 Review
Review

Affiliation:
1Department of Environmental Health Sciences, University at Albany School of Public Health, Rensselaer, NY 12144, USA
ORCID: https://orcid.org/0000-0001-9533-2582
Affiliation:
1Department of Environmental Health Sciences, University at Albany School of Public Health, Rensselaer, NY 12144, USA
2Clinical and Experimental Immunology, Wadsworth Center, New York State Department of Health, Albany, NY 12208, USA
Email: David.lawrence@health.ny.gov
ORCID: https://orcid.org/0000-0002-8940-9640
Explor Neuroprot Ther. 2022;2:141–161 DOI: https://doi.org/10.37349/ent.2022.00025
Received: March 14, 2022 Accepted: May 20, 2022 Published: August 11, 2022
Academic Editor: Raymond Chuen-Chung Chang, The University of Hong Kong, China
The article belongs to the special issue Intervention of Neuroimmune Responses
Autism spectrum disorder (ASD) is a class of neurodevelopmental disorders (NDD) characterized by deficits in three domains: impairments in social interactions, language, and communication, and increased stereotyped restrictive/repetitive behaviors and interests. The exact etiology of ASD remains unknown. Genetics, gestational exposure to inflammation, and environmental stressors, which combine to affect mitochondrial dysfunction and metabolism, are implicated yet poorly understood contributors and incompletely delineated pathways toward the relative risk of ASD. Many studies have shown a clear male bias in the incidence of ASD and other NDD. In other words, being male is a significant yet poorly understood risk factor for the development of NDD. This review discusses the link between these factors by looking at the current body of evidence. Understanding the link between the multiplicity of hits—from genes to environmental stressors and possible sexual determinants, contributing to autism susceptibility is critical to developing targeted interventions to mitigate these risks.
Autism spectrum disorder (ASD) is a class of neurodevelopmental disorders (NDD) characterized by persistent deficits in multiple domains, including deficits in social reciprocity, verbal and nonverbal communication, and deficiencies in skills required for developing, maintaining, and understanding social interactions [1]. In addition to the social and communication deficits, restricted and repetitive patterns of behavior, interests, or activities are required to diagnose ASD [2]. Furthermore, autistic individuals may have comorbidities like gastrointestinal (GI) disorders, epilepsy, immune disorders, sleep disorders, and/or intellectual disability [3]. The spectrum also includes disorders like pervasive developmental disorder-not otherwise specified (PDD-NOS), Asperger syndrome, Rett syndrome, and childhood disintegrative disorder [4]. Although the exact etiology of ASD remains unknown, several etiologic factors have been suggested. These include but may not be limited to genetic, epigenetic, environmental, immunological, and endocrinological factors. In addition, a growing body of evidence highlights the role of the gut microbiome, mitochondrial dysfunction, and endoplasmic reticulum (ER) stress in the molecular pathogenesis of autism.
The complex yet poorly understood interactions between these factors contribute to the neurodevelopmental changes and features characteristic of ASD. Several hypotheses have been proposed to explain the complex interactions between these factors. One such hypothesis is that individuals who develop autism are genetically susceptible [5]. Numerous studies have identified susceptibility genes [6–11]. Another hypothesis is the environmental hypothesis, which suggests that environmental risk factors synergize with gene-associated susceptibilities to create a threshold point for brain dysfunction [12]. Another hypothesis is that inflammatory events during pregnancy may disrupt the normal expression of immune cellular and molecular functions during critical stages of brain development, thereby contributing to the risk for NDD, including ASD [12]. This hypothesis is also known as the maternal immune activation (MIA) hypothesis. As for the gut microbiome, changes in its composition have been hypothesized to influence the development and presentation of symptoms of ASD, and this is referred to as the gut microbial dysbiosis hypothesis [13–17]. Lastly, a more nuanced hypothesis tries to combine all the other hypotheses and, at the same time, introduce a new way of understanding the possible etiology of ASD. Given the apparent male preponderance in the incidence of ASD and NDD, this hypothesis suggests that inherent differences in the neuroimmune system of susceptible developing males versus females increase the risk for dysregulated brain development [18]. This review examines these risk factors and hypotheses by looking at the current body of evidence. Understanding the link between the multiplicity of hits—from genes to environment and possibly sexual determinants, that contribute to susceptibility is critical to developing targeted interventions to mitigate these risks.
Several lines of evidence ranging from epidemiologic to experimental support genetics as a major etiologic factor associated with the development of ASD. For example, some studies have shown the co-occurrence of ASD with rare genetic syndromes such as tuberous sclerosis complex (TSC) and fragile X syndrome (FXS), Rett syndrome, and Angelman syndrome [19]. However, none of these syndromes currently linked to ASD have been proven to cause autism selectively. Instead, each syndrome is characterized by abnormal neuro-behavioral phenotypes, including ASD. Furthermore, the existence of different phenotypic variants in ASD may reflect an interaction between multiple gene loci, as well as the existence of distinct dysfunctional genes within an individual’s genome [19]. For example, a study by Watson et al. [20], identified a set of patients with Angelman phenotypes due to genetic mutations of the X-linked methyl CpG binding protein 2 (MECP2), a protein that is also defective in the Rett syndrome. Although only Rett syndrome is categorized under the autism spectrum, both syndromes have features characteristic of autism and can be termed syndromic ASD. Thus, the common mutation noted in both syndromes suggests a genetic or molecular overlap in biological pathways or brain circuits influencing the different forms of syndromic ASD [21]. Although autism is observed at a higher-than-expected frequency in these rare genetic syndromes, they together account for less than 10% of autism cases [19]. However, because the dysfunctional gene loci in most of these conditions have been identified, the study of these conditions provides substantial insight into potential candidate genes associated with the development of ASD. Such insight is critical for understanding the relationships between subtypes of ASD and identifying common links between them.
Other evidence for the genetic basis of autism comes from twin studies which reveal a 60–90% concordance rate of autism in monozygotic twins (MZT) and 0–24% in dizygotic twins [22, 23]. Not unexpectedly, based on the higher male prevalence of ASD [24], a twin study reported that > 80% of the MZT and the dizygotic twins (DZT) of the same or different sex were males [25]. Interestingly, 77% of both male MZT had ASD, whereas only 50% of both MZT females had ASD; for same-sex DZT, the percentages for both twins having ASD were 31% and 36% for males and females, respectively [26]. The results suggest a moderate genetic heritability and a substantial shared twin environmental component. The elusive current unknown may be the microbiota-gut-brain axis. Although not well investigated, the gut microbiome develops before birth, and it differs between fetal twins based on the placental microbiome [27–29]. The gut microbiome has been reported to influence brain development [30, 31]. Familial studies also demonstrate an 18-fold increase in autism risk among siblings of an autistic child, compared to the general population [32]. Studies also show that the concordance rate for non-twin siblings decreases as the interval between pregnancies increases, suggesting that non-genetic factors, influence the concordance rates [33]. These studies, taken together, suggest a genetic contribution to the etiology of autism.
In recent years, whole-genome studies using novel techniques have allowed the identification of new and essential candidate loci [6–11]. Many of these candidate genes encode proteins involved in neuronal development, regulation of synapse formation and function, ion channels, and many other critical biological pathways [34]. For example, the secreted protein acidic and rich in cysteine-like protein 1 gene (Sparcl1) or high endothelial venule protein (Hevin) gene encodes an extracellular matrix protein that plays an important role in regulating synaptogenesis and synaptic plasticity. Mutation of this gene results in the production of a misfolded HEVIN protein which cannot be transported out of the endoplasmic and has been associated with an increased risk of ASD [35]. A similar mutation has been noted in the Neuroligin3 (NLGN3) gene which also encodes extracellular matrix proteins. The mutation results in the production of defective proteins which are retained in the ER leading to ER stress [36].
Although certain genes have been implicated in playing a role in the etiology of ASD, it is evident that genetic factors alone do not account for all cases of ASD. Genetic syndromes including heritable and de novo mutations account for only 10–20% of ASD [5]. This means that non-genetic factors have a much greater attributable risk in the etiology of autism. ASD does not follow a Mendelian pattern of inheritance, and in non-Mendelian disorders, environmental factors often determine whether individuals who carry susceptibility genes become ill [37]. Therefore, given the dominance of non-genetic factors in the etiology of ASD, an understanding of the exposome becomes critical to unraveling its etiology. The exposome encompasses all the environmental exposures or non-genetic influences that an individual encounters from conception onwards and how these influences affect the individual’s health. The exposome may influence the extent of gene expression; that is, have an epigenetic input, that may ultimately determine the occurrence of ASD in susceptible individuals.
The Centers for Disease Control and Prevention (CDC) reported an increase in autism rates in the United States from 1 in 150 children in 2000 to 1 in 54 in 2016, with the current rate now at 1 in 44 children [24]. While some agree that this represents a true increase in the prevalence of autism, others argue that this may simply reflect an increase in the diagnosis of autism [38].
Nevertheless, these figures have drawn attention to the role of environmental factors in the etiology of autism. Environmental factors in this case refer to anything apart from the genes that are capable of producing a biological or behavioral response including modification of gene expression (epigenetics). It is somewhat synonymous with the exposome.
Several environmental pollutants have been associated with the development of autism. These include chemicals like pesticides, polychlorinated biphenyls (PCBs), perfluoroalkyl and polyfluoroalkyl substances (PFAS), air pollutants [e.g., diesel exhaust particles, and nitrogen dioxide (NO2)], heavy metals, phthalates, medications (e.g., valproate and thalidomide), and even food allergens (e.g., gluten and casein) [38–42]. Some of these chemicals are known to cross the placental barrier and exert neurotoxic effects on the developing fetal brain [42–45]. When it comes to environmental factors such as smoking, studies exploring the association between maternal smoking and ASD in offspring have reported varying results. For example, one meta-analysis of 15 studies on maternal prenatal smoking and ASD risk in offspring using a random-effects model, found no evidence of an association between the two [summary odds ratio (OR) 1.02, 95% confidence interval (CI): 0.93–1.12] [46]. Another meta-analysis of 22 observational studies using the same model also reported no significant association between smoking during pregnancy and offspring with ASD [47]. However, when population-level smoking metrics (matching pooled ORs with smoking prevalence among adult males in each country) were applied, the risk for autism was significantly associated with maternal smoking [47]. High perinatal alcohol exposure has been associated with NDD such as fetal alcohol syndrome [48–50]. However, studies investigating the relationship between maternal alcohol consumption and ASD have also yielded conflicting results [51–53].
Environmental factors are not limited to chemicals and drugs but include parental age, perinatal risks, nutrition, maternal illnesses, and extreme psychosocial factors [54]. For example, the BTBR T+ Itpr3tf/J (BTBR) mice are an experimental model for behaviors that closely resemble autism, and this strain has aberrant autoimmunity regarding the production of immunoglobulin G (IgG) antibodies to the brain antigens [55]. When BTBR IgG is injected into pregnant C57BL/6J (B6) mice (a strain with normal behavior), the B6 adult offspring have more ASD-like behaviors [56]. Similarly, when B6 embryos are implanted into BTBR females, the B6 adult offspring have behaviors shifted toward more ASD-like behaviors; in contrast, implanting BTBR embryos into B6 pregnant females improved the adult behaviors of the BTBR adult offspring [56]. Furthermore, implanting BTBR embryos into B6 females lessened the ASD-like behaviors of BTBR offspring [55]. Interestingly, the maternal stress from intraperitoneal injections of sterile saline into the pregnant B6 mice also influenced the behaviors of the B6 offspring as adults [56]. These studies suggested that the maternal environment can partially overcome the genetic influences suggesting both genetics and environment are influential. Maternal IgG antibodies, cytokines, and other organ factors including those unique to the placenta are transported into the fetus and may affect neural development. Antibodies to numerous antigens have been implicated in the development of ASD [57–59]. Factors present in pregnant BTBR mice influence the offspring’s behavior even when the offspring have the background genetics of a mouse strain with normal behavior and vice versa. It also suggests that physical stress itself can be a modifying factor especially if such stress comes during daylight times since mice are nocturnal. However, it is essential to note that although the BTBR mouse is used as an autistic model, it does not entirely depict human ASD, in that incidence of autistic behavior in female BTBR mice is equivalent to the incidence in males. However, this mouse model clearly supports the fact that environmental factors synergize with genetic and other factors to trigger the development of ASD.
Environmental factors can be causal if they are harmful and precede ASD, mediating if they influence the expression of autism susceptibility genes, moderating if they impact the severity of autism, and protective if they decrease the risk of ASD [60]. The extent to which environmental factors mediate, moderate, or protect against autism is still the subject of much debate and research. However, an understanding here is also critical to deciphering the complex etiology of autism.
The role of MIA in the development of ASD is an area of growing research interest. The first clue linking MIA to the development of autism came by way of a 1971 study which showed that children exposed to rubella in the first trimester of pregnancy had an increased incidence of congenital rubella syndrome (CRS), and as much as 8–13% of these children were later diagnosed with ASD [61–63]. A similar association is seen between influenza infection during pregnancy and schizophrenia in the offspring of affected women [64]. This association is not limited to viral infections as it has also been demonstrated following bacterial infections [65].
Researchers suggest that MIA or the generation of maternal inflammatory response to infections, rather than the infections themselves, is to blame for the adverse autistic phenotype [66]. The BTBR mouse strain has an autoimmune phenotype, which might be considered a genetically spontaneous form of MIA. They express high serum IgG levels, IgG antibodies to brain antigens, and high IgG levels in the brain. They have high numbers of plasma cells in the blood and spleen, an increased proportion of T helper type 2 (Th2) cells, and elevated peripheral blood neutrophils all of which are suggested to promote humoral immunity, and may represent a heightened inflammatory state [55, 67]. In addition, the BTBR mouse strain displays autism-like behaviors and is often used as an autism model. This relationship between immune system dysregulation and ASD-like behaviors noted in the BTBR mouse parallels human research reporting immune system dysregulation in autistic persons [65, 68–70].
While the exact mechanisms by which MIA results in these autistic phenotypes remain unclear, it is accepted that the induction of maternal cytokine response and the subsequent downstream cellular and molecular events are responsible for the maladaptive neurodevelopmental changes [71]. Cytokine positive or negative influences on nervous system development and functions can be substantial, and the cytokines may be centrally and/or peripherally produced and can be produced by multiple types of immune cells as well as neurons, themselves [6, 71].
Polyinosinic:polycytidylic acid (poly I:C) and lipopolysaccharide (LPS) are viral and bacterial mimics respectively. They stimulate pattern recognition receptors (PRRs), Toll-like receptor 3 (TLR3), and TLR4, respectively, which induce proinflammatory cytokine levels in the maternal serum, amniotic fluid, and placenta [72–74]. These proinflammatory cytokines have been detected at elevated levels in the fetal brain following MIA with poly I:C or LPS [72–74]. In this regard, one of the most studied cytokines is interleukin-6 (IL-6). IL-6 is a multifunctional inflammatory cytokine produced by lymphoid and non-lymphoid cells [75]. It is a polypeptide mediator regulating the immune response, the acute phase reaction, and hematopoiesis [75].
In murine models, the induction of MIA using a viral mimic-synthetic double-stranded RNA (poly I:C) resulted in an increase in maternal IL-6 messenger RNA (mRNA) and IL-6 protein in the placenta and elsewhere in the maternal system [76]. In addition, placentas from the MIA mothers exhibited increases in CD69+ decidual macrophages, granulocytes, and uterine natural killer (NK) cells, indicating elevated early immune activation [76]. The overall effect was that offspring of the immune-activated dams displayed behavioral features reminiscent of human autism [77–83]. Interestingly, when pregnant dams were injected with poly I:C at mouse embryonic developmental stage 12.5 (E12.5), the offspring as adults exhibited ASD-like behavior, and IL-6 had been generated in the brain. However, when poly I:C was injected into pregnant dams lacking IL-6 receptor (IL-6R) on trophoblasts (placenta), IL-6 was still generated; however, neuroinflammation did not occur and the offspring as adults exhibited normal behavior [78]. Conversely, induction of MIA via the injection of poly I:C in IL-6 knock-out mice did not result in many of the behavioral changes noted in the offspring of wild-type mice after MIA [81]. Additionally, coadministration of an anti-IL-6 neutralizing antibody in the poly I:C model of MIA prevents the behavioral deficits caused by poly I:C administration and normalizes the associated changes in gene expression in the brains of adult offspring [81].
These studies indicate that MIA induces maternal IL-6, which can be transported into the fetus, but if the placental trophoblasts lack IL-6Rα the MIA-generated IL-6 may directly affect the placenta influencing placental functions that may allow the greater entrance of maternal factors to enhance fetal neuroinflammation and processes affecting ASD. In addition, the placenta itself has been shown to develop a strong immunological and morphological reaction to the MIA and these may contribute to the maladaptive fetal brain changes [5, 76, 84–86].
These studies support the importance of the maternal-placental-fetal interface in the health of the offspring. Besides increasing IL-6 in the fetal brain, MIA increases the expression of IL-4 and interferon gamma-induced protein 10 (IP-10 or CXCL10), and interestingly, normal baseline levels of maternal IL-6 are needed to maintain the fetal brain levels of other cytokines such as IL-2 and IL-12p40/p70 [84]. Additional cytokines of interest include IL-1β, IL-4, IL-6, IL-8, interferon-gamma (IFN-γ), and transforming growth factor-beta (TGF-β). Their functions and their potential role in the development of ASD are documented in Table 1.
Some cytokines of interest in autism [87]
| Cytokine | Function | References |
|---|---|---|
| IL-1β | IL-1β is an inflammatory cytokine expressed very early in immune responses. It propagates tissue inflammation by activating local immune cells and the vascular endothelium. IL-1β also stimulates IL-6 production. IL-1β can cross the blood-brain interface and stimulate its own expression in the hypothalamus, resulting in altered neuroendocrine activity including fever and sickness behavior. IL-1 and its receptors are found throughout the nervous system during critical developmental periods. It appears to have a role in both central nervous system (CNS) pathology and healing. | [69, 87–97] |
| IL-4 | IL-4 is a cytokine that activates Janus kinase/signal transducer and activator of transcription (JAK/STAT; STAT6), mitogen-activated protein kinase (MAPK), and phosphoinositide 3 (PI3) kinase signaling pathways. It induces “alternatively activated macrophages that promote tissue repair, activate basophils and mast cells, and promote B-cell isotype switching towards IgG1 and IgE. IL-4 also participates in the antihelminth immune response by inducing gut epithelial cell turnover. In addition, it plays a role in allergy and asthma-related immune responses. During brain development, IL-4 promotes the formation of oligodendrocytes among neuronal progenitor cells. In later stages of neurodevelopment, IL-4 alters the formation of synapses, leading to an increase in the proportion of gamma-aminobutyric acid (GABAergic) synapses in cell cultures. | [70, 87, 98–107] |
| IFN-γ | IFN-γ is a type II produced primarily by T cells and NK cells during cell-mediated immune responses. Its main function is to activate macrophages and participate in the immune response against viral infections. It also signals through the JAK/STAT (STAT1) and MAPK pathways. IFN-γ and IL-4 offset each other’s activity through Th1/Th2 interactions, so a dysfunction/imbalance in one cytokine often affects the other. IFN-γ also influences dendritic morphology and synaptogenesis, resulting in long-term changes in neuronal connectivity and communication. | [87, 108–116] |
| TGF-β | TGF-β is a superfamily of proteins that includes growth differentiation factors, bone morphogenic proteins (BMPs), activins, and inhibins. They function to maintain immune homeostasis, direct lymphocyte differentiation, and orchestrate aspects of embryonic development. They exist in three isoforms, each with overlapping roles. TGF-β1 is largely immune-suppressive, limiting excessive T cell activity and inflammation. It is also involved in neuronal migration, survival, and synaptogenesis. | [117–120] |
A common characteristic in the activity of the cytokines mentioned above is their ability to alter synapse formation. There are immune synapses [121], neuronal synapses [122], and neural-immune synapses [123, 124]. The synapses seem to be the principal meeting point between the immune and the nervous systems, and there is increasing evidence highlighting abnormalities in neural synaptogenesis as potential factors underlying the etiology of ASD. Microglia—the resident immune cells of the CNS, have also been implicated in normal synaptogenesis and the formation of neural networks in the developing brain [125–128]. Evidence suggests that abnormalities in microglia play a role in ASD [126].
Microglia alterations have been detected in postmortem autistic brain tissue and animal models of ASD [106]. Some detected alterations include somal enlargement, process retraction and thickening, and extension of filopodia from processes [129]. These alterations indicate microglial activation. Microglia activation might be triggered primarily environmentally, with the response likely contingent on specific gene-environment interactions [129]. Furthermore, microglial activation may be present throughout the life span in many patients with autism, including at an early, developmentally critical age [129, 130]. To better understand the origin and effects of this phenomenon, it will be necessary to quantitatively determine whether microglial activation occurs alongside other cellular, neuroimmune, and genetic abnormalities associated with autism.
While microglial activation may be associated with the development of ASD, other studies suggest a similar association with microglia depletion. One animal study revealed that microglia depletion in mice altered synaptic structural plasticity (microglia spine formation and elimination) during the late postnatal period (P19) and in young adulthood (P30) [131]. In addition, mice depleted of microglia showed deficits in multiple learning tasks and significantly reduced motor-learning-dependent synapse formation [131]. The same study showed that the removal of brain-derived neurotrophic factor (BDNF) from microglia essentially replicated the effects of microglia depletion [131]. In addition, microglial BDNF increases neuronal tropomyosin-related kinase receptor B phosphorylation, a key mediator of synaptic plasticity [131, 132]. Together, these findings indicate that microglia serve essential physiological functions in learning and memory (two domains that are largely impaired in autism) by promoting learning-related synapse formation through BDNF signaling [131].
Although most research into the altered immune function in autism has focused on the inflammatory role of microglia in destroying already developed neural networks, the critical non-inflammatory role of microglia in normal neurodevelopment is increasingly being recognized as an essential mechanistic pathway for the development of ASD [126]. This recognition exemplifies the importance of cellular homeostasis for healthy neural development.
The most significant comorbidities associated with ASD are GI comorbidities [133]. Compared to the general population, autistic individuals have been shown to have a higher frequency of GI problems [134]. GI problems also appear to affect the severity of autistic symptoms [135]. Many of these problems appear to be influenced by the gut microbiome. The gut microbiome is a term that describes the diversity of microorganisms living in the gut, including their genome and their interaction with the host [136]. It is a fundamental part of human physiology and studies have shown that changes in the gut microbiome can significantly alter several aspects of human physiology including immune, GI, and brain functions (the microbiota-gut-brain-axis) [17, 137–144]. Studies have demonstrated that gut microbiome dysbiosis can directly influence behavioral and neuropathological endophenotypes of human autism [17, 145–148]. Several mechanisms and pathways by which the gut microbiota may bring about these changes are being explored including their influence on neurogenesis, neuroimmune pathways, and the metabolism of compounds like tryptophan and serotonin [133]. Tryptophan is the sole precursor of serotonin which is an important neurotransmitter involved in mood, behavior, and cognition, all of which are affected in autism [149]. Other neurotransmitters affected by gut microbiota include γ-aminobutyric acid and norepinephrine which also influence various aspects of behavior [149]. The effects of GI microbiota on neurogenesis have been demonstrated in murine studies where germ-free (GF) mice (i.e. mice that are microbiota deficient from birth) showed significant alterations in the morphology of neurons in the dorsal hippocampus compared to conventionally colonized (CC) mice. Accordingly, the GF mice exhibited deficits in social cognition, altered stress hormone signaling as well as anxiety-like behaviors, features which are reminiscent of human autism [150]. These findings suggest that the gut microbiota play a significant role in the development of normal gross morphology of the amygdala and hippocampus and that absence may contribute to neural remodeling which may ultimately result in maladaptive stress responsivity and behavioral profile [150].
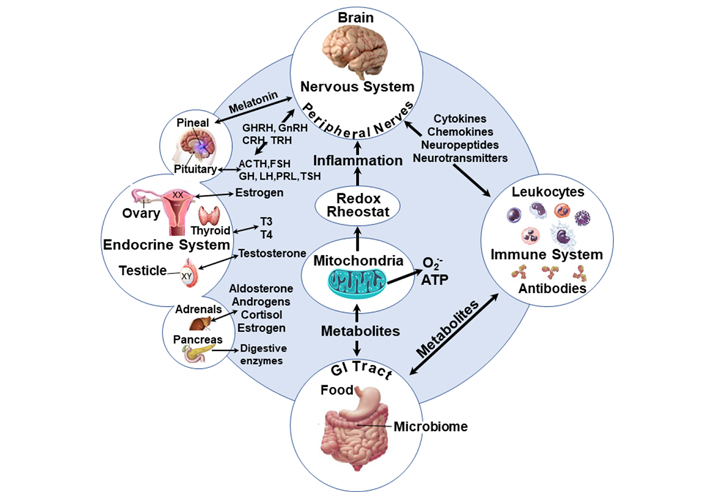
Schematic of the inter-organ interactions and the central involvement of mitochondrial functions. Health disparities, which can differentially influence the development of autism, may also affect the level of oxidative stress leading to more cellular damage and alteration of the immune neuroendocrine network [151, 152]. Mitochondria activity is posited to be centrally implicated in the development of ASD since mitochondria and their effect on oxidative stress affect ASD [153, 154] and the redox rheostat [155, 156] which increases with SDOH. Furthermore, male embryos seem to grow faster due to mitochondrial function [156]. SDOH: social determinants of health; GHRH: growth hormone-releasing hormone; GnRH: gonadotrophin- releasing hormone; CRH: corticotrophin-releasing hormone; TRH: thyrotropin-releasing hormone; ACTH: adrenocorticotropic hormone; FSH: follicle-stimulating hormone; GH: growth hormone; LH: luteinizing hormone; PRL: prolactin; TSH: thyroid-stimulating hormone; T3: triiodothyronine; T4: thyroxine
The communication between the gut microbiota and the brain is bidirectional and the inter-organ bidirectional regulatory effects may be functionally or dysfunctionally linked through mitochondria [157], redox mechanisms, and metabolic activities (Figure 1).
Mitochondrial dysfunction can lead to heightened oxidative stress and inflammation as reported for ASD [158] and depression [159]. Additionally, some of the organ activities may consist of sex-specific elements, and it has been suggested that sex differences exist for mitochondrial activities [160, 161] and levels of the antioxidant glutathione in the brain [162].
ASD, like many other NDD, is known to disproportionately affect males compared to females, and accordingly, sex-specific factors have been hypothesized to increase male risk and female protection for ASDs [163]. A 2018 analysis of medical and school records of 8-year-old from eleven ASD monitoring sites across the US showed that ASD was 4.2 times as prevalent among males as among females [24]. It has been hypothesized that these epidemiological data could reflect a bias in diagnostic criteria, probably due to different behavioral phenotypes noted in males versus females [164–166]. For example, one study showed that females exhibited better daily living skills compared to males and while these results were not statistically significant, the study suggests that sex might moderate how behavioral symptoms are expressed [167].
Although sex differences have been documented in numerous diseases including ASD, the reasons for these differences are not well understood. However, some explanations that have been posited for the male bias noted in the prevalence of autism include the following: males and females have characteristics inherent in their biological makeup that make them differentially susceptible to the development of ASD. The different sex hormones present in males versus females may influence immune responses towards either cell-mediated or humoral immunity. For instance, estrogen and progesterone increase Th2 cell activity, whereas testosterone generally suppresses their activity [168]. Immune cells are known to express sex hormone receptors, and because females and males have differential expression of these receptors, this may partially explain the greater prevalence of certain types of disease in the different sexes. For instance, females have a greater prevalence of autoimmune diseases, while males suffer more from cardiovascular and neuropsychiatric diseases [151]. The differential response of immune cells to sex hormones may also partially explain, the differential responses to MIA noted in males versus females. Following MIA, ultrasonic vocalizations (USVs) emitted during social interactions of rats were only affected in males regarding frequency and duration; however, MIA did not differentially affect female and male repetitive behaviors [169]. Another study analyzing transcriptional changes in mouse fetal brain in response to MIA showed that MIA activated the integrated stress response (ISR) in male, but not female, MIA offspring [170]. They also discovered that the ISR occurred in an IL-17a-dependent manner, which reduced global mRNA translation and altered nascent proteome synthesis [170]. Additionally, blockade of the ISR prevented behavioral abnormalities as well as increased cortical neural activity in MIA male offspring [170]. This data suggests that a sex-specific response to MIA results in the ISR which in turn leads to the associated sex-specific neurodevelopmental changes. Lastly, it is known that fetal testosterone signals brain masculinization during a restricted critical period. It has been suggested that this restricted window of brain masculinization may coincide with a crucial period during which MIA negatively impacts fetal brain development, thus explaining the higher prevalence of autism noted in males [18].
Another area where sex differences have been noted is in the structure and function of the microglia. Microglia number, morphology, and immune molecule load are influenced by hormonal factors among other things [171]. Animal studies have shown both a region-specific increase in the number and a change in the morphology of microglia under the influence of testosterone [172]. These findings are most prominent in the hippocampus and amygdala-brain regions which have also been implicated in ASD [172, 173]. Animal studies have also identified sex differences in the microglia transcriptome [174]. The exact mechanisms through which sex differences in microglia morphology and function influence the etiology of autism are unclear. However, given the possible role of the microglia in general in the etiology of ASD, it is safe to hypothesize that sex-specific differences in microglia activity contribute to the sex-specific differences noted in both prevalence and manifestation of ASD.
The sexual dichotomy is also evident in social interactions, leading to the observation by researchers that “people with autism simply match an extreme of the normal male profile, with an extraordinarily intense drive to systemize and an unusually low drive to empathize [175, 176]. Systemizing consists of developing rules or principles to facilitate understanding complex systems (e.g., machinery, weather patterns) and underlies the capacity to understand and manipulate objects in one’s physical environment [176, 177]. Systemizing is traditionally more characteristic of males. Empathizing consists of perceiving and emotionally responding to various mental states in others and underlies the capacity to understand and interact with individuals in one’s social environment [176, 177]. Empathizing is traditionally associated with females.
In autistic individuals, the increased tendency to systemize manifests as an increased affinity for predictable phenomena. Unpredictable phenomena like social interactions create anxiety, resulting in a desire to impose predictability and ‘sameness’ by tantrums and insistence on repetition.
This theory is known as the “extreme male brain” theory [176]. This theory points to the possibility that an alteration during sexual differentiation of the brain may be responsible for the “extreme” male phenotype noted in autism. This may result from the actions of hormonal factors like fetal testosterone [176, 178]. Fetal testosterone is believed to influence the organization of brain symmetry, which may play a role in shaping sexual dimorphism in cognition and behavior [178]. The hypothalamus and amygdala are two critical regions of the brain where fetal testosterone results in sexual dimorphism [179]. Through the release of corticotrophin-releasing factor (CRF), the hypothalamus regulates the release of oxytocin, which in turn plays a vital role in the sexually dimorphic aspect of human social reciprocity and interactions-behaviors which are deficient in people with autism [180]. Low blood levels of oxytocin are reported in autistic patients [181]. Studies have shown that administering oxytocin improves several aspects of social interaction in patients with autism [181]. One role of the amygdala is interpreting sensory and emotional information and mediating resultant behaviors. It is particularly active during fearful events. Changes in the function and neuropathology of the amygdala have been noted in autism [173]. Other reasons for the “extreme male brain” include the epigenetic effect on X chromosome genes and the male-limited expression of genes on the Y chromosome [178, 182].
The higher prevalence of ASD in males may also relate to sex differences in rates of development and antigenicity, which interferes with the usual establishment of maternal immune tolerance to the developing fetus. Male embryos express the sex-determining region Y protein (Sry) gene before implantation [183], male embryos grow faster than female embryos allowing faster implantation [184, 185], which has been associated with ‘Growth factor Y’ on the Y chromosome [186]. The blastocyst before implantation also expresses zinc finger Y-chromosomal protein (Zfy) and a histocompatibility Y (H-Y) antigen (Smcy) [186]. The accelerated male embryo growth may penetrate the uterine wall to greater depth upon implantation. The maternal immune system initially aids implantation, but it needs to shift away from cell-mediated immunity with inflammatory products toward a more anti-inflammatory and antioxidative condition, which should occur early in the 1st trimester and be maintained throughout the pregnancy until late in the 3rd trimester at the normal time for parturition/birth. With more rapid embryo growth, a subtle precocious implantation difference may preempt an adequate maternal immune shift, and since cytotoxic T cells and antibodies to male antigens are possible [187–191], there may be male-specific immunity leading to the release of damage-associated molecular patterns (DAMPs) activating more inflammation and interfering with the immune tolerance. As mentioned earlier, it is known that mothers of children with ASD have more antibodies that react with brain antigens such as anti-Caspr2 and anti-Pentraxin2 [192, 193]. This may be an additional hypothesis for the male prevalence.
Additional insights into the male bias in autism come from a completely different and somewhat controversial quarter—the maternal immune hypothesis of male homosexuality [18]. According to this hypothesis, maternal anti-H-Y antibodies cross the placental barrier to the fetus, affecting several aspects of sexual differentiation in the fetal brain, with the result that each succeeding male fetus is exposed to increasing effects of such maternal activation of memory lymphocytes [18]. Sexual orientation has been shown to positively correlate with an individual’s number of older brothers, with each additional older brother increasing the odds of homosexuality by approximately 33% [194]. This association was not found with an increasing number of older sisters. Furthermore, studies investigating the association between MIA and male homosexuality reveal that women with gay sons have higher titers of antibodies against genes encoding proteins integral to male brain development [195]. As mentioned earlier, several studies have reported a link between MIA and autism, and some studies report an increased incidence of non-heterosexual behaviors (including homosexuality, bisexuality, and asexuality) among autistic individuals [196]. However, a common mechanistic pathway uniting ASD, MIA, and sexual preferences remains to be seen. The existence of such a pathway may be critical to deciphering the etiology of ASD. Additionally, if ASD is indeed a manifestation of an “extreme male brain”, it will be interesting to study how this theory impacts females with autism. Are females with autism more likely to be masculine in their behavior? Is there an increased incidence of autism among female-to-male transgender individuals? All these questions remain to be answered.
While all mechanisms above seem plausible, there is no clear epidemiological or clinical data to suggest that they selectively increase the autism risk for males compared to females. Furthermore, the occurrence of ASD in females suggests that other factors are at play.
From the information above, the commonalities between these risk factors are evident. It is known that the immune organs and endocrine glands are innervated by the nervous system. Additionally, hormonal signals from the endocrine system influence both the nervous and immune systems in a sex-specific manner. These mechanisms are in turn influenced by genes whose expression is ultimately modified by environmental exposures. Illustrated in Figure 1 above, is the interplay between some of these factors and how they may ultimately influence brain function. Although a common pathway unto which all these risks converge remains to be clearly defined, the interconnectedness of all these risk factors makes the goal of identifying a common mechanistic pathway plausible.
In the meantime, understanding the commonalities and differences between the multiplicity of hits that contribute to susceptibility is critical to the development of interventions aimed at mitigating these risks.
ASD: autism spectrum disorder
BDNF: brain-derived neurotrophic factor
ER: endoplasmic reticulum
GI: gastrointestinal
IFN-γ: interferon-gamma
IgG: immunoglobulin G
IL-6: interleukin-6
ISR: integrated stress response
MIA: maternal immune activation
MZT: monozygotic twins
NDD: neurodevelopmental disorders
poly I:C: polyinosinic:polycytidylic acid
STAT: signal transducer and activator of transcription
TGF-β: transforming growth factor-beta
All authors contributed to the conception of this study. SO performed the database search and wrote the first draft of the manuscript; DAL contributed to sections of the manuscript including the conceptualization and drawing of Figure 1. All authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
SO received partial support from the National Institute of Environmental Health Sciences (NIEHS) award RO1ES025584 to DAL. The funders had no role in the study design, data collection, analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 10438
Download: 96
Times Cited: 0
Haruka Sawamura ... Satoru Matsuda
Fábio José Coelho Souza-Junior ... Sabrina Francesca Lisboa
Niklas Frank ... Carola Y. Förster
Mydhili Radhakrishnan ... Sumana Chakravarty
