 Original Article
Original Article

Affiliation:
Department of Biotechnology and Genetic Engineering, Philadelphia University, Amman 19293, Jordan
Email: tfroukh@philadelphia.edu.jo
ORCID: https://orcid.org/0000-0002-2862-0733
Affiliation:
Department of Biotechnology and Genetic Engineering, Philadelphia University, Amman 19293, Jordan
Explor Neuroprot Ther. 2026;6:1004161 DOI: https://doi.org/10.37349/ent.2026.1004161
Received: November 06, 2025 Accepted: May 14, 2026 Published: July 02, 2026
Academic Editor: Vladimir Josef Balcar, The University of Sydney, Australia
The article belongs to the special issue Advances in the Pathogenesis, Diagnosis and Treatment of Attention Deficit Hyperactivity Disorder
Aim: Identify new genetic variants linked to neurodevelopmental disorders to be used in routine genetic diagnostics.
Methods: Whole exome sequencing (WES) was used for ten unrelated families from Jordan characterized by one or two affected members with neurodevelopmental disorders (NDDs), in which the parents are consanguineous in nine families.
Results: Ten variants are identified in this study; one variant per family. Eight variants are identified by cross-link with the pathogenic variants in ClinVar and in our in-house database, and two variants are identified for the first time. Nine of the total identified variants are homozygous in the nine consanguineous families and one variant is heterozygous in the non-consanguineous family. Three of the identified variants were previously reported in unrelated Jordanian families compared to the families in this study. These three variants are NM_018359.5:c.344T>A;p.Val115Glu in the gene UFSP2, NM_000487.6:c.256C>G;p.Arg86Gly in the gene ARSA and NM_017882.3:c.144G>A;p.Trp48* in the gene CLN6. The variants that are identified for the first time are splicing variants; splice donor variant NM_000433.4:c.366+1G>C in the gene NCF2 and splice acceptor variant NM_000191.3:c.498-1G>A in the gene HMGCL.
Conclusions: The results of this study stress the importance of continuous research using WES for NDDs in Jordan to identify new variants.
Neurodevelopmental disorders (NDDs) are a diagnostic category addressed in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5). This category includes a group of disorders that commonly start in childhood and can be chronic, persisting for life. The features commonly observed in patients with NDDs include global developmental delay (GDD), intellectual disability (ID), attention deficit hyperactivity disorder (ADHD), autism spectrum disorder (ASD), communication disorders (CDs), learning disorders (LDs), motor disorders (MDs), and epilepsy. Because the majority of NDDs are caused by genetic factors, genetic diagnostic procedures—including whole genome/exome sequencing (WGS/WES) are generally conducted for patients with NDDs [1].
Jordanian clinicians face difficulties in diagnosing patients with NDDs and therefore request WGS/WES. Philadelphia University receives these requests and provides free service on a scientific basis. In 2024, ten unrelated Jordanian patients were referred to Philadelphia University, and we present in this study their WES analysis.
Ten unrelated Jordanian families with one or more patients with NDDs were included in this study. Ethical approval was obtained from the Institutional Review Board (IRB) of Philadelphia University in Jordan (IRB number 2/1/2024-2025). All participants provided signed informed consent to publish the results of their exome sequences anonymously. Peripheral blood was collected from all patients and family members in EDTA tubes and total genomic DNA was extracted using the QIAGEN FlexiGene DNA kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol. Exomes were captured using Agilent SureSelect V8. The sequencing platform was Illumina NovaSeq 6000. The raw data output was 9 GB per sample with an average coverage of 100×.
All bioinformatics analyses were conducted using the pipeline implemented at Philadelphia University, Amman, Jordan. The generated paired-end reads were trimmed using Trimmomatic-0.39 [2] and aligned using BWA [3] to the GRCh38-full analysis list (containing the alternative locus scaffolds in addition to all sequences present in the non-alternative analysis set) plus human decoy sequences from hs38d1 (GCA_000786075.2). Picard [4] was used to mark duplicates, and ABRA [5] was used to realign indels. Variants were called and normalized using BCFtools [6, 7]. Variants were annotated using SnpEff [8] and SnpSift [9]. All annotations were conducted based on the latest human genome release, GRCh38.p14.
Only high-quality variants were retained with a read depth (DP) greater than or equal to 10, Phred-scaled quality score (QUAL) greater than or equal to 30, base quality (BQ) greater than or equal to 30, and mapping quality (MQ) greater than or equal to 40. Variants were annotated using the following public databases: OMIM (release 2862025) [10, 11], ClinVar (release 20250615) [12], dbSNP (build_ID 157) [13], 1000 Genomes Project [14], TOPMed Freeze8 [15], gnomAD joint (exome and genome) v4.1.1 [16], ALFA total (20241028) release 4 [17], RegeneronME (around 983,578 exomes) [18], Turkish Genome Project [19], and Qatari [13]. SIFT, Polyphen, and PROVEAN were used to predict the functional effect of missense variants [20–22]. Variants were filtered, prioritized, and identified as described previously [23, 24].
Two deep learning in silico prediction tools were used to assess the consequences of splice variants using the web service (https://spliceailookup.broadinstitute.org/): SpliceAI and Pangolin [25, 26].
In this study, we examined ten unrelated families with one or more affected individuals with NDDs from Jordan. All affected individuals underwent clinical evaluations in either small clinics or hospitals. The pedigrees in Figure 1 show eight families with one affected individual, one family with two affected individuals, and one family with four affected individuals. In family 1, the affected individual passed away shortly after recruiting the family, while in family 5, three of the affected individuals had passed away before being recruited into this study. The various phenotypes observed for each affected individual are summarized in Table 1.
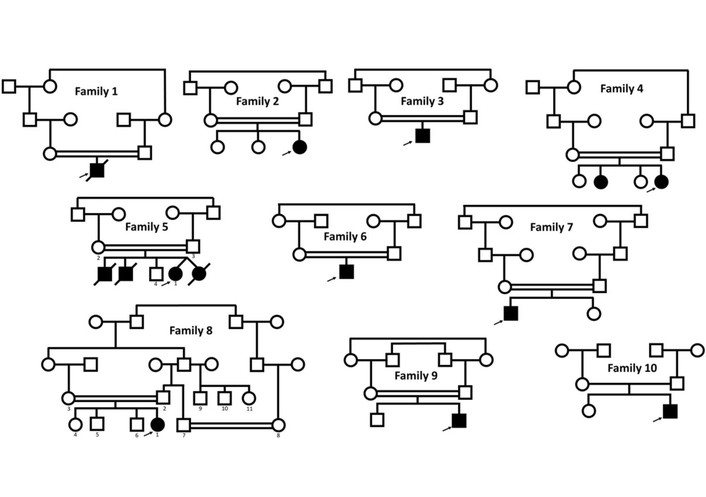
Pedigrees of the families included in this study. Arrows indicate the patient underwent WES. Double horizontal lines reflect consanguineous marriage.
Phenotypes of the patients included in this study.
| Family# | Patient’s age at recruitment time | Phenotypes |
|---|---|---|
| 1 | 2 years | Microcephaly, hearing disability, seizure |
| 2 | 2 years | Leukodystrophy, neuroregression, muscular regression |
| 3 | 2 years | Developmental delay, seizure |
| 4 | 10 years | Neuroregression, muscular regression |
| 5 years | ||
| 5 | 7 years | Recurrent vomiting and organic acidemia, family history of death shortly after birth |
| 6 | 3 years | Congenital cataract and developmental delay |
| 7 | 5 months | Moderate thrombocytopenia, unilateral missing radius |
| Absent left thumb, right rudimentary thumb and single kidney | ||
| 8 | 1 year | Skin lesion, dermatitis of immunodeficiency |
| 9 | 1 month | Multiple long bone fractures and deformities |
| 10 | 7 years | Developmental delay, ADHD |
Likely causative variants were identified for all patients (Table 2). Nine of the variants were homozygous and segregated with the disease phenotype in all family members, while one variant was de novo in one family.
The likely causative variants identified in all families.
| Family# | Parent’s relatedness | Identified variant | Allele frequency | Genotype in the patient | Gene symbol | OMIM disease name | OMIM disease# | Inheritance pattern* | |
|---|---|---|---|---|---|---|---|---|---|
| In-house* | gnomAD | ||||||||
| 1 | First cousins, once removed (grandmother’s of the mother is the father's aunt) | NM_000218.3:c.1552C>T;p.Arg518* | 0.004274 | 0.00014313 | Homozygous | KCNQ1 | Jervell and Lange-Nielsen syndrome | 220400 | AR |
| 2 | First cousins | NM_000487.6:c.256C>G;p.Arg86Gly | 0.008547 | 0.000000622939 | Homozygous | ARSA | Metachromatic leukodystrophy | 250100 | AR |
| 3 | First cousins | NM_018359.5:c.344T>A;p.Val115Glu | 0.012821 | 0.0000713148 | Homozygous | UFSP2 | Condition developmental and epileptic encephalopathy 106 | 620028 | AR |
| 4 | First cousins, once removed (grandmother’s of the mother is the father's uncle) | XM_047432780.1:c.240G>A;p.Trp80* | 0.008547 | – | Homozygous | CLN6 | Ceroid lipofuscinosis, neuronal, 6A | 601780 | AR |
| 5 | First cousins | NM_000191.3:c.498-1G>A | 0.004274 | – | Homozygous | HMGCL | HMG-CoA lyase deficiency | 246450 | AR |
| 6 | First cousins | NM_000154.2:c.678delT;p.Asn226fs | 0.004274 | – | Homozygous | GALK1 | Galactokinase deficiency with cataracts | 230200 | AR |
| 7 | First cousins | NM_022725.4:c.109dupT;p.Trp37fs | 0.004274 | – | Homozygous | FANCF | Fanconi anaemia, complementation group F | 603467 | AR |
| 8 | First cousins | NM_000433.4:c.366+1G>C | 0.004274 | – | Homozygous | NCF2 | Chronic granulomatous disease 2 | 233710 | AR |
| 9 | Double first cousins | NM_006371.5:c.18_25delGGGGGCCG;p.Ala10fs | 0.004274 | 0.00000273746 | Homozygous | CRTAP | Osteogenesis imperfecta, type VII | 610682 | AR |
| 10 | No relatedness | NM_001244810.2:c.1241delT;p.Leu414fs | 0.00213675 | 0.000000619546 | Heterozygous | FOXP1 | Intellectual developmental disorder with language impairment, with or without autistic features | 613670 | AD |
AR: autosomal recessive; AD: autosomal dominant. –: No record in gnomAD; *In-house: Philadelphia University/Jordan on October, 2025.
We identified four variants in four families that were not previously reported in-house but were previously reported in ClinVar as pathogenic variants. These four variants are frameshift: (NM_000154.2:c.678delT,p.Asn226fs in the gene GALK1; NM_001244810.2:c.1241delT;p.Leu414fs in the gene FOXP1; NM_022725.4:c.109dupT;p.Trp37fs in the gene FANCF; and NM_006371.5:c.18_25delGGGGGCCG;p.Ala10fs in the gene CRTAP). We also identified one variant is previously reported in-house and is also previously reported in ClinVar as pathogenic (NM_017882.3:c.144G>A;p.Trp48* in the gene CLN6). Additionally, we identified two variants in two families previously reported in ClinVar as likely pathogenic: a stop-gain variant in the gene KCNQ1 (NM_000218.3:c.1552C>T;p.Arg518*) that is not previously recorded in-house and a missense variant in the gene ARSA (NM_000487.6:c.256C>G;p.Arg86Gly) that is previously recorded in-house. One missense variant in UFSP2 (NM_018359.5:c.344T>A;p.Val115Glu) is reported in ClinVar with conflicting classifications of pathogenicity but is considered in this study as likely pathogenic.
Two novel splice-site variants were identified that are not recorded in any public database: a splice acceptor variant in HMGCL (NM_000191.3:c.498-1G>A; Figure 2) and a splice donor variant in NCF2 (NM_000433.4:c.366+1G>C; Figure 3).
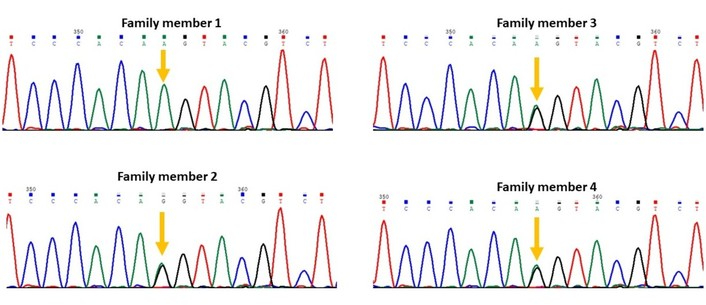
Segregation of the variant NM_000191.3:c.498-1G>A in the gene HMGCL in family 5; the variant is homozygous in the patient (family member 1), heterozygous in all other healthy family members.
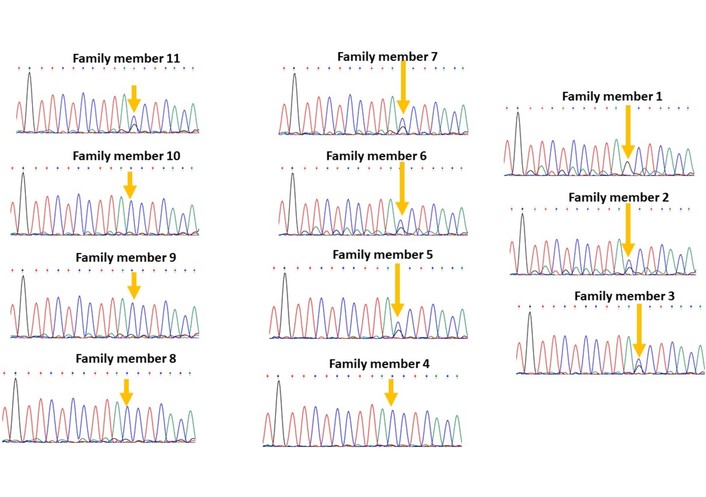
Segregation of the variant NM_000433.4:c.366+1G>C in the gene NCF2 in family 8; the variant is homozygous in the patient (family member 1), heterozygous in healthy family members 2, 3, 5, 6, 7, 11, and wild type in healthy family members 4, 8, 9, 10.
Based on SpliceAI analysis, the variant NM_000433.4:c.366+1G>C revealed a high-confidence prediction for the loss of the splice donor site (score = 1), and it is predicted to result in the gain of a cryptic donor site (score = 0.79) located 6 bp downstream of the variant. Similarly, Pangolin supported the results of SpliceAI with a significant splice loss score of 0.87 and a significant splice gain score of 0.68 at 6 bp downstream of the variant. For the variant NM_000191.3:c.498-1G>A, SpliceAI provided a high-confidence prediction for the loss of the splice acceptor site (score = 1). The analysis also suggested a potential loss of the donor site with (score = 0.87) and a minor gain of cryptic acceptor site (score = 0.21) at 2 bp upstream of the variant. Pangolin yielded results consistent with SpliceAI, with a score of 0.87 for the loss of a splice acceptor variant and a score of 0.4 for gaining a new splice acceptor variant at 2 bp upstream. The predicted scores by SpliceAI and Pangolin strongly suggest major disruption of the normal splicing for the HMGCL and NCF2 messenger RNAs in patients homozygous for these two variants.
NDDs are heterogeneous disorders associated with a wide range of phenotypes. Multiple research projects have been conducted to identify the genetic causes of NDDs in Jordan [23]. The investigated patients in this study exhibited ten genetic variants in ten different genes, which are considered as causatives for the studied conditions. Eight of these variants were previously reported, and two are novel.
The identified variant in Family 1 leads to a premature stop codon in the gene transcript, which is predicted after then to undergo nonsense-mediated decay, resulting in a truncated or absent voltage-gated potassium channel. The normal role of these channels is to maintain the membrane potential, regulate cell volume, and modulate the electrical excitability in neurons [27]. The variant is reported in the literature as a founder mutation in the Swedish population, in which heterozygous carriers of the variant are clinically asymptomatic, while homozygous individuals exhibit long QT intervals on EKG; Jervell and Lange-Nielsen syndrome (OMIM # 220400) [28]. The data for this family align with these reports; the patient presents with deafness, syncope, seizures, and a QTc of 0.57 milliseconds based on EKG. This indicates that the variant is very likely to be associated with disease.
In Family 2, the identified variant is in a gene that codes for the enzyme arylsulfatase A, a lysosomal enzyme that hydrolyzes cerebroside sulfate [29]. The identified missense variant Arg86Gly, changes the basic polar amino acid to a neutral nonpolar amino acid, which is considered to be pathogenic despite the absence of conclusive evidence [30]. This is because (1) the variant was identified previously in Jordan in an unrelated family with metachromatic leukodystrophy phenotypes, and (2) other variants at the same amino acid position were determined to be pathogenic with conclusive evidence [31–33]. Pathogenic variants in this gene are likely the cause of metachromatic leukodystrophy (OMIM # 250100).
In Family 3, the identified variant is in the protein-coding gene UFSP2, a thiol protease that mediates the processing of the precursor of UFM1 and deconjugates UFM1 from its target proteins (deUFMylation) [34]. Decreased function of UFSP2 due to a homozygous loss-of-function variant will increase the abundance of UFMylated targets and cause the condition, developmental and epileptic encephalopathy 106 (OMIM # 620028) [35]. The identified variant in this study is considered pathogenic because it has been previously reported with conclusive evidence in 8 families from Pakistan and Afghanistan with developmental and epileptic encephalopathy 106 [35].
The identified variant in Family 4 leads to a premature stop codon in the gene transcript, which is predicted after then to undergo nonsense-mediated decay, resulting in a truncated or absence of the ceroid-lipofuscinosis neuronal protein 6 (CLN6; OMIM # 601780). CLN6 functions as an obligate partner in the recruitment of newly synthesized lysosomal enzymes in the endoplasmic reticulum for posttranslational modification [36]. CLN6 deficiency leads to a depletion of various lysosomal enzymes from the lysosomal compartment and thus causes the autosomal recessive condition ceroid lipofuscinosis, neuronal, 6A.
In Family 5, a novel splice acceptor variant was identified in the gene HMGCL. This gene encodes the enzyme 3-hydroxy-3-methylglutaryl coenzyme A lyase that catalyzes the cleavage of HMG-CoA into acetoacetic acid and acetyl-CoA, which is considered a key step in ketogenesis and the terminal step in leucine catabolism [37]. Pathogenic mutations in this gene lead to a deficiency in the enzyme and are associated with the condition HMG-CoA lyase deficiency (OMIM # 246450), which is characterized by metabolic acidosis, hypoglycemia, and elevated urinary organic acid metabolites [38]. The main clinical signs of irritability, lethargy, coma, and vomiting were exhibited in the studied patient. Moreover, the evidence from SpliceAI and Pangolin suggests a loss-of-function mechanism due to the abolition of the normal splice acceptor site.
The identified variant in Family 6 leads to a premature stop codon in the gene transcript, which is predicted after then to undergo nonsense-mediated decay, resulting in a truncated or absence of the galactokinase. Galactokinase catalyses the phosphorylation of galactose, the first step in galactose metabolism, and the faultiness of this function leads to the condition galactokinase deficiency with cataracts (OMIM # 230200), which is characterized by galactitol accumulation in the lens if the affected individual is not given a lactose-free diet [39].
In Family 7, the identified variant leads to a premature stop codon in the gene transcript, which is predicted after then to undergo nonsense-mediated decay, resulting in a truncated or absent protein FANCF. The protein FANCF serves with other proteins a nuclear function to maintain genomic integrity, especially in DNA repair [40]. The lack of this function is associated with hypersensitivity to DNA-damaging agents, chromosomal instability, and defective DNA repair. When the affected cells are in bone marrow, anaemia, leukopenia, and thrombopenia will be the result, which are features of the condition Fanconi anaemia, complementation group F (OMIM # 603467) [41].
In Family 8, the splice donor variant was identified for the first time in the gene NCF2. This gene codes a neutrophil cytosolic factor-2, a component of the NADPH oxidase complex that mediates the transfer of electrons from cytosolic NADPH to O2 to produce the superoxide anion (O2–) [42]. Improper or the absence of these functions leads to an impaired ability of phagocytes to mount a burst of reactive oxygen species in response to pathogens [43]. The predictions of SpliceAI and Pangolin indicate that the variant is likely to disrupt the normal splicing and the inclusion of intronic sequence or exon skipping, leading to a frameshift and premature stop codon. Pathogenic variants in this gene are likely causative of the condition chronic granulomatous disease 2 (OMIM # 233710).
In Family 9, the identified variant leads to a premature stop codon in the gene transcript, which is predicted after then to undergo nonsense-mediated decay, resulting in a truncated or absence of the cartilage associated protein, which is necessary for efficient 3-hydroxylation of fibrillar collagen prolyl residues [44]. Improper function of this protein leads to osteogenesis imperfect type VII (OMIM # 610682), a connective tissue disorder characterized by bone fragility and low bone mass, which matches the phenotype of the studied patient [45].
In Family 10, the parents of the patient are not consanguineous. A de novo truncating variant NM_001244810.2:c.1241delT;p.Leu414fs was identified in the gene FOXP1, causing the autosomal dominant condition intellectual developmental disorder with language impairment with or without autistic features (OMIM # 613670). FOXP1 is a transcriptional repressor and forms homodimer or heterodimer with subfamily members, organizing motor axon projection and establishment of motor pool [46]. Because FOXP1 controls the development process, it was hypothesized that disruption in this gene causes developmental conditions that are associated with language impairment, ID, and ASDs [47].
The term NDDs is broad. Based on the identified genetic variants and the associated genetic disorders, the patients of this study can be grouped into six subcategories: CD (family 1), ASD (family 10), ID (family 7), MD (families 2 and 3), neurocognitive disorders (families 4 and 5), and specific LD (families 6, 8, and 9).
Despite the significant findings regarding the genetic variants in this study, several limitations should be addressed. First, due to the lack of specialized genetic medical institutes in Jordan, the phenotypic characterization of the patients was limited, and standardized neurodevelopmental assessments were absent. Second, this study relied on computational validation instead of experimental validation for novel splicing variants. Future functional studies are warranted to definitively confirm the impact of the identified splicing variants on mRNA.
This study emphasizes the use of WES to identify the genetic bases of rare diseases in Jordan. The identified cause in family 6 uncovers the importance of early diagnosis to avoid progression of the phenotypes in the patient by managing food and diet at early stages of life. Identification of pathogenic variants enables families to plan subsequent pregnancies with a reduced risk of NDDs.
ASD: autism spectrum disorder
CDs: communication disorders
ID: intellectual disability
IRB: Institutional Review Board
LDs: learning disorders
MDs: motor disorders
NDDs: neurodevelopmental disorders
WES: whole exome sequencing
WGS: whole genome sequencing
The authors thank all the families who participated in this study.
TF: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Formal analysis, Visualization, Funding acquisition. EA: Investigations, Writing—review & editing. Both authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
Ethical approval was obtained from the Institute Review Board (IRB) of Philadelphia University in Jordan (IRB number 2/1/2024-2025) and complies with the Declaration of Helsinki.
Signed informed consents were obtained from all participants in this study. Consents were signed by the parents for all children below 16 years.
Consents were obtained from all families in this study to publish their data anonymously.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
This study was funded by the Deanship of Scientific Research and Graduate Studies at Philadelphia University in Jordan. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 503
Download: 8
Times Cited: 0
Montserrat Gerez-Malo ... Carlos Acosta
Yvette Hus, Osnat Segal
Rosa Angela Fabio ... Pina Filippello
Daniela Smirni ... Michele Roccella
Blandine French ... Amanda Kirby
