflammatory and neuroprotective properties [160–162], and its effects can be mediated, for example, by activation of CB2, TRPV1, and PPARα [157, 159].
eCBs are synthesized by several cells and act on different brain cells, including neurons and microglia [163, 164]. Therefore, the widespread localization of the eCB system molecules in brain cells and its multi-target actions allows the control of many functions, from controlling local cellular actions to big circuits involved in behavior.
CB1Rs are expressed in the periphery, but mostly in the CNS; they are found in telencephalic and cerebellar regions, mostly in neurons, but also in glial cells [165–167]. They are the most expressed G-protein coupled receptors in the brain [165–167]. The neuronal CB1 expression is generally located at presynaptic elements, where they are coupled to Gi protein [165, 166]. Therefore, neuronal CB1 activation usually inhibits neurotransmitter release [165, 167, 168]. In the striatum, CB1R is expressed by parvalbumin- positive interneurons, whereas in the cerebral cortex, hippocampus, and amygdala they are predominately, but not exclusively, expressed by cholecystokinin (CCK)-positive interneurons [165]; they can also be expressed by glutamatergic neurons [169]. CB2Rs are mostly expressed by immune cells in the periphery, but also by microglia in the brain [170]. However, there is evidence of neuronal expression in the postsynaptic neurons, where its activation could hyperpolarize cells and inhibit signal transmission [171, 172]. Their expression is much lower than that of CB1 and it can be increased by several stimuli [170, 173].
Stress exposure affects the eCB system in the brain. For instance, acute or chronic homotypic stress activate FAAH and reduce AEA levels; in contrast, 2-AG levels are increased [174]. Repeated homotypic stress potentiates these effects on eCB levels, and reduces the expression of CB1 in most brain areas, such as hippocampus and amygdala, whereas increases it in the PFC [174]. Impaired CB1 signaling by AEA result in a lack of adaptation to repeated stress and consequently, behavioral changes [175], and impaired CB2 signaling could contribute to neuroinflammation [173]. Data regarding heterotypic stressors on eCB levels are less consistent [176]. eCB tone also controls the HPA axis. For example, CB1 antagonists increase circulating levels of adrenocorticotropic hormone (ACTH) [176, 177].
The presence of eCB signaling in stress-sensitive nuclei, such as hypothalamic and upstream limbic structures (amygdala, hippocampus, and PFC) suggests it plays an essential role in regulating the stress’s neuroendocrine and behavioral effects [178]. The amygdala is one of the primary limbic structures involved in activating the HPA axis in response to stressful stimuli. In contrast, hippocampus and PFC have been identified as inhibitors of the HPA axis and are also involved in glucocorticoid-mediated negative feedback [179]. Consequently, adequate eCB signaling in the limbic system is essential to mitigate the consequences of aversive stressful situations, as extensively reported [29].
The interplay between eCB, stress, and the inflammatory system has gained much attention in the neuroimmune area. Several works support that the anti-stress effects of eCBs in animal models could involve the anti-inflammatory properties of these compounds [35–37, 167]. For instance, subchronic stress in mice induced an increase in the pro-inflammatory profile in the frontal cortex, which was attenuated by pharmacological activation or overexpression of CB1Rs or CB2Rs [35, 36]. Moreover, overactivation of microglial cells to LPS stimulation, the anxiogenic response, and sensitized conditioned fear response after repeated stress exposure in mice were attenuated or prevented by a non-selective CB1/CB2 agonist during stress [37]. Accordingly, eCBs can attenuate behavioral responses in stressful situations and limit the inflammatory response to different stimuli, acting as a buffer system against stressors [29, 39].
Several data also support an interplay between cannabinoid receptors and the TLR4 pathway [180–183]. For example, TLR4 and CB2 are colocalized in peritoneal macrophages; exposure to LPS or to a CB2 antagonist decreased their interaction, whereas 2-AG increased it, suggesting CB2 activation by 2-AG could dampen TLR4 signaling [180]. Corroborating this idea, a recent study in mast cells indicated that persistent activation of TLR4 by LPS engages the eCB 2-AG, which activates CB2 and attenuates inflammatory response [181]. Moreover, several other in vitro studies demonstrated that inflammatory effects of TLR4 activation are attenuated by cannabinoid receptor agonists [181–183].
Altogether, the data mentioned above support the hypothesis that activation of the TLR4 pathway in microglia by stress mobilizes 2-AG, which would activate CB2 as a protective mechanism to attenuate further microglia activation and its consequences. However, considering that 2-AG is a full agonist at CBs receptors, exposure to increased 2-AG levels during repeated stress can result in CB1 downregulation, and decreased CB1 signaling impairs control of synaptic neurotransmission [174], resulting in behavioral changes. Moreover, we speculate if increased 2-AG during repeated stress could somehow impair neuronal CB2 signaling, contributing to impaired synaptic transmission.
Additional to CB1Rs/CB2Rs, behavioral, neuroprotective, and anti-inflammatory effects of eCBs in stress situations can be further mediated by nuclear PPARs, mainly PPARα and PPARγ [164]; the last one has the highest expression levels in the CNS, in different cell types [184]. PPARs act as transcription factors by binding to specific DNA regions and regulating gene expression [165]. PPARα KO mice did not demonstrate anxiety or depressive-like behaviors but showed increased fear learning [170]. Moreover, exposure to CUMS increased PPARα protein and mRNA in the hippocampus. It also induced behavioral changes, HPA axis activation, oxidative parameters, and reduced neurotrophic factors in the hippocampus. These changes were blocked by the eCB PEA in a dose-dependent manner, and the PEA effect was attenuated or prevented by an antagonist of PPARα, MK886 [169].
Corroborating a potential protective role also for PPARγ receptors, animals with PPARγ deletion in neurons show an exacerbated anxiogenic effect after acute stress, similar to a PPARγ antagonist [185]. Moreover, exposure to acute restraint stress (6 eCB) in rats increased the expression of PPARγ in the PFC [186, 187], and activation of these receptors attenuated stress-induced neuroinflammation, oxidative/nitrosative consequences in the PFC [167], and the anxiogenic effect [171]. Reduced NMDA signaling and increased glutamate uptake by glia could be involved in those effects [166]. Contrary to acute, repeated (4 days to 7 days) homotypic stress reduced PPARγ levels in the PFC [2] or amygdala [168]. These studies did not evaluate PPARα involvement, nor if eCBs attenuated the observed changes.
Interestingly, PPAR activation can interfere with proinflammatory actions of HMGB via NFκB inhibition and also inhibits HMGB transcription [188]. Therefore, PPAR could regulate and be regulated by TRL4 signaling. For instance, the reduced PPARγ levels in the PFC after repeated stress was not observed in TLR4 KO mice [2] and was attenuated by repeated treatment with a microglia inhibitor during stress [168]. Similarly, in macrophages, LPS inhibited PPARγ mRNA synthesis through a NFκB-dependent mechanism, which was not observed after TLR4 pharmacological inhibition or in TLR4 KO mice [189]. We suggest that increased PPARγ transcription levels by acute stressors could be a protective mechanism after TRL4 pathway activation, which could parallel the initial changes in the eCB signaling in the brain [176] to promote stress habituation (Figure 3A). However, after prolonged or repeated exposure to the same kind of stress, this mechanism would be dysregulated, with the intensification of changes in the eCB system [176], increased TLR4 signaling, and decreased levels of PPARγ, which could contribute to behavioral consequences of stress (Figure 3B). Although there are still few studies evidencing a direct relationship between stress, eCB system, and TLR4 in the brain, the in vitro and in vivo evidence of anti-inflammatory effects of eCBs mentioned above and others [190–192], including with potential involvement of TLR4 [2, 180, 181, 185], suggest that these mechanisms are important in stress-related disorders and their modulation could be beneficial in these disorders, such as MDD.
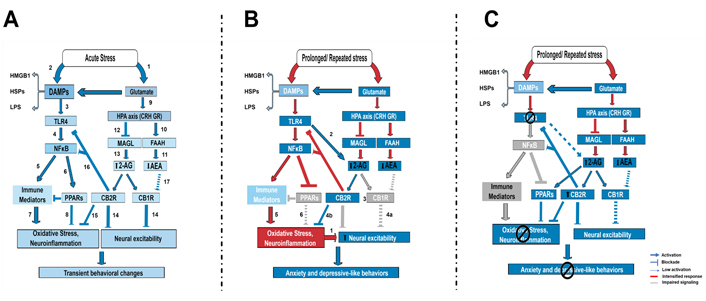
Schematic representation of TLR4 and eCB signaling interaction in the modulation of behavioral response to stress exposure. A) Short-term acute stress, including LPS, increase glutamate release (1) and release DAMPs (2), which can activate microglia. Microglia activation can result from the activation of TLR4 signaling. DAMPs such as HMGB activate TLR4 receptors (3) in brain areas, such as the PFC, resulting in NFκB activation (4), transcription of proinflammatory-related genes (5), and also PPARs (6). PPARs can be activated by fatty acids, including its derivatives, such as the eCBs 2-AG, PEA, and oleoylethanolamide (OEA, not shown). These mechanisms have opposite effects on oxidative stress and neuroinflammation induction, with immune mediators increasing them (7), whereas PPAR activation decreases it (8). Glutamate, also considered a DAMP, induces HPA axis activation (9), which consequently impacts the eCB signaling: CRH release increases FAAH activity (10) and decreases AEA levels (11), and glucocorticoid receptor (GR) activation by corticosterone could decrease MAGL activity (12) and increase 2-AG levels (13) in the brain. 2-AG is a full agonist at CB receptors, whereas AEA is a partial agonist. 2-AG action at CB1 and CB2 modulates neuronal excitability (14) and neuroinflammation (15), respectively, including by limiting TLR4 activation (16). AEA signaling is attenuated (17). The resultant expected effect would be transient behavioral changes, stress habituation, and coping behavior; B) under prolonged or repeated stress exposure, the TLR4 pathway activation and glutamate release are exacerbated. The proinflammatory profile is increased and PPARγ expression is suppressed, which contributes to increasing oxidative stress and neuroinflammation, contributing to increased neuronal excitability (1). The eCB changes induced by stress are also exacerbated, with even lower levels of AEA and higher levels of 2-AG. Persistent TLR4 activation also recruits 2-AG (2). High levels of 2-AG can contribute to the downregulation of CB1Rs (3), impairing the control of neuronal excitability via CB1 (4a), but maintaining activation of microglia CB2Rs (4b). CB2R could be downregulated in neurons (no literature report about that), contributing to neuronal excitability (?). The higher oxidative stress/neuroinflammation (5) resultant from high TLR4 activation and decreased PPAR (6), along with impaired neuronal CB1 signaling (4) could contribute to higher neuronal excitability, impaired coping, and behavioral dysfunction; C) after pharmacological or genetic blockade of TLR4, the impact on eCB signaling and PPARγ expression could be restored. TLR4 blockade can prevent impairment in microglia PPARs signaling by decreasing the NFκB pathway, and decreasing oxidative damage and neuroinflammation. This blockade can contribute to decreasing the impact on the eCB signaling, which can now control the neuroimmune response by activating PPARs and CB2 or buffer the neuronal activity by acting on neuronal CB receptors
Thus, our current working hypothesis is that TLR4 pathway activation by DAMPs after acute or repeated homotypic stress exposure differently impacts eCB signaling through CB1, CB2, and PPARγ effects, influencing behavioral response and stress habituation (Figure 3). A protective mechanism induced by TLR4 blockade after acute stress can be mediated by intensifying eCB signaling at PPARγ and CB1Rs/CB2Rs, contributing to stress habituation. After prolonged or repeated stress, TLR4 blockade could prevent impairment in PPARγ expression and limit increased levels of 2-AG during stress, potentially attenuating effects in the eCB signaling. The eCBs could then act via PPARγ, CB1, and CB2 to counteract stress effects (Figure 3C). Considering that several studies evaluating neuroprotective and anti-inflammatory effects of cannabinoids, including AEA, 2-AG, PEA, OEA, synthetic agonists, and cannabidiol, in models of Alzheimer, multiple sclerosis, drug abuse, and cognition, for example, indicate that their effects are mediated by CB1/CB2 and PPAR receptors (for review, see [193]), adequate signaling via these receptors, and maybe others, can be essential for the overall anti-stress effects of eCBs in inflammatory conditions and psychiatric disorders.
In summary, dysfunctional eCB signaling under stressful situations can contribute to increased neuronal excitability and facilitation of the inflammatory effects of stress in neuronal circuits. Since there is a direct connection between eCB signaling during stress and the immune system, with crosstalk between TLR4 and eCB system, possibly involving different receptors, we propose that inhibiting TLR4 in microglia would facilitate stress adaptation by decreasing the TLR4 pathway activation itself, but could also do so by regulating the eCB synthesis and signaling in microglia and neurons. These eCBs would act on microglia receptors, contributing to decreasing neuroinflammation, but could also regulate neuronal excitability by activating neuronal receptors. Therefore, the possible beneficial effects of inhibiting TLR4 signaling in stress could be, in part, through the facilitation of eCB signaling, mainly by CB1, CB2, and PPARs.
The current available therapy to treat neuropsychiatric disorders still faces a lack of efficacy or refractoriness. These problems are probably related to the complex neurobiology of these disorders. For instance, several pieces of evidence indicate that some, but not all, individuals suffering from psychiatric disorders have a proinflammatory profile. Some data indicate that this profile is related to symptom severity and could predict resistance to conventional antidepressant treatment [88, 194–196]. Moreover, several data show the antidepressant effects of anti-inflammatory drugs (for review, see [196]). Therefore, targeting immune system mechanisms could improve symptoms, allowing some patients to respond to treatment.
Based on the evidence discussed in this review, the overactivation of the TLR4 pathway by stress exposure and signs of its alteration in psychiatric patients indicate that it could contribute to neuropsychiatric disorders. Modulating the TLR4 pathway is expected to decrease the NFκB activation and the NLRP3 inflammasome pathway, attenuating the expression of proinflammatory cytokines. Therefore, the imbalance in this system can trigger deleterious processes in the body by increasing the inflammatory response. Accordingly, modulation of this pathway could be a promising therapeutic strategy for those diseases.
Despite several studies proposing the modulation of this pathway to counteract stress effects, as discussed in this review, many aspects related to neuroinflammation remain unclear, and it is unlikely that one singular mechanism would promote clinically relevant effects. However, considering the potential relationship between the TLR4 pathway activation and eCB system actions, modulation of the TLR4 pathway could directly modulate its pathway and implicate eCB signaling, amplifying its potential effects.
2-AG: 2-arachidonoylglycerol
AEA: anandamide
AP-1: activator protein 1
BDNF: brain-derived neurotrophic factor
CB1: cannabinoid type 1
CB2Rs: cannabinoid type 2 receptors
CMS: chronic mild stress
CNS: central nervous system
COX-2: cyclooxygenase-2
CRH: corticotropin-releasing hormone
CRS: chronic restraint stress
CUMS: chronic unpredictable mild stress
CX3CR1: C-X3-C motif chemokine receptor 1
DAMPs: damage-associated molecular patterns
eCB: endocannabinoid
EPM: elevated plus-maze
FAAH: fatty acid amide hydrolase
fr-HMGB1: fully reduced high mobility group box 1
FST: forced swim test
GM-CSF: granulocyte-macrophage colony-stimulating factor
GR: glucocorticoid receptor
HMGB1: high mobility group box 1
HPA: hypothalamus-pituitary-adrenal
HSPs: heat shock proteins
Iba-1: ionized calcium binding adaptor molecule 1
IDO: indoleamine 2,3-dioxygenase
IFNα: interferon α
IkB: inhibitor of nuclear factor kappa B
IKK: inhibitor of nuclear factor kappa B-kinase
IL-1β: interleukin 1β
iNOS: inducible nitric oxide synthase
IRAK: interleukin-1 receptor-associated kinase
IRF3: interferon regulatory factor 3
KO: knockout
LPS: lipopolysaccharide
MAGL: monoacylglycerol lipase
MAPK: mitogen-activated protein kinase
MD-2: myeloid differentiation factor 2
MDD: major depressive disorder
mRNA: messenger RNA
MyD88: myeloid differentiation factor 88
NFκB: nuclear factor kappa B
NLRP3: nucleotide oligomerization domain-like receptor protein 3
NMDA: N-methyl-D-aspartate
OFT: open field test
PAMPs: pathogen-associated molecular patterns
PEA: palmitoylethanolamide
PFC: prefrontal cortex
PGE2: prostaglandin E2
p-IkBα: phosphorylated inhibitor of nuclear factor kappa B α
p-NFκB: phosphorylated nuclear factor kappa B
PPARα: peroxisome proliferator-activated receptor α
PRR: pattern recognition receptor
PVN: paraventricular nucleus of the hypothalamus
SPT: sucrose preference test
TAK1: transforming growth factor β-activated kinase 1
TIRAP: Toll/interleukin-1 receptor domain-containing adapter protein
TLRs: Toll-like receptors
TNF-α: tumor necrosis factor α
TRAF6: tumor necrosis factor receptor-associated factor 6
TRAM: Toll/interleukin-1 receptor-domain-containing adapter-inducing interferon-β-related adaptor molecule
TRIF: Toll/interleukin-1 receptor-domain-containing adapter-inducing interferon-β
TST: tail suspension test
FJCSJ and LCC conceptualized the idea about this topic, created the figures and the Table. FJCSJ, LCC, and SFL searched the literature and wrote the original draft. SFL supervised FJCSJ and LCC writing, and performed the review and editing. All authors contributed to the final manuscript and approved the final version of the manuscript.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Sabrina Francesca Lisboa receives fellowship from The São Paulo Research Foundation- FAPESP [2017/19731-6], National Council for Scientific and Technological Development-CNPq [420818- 2018-9], and L’Oreal/Brazilian Academy of Sciences-For Women in Science. Fábio José Coelho Souza-Junior receives a fellowship from Coordination for the Improvement of Higher Education Personnel-CAPES [88887.510048/2020-00]. Laura Colete Cunha received undergraduate student fellowships from CNPq [166068/2020-9, 2019-1444, 2021-1306]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Haruka Sawamura ... Satoru Matsuda
Sarah Otaru, David A. Lawrence
Niklas Frank ... Carola Y. Förster
Mydhili Radhakrishnan ... Sumana Chakravarty
