 Review
Review

Affiliation:
1School of Biotechnology and Biomolecular Sciences, Faculty of Science, University of New South Wales, Sydney, New South Wales 2052, Australia
ORCID: https://orcid.org/0000-0002-5206-0887
Affiliation:
2School of Pharmacy, Faculty of Medicine and Health, University of Sydney, Sydney, New South Wales 2006, Australia
ORCID: https://orcid.org/0000-0002-2732-895X
Affiliation:
2School of Pharmacy, Faculty of Medicine and Health, University of Sydney, Sydney, New South Wales 2006, Australia
Email: ingrid.gelissen@sydney.edu.au
ORCID: https://orcid.org/0000-0002-3961-2288
Affiliation:
1School of Biotechnology and Biomolecular Sciences, Faculty of Science, University of New South Wales, Sydney, New South Wales 2052, Australia
Email: aj.brown@unsw.edu.au
ORCID: https://orcid.org/0000-0002-4475-0116
Explor Neuroprot Ther. 2022;2:1–27 DOI: https://doi.org/10.37349/ent.2022.00015
Received: October 26, 2021 Accepted: December 01, 2021 Published: January 05, 2022
Academic Editor: Ta-Yuan Chang, Geisel School of Medicine at Dartmouth, USA
The article belongs to the special issue Cholesterol Dyshomeostasis in Neurological Diseases
The cholesterol is a vital component of cell membranes and myelin sheaths, and a precursor for essential molecules such as steroid hormones. In humans, cholesterol is partially obtained through the diet, while the majority is synthesized in the body, primarily in the liver. However, the limited exchange between the central nervous system and peripheral circulation, due to the presence of the blood-brain barrier, necessitates cholesterol in the brain to be exclusively acquired from local de novo synthesis. This cholesterol is reutilized efficiently, rendering a much slower overall turnover of the compound in the brain as compared with the periphery. Furthermore, brain cholesterol is regulated independently from peripheral cholesterol. Numerous enzymes, proteins, and other factors are involved in cholesterol synthesis and metabolism in the brain. Understanding the unique mechanisms and pathways involved in the maintenance of cholesterol homeostasis in the brain is critical, considering perturbations to these processes are implicated in numerous neurodegenerative diseases. This review focuses on the developing understanding of cholesterol metabolism in the brain, discussing the sites and processes involved in its synthesis and regulation, as well as the mechanisms involved in its distribution throughout, and elimination from, the brain.
Cholesterol plays important structural and functional roles in the brain where it comprises approximately a quarter of the body’s total cholesterol pool [1]. It is a crucial molecule for cell membrane formation and flexibility, lipid raft assembly, as well as biochemical processes such as glucose transport and inflammatory signaling [2–4]. In addition, cholesterol can be oxidized into oxysterols, which serve various functions including mediating sterol export [5].
Brain cholesterol is acquired and regulated in a different manner to that in peripheral tissues [6]. In the periphery, 70% of the cholesterol is synthesized de novo in the liver, with the remaining 30% obtained through dietary intake. This cholesterol pool is subsequently distributed throughout the body via lipoprotein-mediated lipid transfer [7]. However, in the brain negligible cholesterol can be sourced from circulating cholesterol lipoproteins owing to the restrictive nature of the blood-brain barrier and blood-cerebrospinal fluid (CSF) barriers, and so most is synthesized locally within the organ itself [8]. Brain cholesterol accumulates during early development [9], however cholesterol turnover can be very slow in adult brains with a half-life of 0.5 to 5 years compared to just days in circulation [6, 10]. Brain cholesterol concentration remains relatively constant under normal conditions, with a small fraction replaced via de novo synthesis [11]. However, cholesterol levels may be altered in situations of disease and aging.
Mechanisms are in place to ensure that there is an adequate supply of cholesterol for the functioning of the central nervous system (CNS) and to prevent cholesterol accumulation and associated lipotoxicity [12]. Regulation of cholesterol homeostasis alters during vertebrate development, as the requirements for the sterol for growth and maintenance of various cellular structures may change throughout life. Consequently, cholesterol synthesis and turnover must be tightly regulated to ensure steady levels of sterol in the brain.
Cholesterol biosynthesis is a resource-intensive process as the precursor Acyl-coenzyme A (CoA) is converted into the final sterol by the action of 23 enzymes. The endoplasmic reticulum is the main site for cholesterol synthesis and the location for most of the biosynthetic enzymes. The process can be divided into the early stage, where acetyl-CoA is converted into the first sterol, lanosterol, followed by the post-lanosterol stage which consists of two possible pathways (Figure 1).
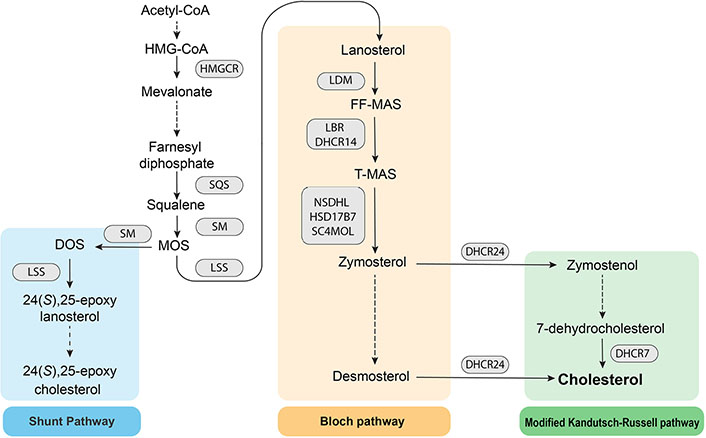
Schematic of the cholesterol biosynthesis pathway. HMG: 3-hydroxy-3-methylglutaryl; SQS: squalene synthase; SM: squalene monooxygenase; MOS: monooxidosqualene; HMGCR: HMG-CoA reductase; LSS: lanosterol synthase; DOS: dioxidosqualene; LDM: lanosterol 14α-demethylase; FF-MAS: follicular fluid meiosis-activating sterol; LBR: lamin B receptor; DHCR14: 14-dehydrocholesterol reductase; T-MAS: testis meiosis-activating sterol; NSDHL: NAD(P) dependent steroid dehydrogenase-like; HSD17B7: hydroxysteroid 17β dehydrogenase 7; SC4MOL: sterol-C4-methyl oxidase like
HMGCR is the first rate-limiting enzyme and the pharmacological target for statins, which are used clinically to lower plasma cholesterol levels. HMGCR catalyzes the reduction of HMG-CoA to mevalonate. Downstream of HMGCR, SQS catalyzes the conversion of farnesyl diphosphate to squalene, the first step specific to sterol biosynthesis. Squalene is subsequently converted into squalene 2,3-epoxide by the second rate-limiting enzyme, SM [13]. Post-lanosterol, sterol intermediates proceed through either the Bloch pathway or the modified Kandutsch-Russell pathway (K-R), which both ultimately yield cholesterol. DHCR24 may act on any intermediate in the Bloch pathway to shuttle intermediates to the modified K-R, bypassing upstream and downstream entry points. The partial inhibition of LSS enables accumulation of MOS, which funnels intermediates into the Shunt pathway, in which SM acts a second time on MOS to yield DOS. LSS then catalyzes DOS into 24(S),25-epoxylanosterol which is ultimately converted into 24(S),25-epoxycholesterol (24,25-EC).
Feedback regulation of cholesterol synthesis is exquisitely controlled at the transcriptional level by sterol responsive element-binding protein (SREBP) and liver X receptor (LXR) transcription factors [14, 15], and at the post-translational level by the ubiquitin-proteasome system [16–19]. Furthermore, several mechanisms operate to remove excess cholesterol from the brain, including intracellular storage, conversion to oxidized metabolites, and efflux into the circulation. Collectively, these mechanisms prevent cholesterol dyshomeostasis within the brain, which can be a key pathological feature in genetic conditions such as Smith-Lemli-Opitz syndrome, as well as neurodegenerative diseases such as multiple sclerosis, Alzheimer’s disease, and Parkinson’s disease [20–22]. Hence, understanding the metabolism of cholesterol within the brain is crucial for recognizing its significance in the maintenance of brain health and for discerning its role in the pathophysiology of various brain diseases.
In this review, we firstly examine cholesterol synthesis and the preference for the modified Kandutsch-Russell versus the Bloch branches of the cholesterol synthesis pathway in various brain cell subtypes. Secondly, we discuss the cholesterol transport mechanisms and the role of ATP-binding cassette (ABC) transporters in cholesterol distribution. Next, we discuss the transcriptional regulation of cholesterol and its implications for brain health. Finally, we highlight the role of oxysterols in health and disease and the mechanisms for cholesterol clearance from the brain.
The brain contains a diverse population of cells, including neurons and various glial cells—astrocytes, oligodendrocytes, and microglia—each with distinct cholesterol requirements and manufacturing capabilities. Early work with cultured cells derived from animal models indicates that neurons predominantly derive their cholesterol from glial cells. Cultured glial cells from chicken embryos are able to manufacture two- to three-fold more cholesterol than neurons [23], and glial cells transfer cholesterol to other cells via ABC transporters (discussed in detail below) [24, 25]. In the absence of glial cells, rat neurons synthesize sterols at a lower rate and are unable to compensate for the cholesterol deficit [24]. However, neurons in glial co-cultures drastically downregulate the expression of the early cholesterol biosynthesis pathway enzyme, SQS, suggesting the reduced capacity for cholesterol synthesis [24, 26]. Neurons in culture rarely esterify newly synthesized cholesterol for storage, and their cholesterol demand is met by exogenous uptake [24, 25].
The brain appears to switch between the use of the post-lanosterol Bloch and K-Rs (summarized in Table 1). Curiously, in developing brains from all mammalian species reported, including humans, desmosterol transiently accumulates to constitute up to 30% of total brain sterols [27]. The post-translational repression of DHCR24 by progesterone is proposed to cause the desmosterol build-up, to facilitate the enrichment and distribution of membrane sterols in the developing brain [27]. Indeed, sterol profiles of embryonic mice cells indicate desmosterol and other Bloch sterol accumulation in neurons and astrocytes [28]. By contrast, in vitro work showed that post-natal rat neurons accumulate 7-dehydrocholesterol and lathosterol, precursors of the K-R, whilst astrocytes retain Bloch intermediates, namely the immediate cholesterol precursor, desmosterol [24]. Oligodendrocytes and microglia seem to favor the use of the K-R, as these cells derived from post-natal rats have higher proportions of lathosterol and 7-dehydrocholesterol than astrocytes and neurons [24]. Evidently, there is a dynamic exchange in preference for these two pathways within the brain throughout the life of a vertebrate. The characteristics of cholesterol production in brain cells have been studied in specific cell types, with findings summarized in Table 1 and discussed in detail below.
Summary of cholesterol synthesis in brain cell subtypes
| Cell type | Subtypes | Function of cell | Function of cholesterol | Cholesterol synthesis | K-R or Bloch pathway? |
|---|---|---|---|---|---|
| Neurons | Subtypes many | Processing and transmission of cellular signals | Regulates membrane fluidity, required for cell membranes and myelin sheaths [29]. Present in lipid rafts and facilitates ion channel function, neuron receptor localization, neurotransmitter transport, and cellular growth and development [2, 26, 29]. | High during embryonic development [28, 30]. Low during adulthood, when cholesterol is mainly sourced from astrocytes [26, 31]. | Both [24, 28] |
| Glial cells | Astrocytes | Maintenance of CNS homeostasis, provision of biochemical and nutritional support to neurons and blood-brain barrier, synaptic transmission, immune function | Required for cell membrane fluidity regulation, lipid raft formation, and carbohydrate metabolism [32]. Cholesterol is exported to other brain cells [32, 33]. | Low during embryonic development [28]. High after birth [26, 34]. | Bloch [24] |
| Oligodendrocytes | Synthesis and maintenance of myelin sheaths to insulate neuronal axons for faster signal transmission | Required for synthesis of myelin sheaths [35–37]. | High after birth and in early childhood [9, 35]. Dynamic during adulthood. Cholesterol is sourced locally and from astrocytes [38, 39]. | Possibly K-R [24] but requires further study | |
| Microglia | Brain macrophage; immune function and injury repair | Lipid composition modulates microglial function in phagocytosis, immune surveying, synapse pruning [33, 40, 41]. Desmosterol (cholesterol precursor) activates LXR signaling to resolve inflammation and promote oligodendrocyte maturation [42]. | Synthesis rate low, sourced from astrocytes [43]. | Possibly K-R [24] but requires further study |
Note that many neuronal subtypes are present in the brain, but for the purpose of this review the main role of neurons and their cholesterol synthesis is discussed
In neurons, cholesterol is an important component of myelin and the membranes of synapses, dendrites, and axons, where it regulates membrane fluidity [29]. Neurons can synthesize their own cholesterol to a limited extent. Murine neurons express mRNA transcripts and proteins of components of the cholesterol synthesis pathway, including those for the early enzyme SQS, the post-lanosterol enzymes NSDHL and LDM, and the terminal enzyme DHCR24 [24, 42]. Neuron-rich areas of the mouse hippocampus express higher amounts of cholesterol biosynthetic enzyme mRNA transcripts than astrocyte-rich areas [44], although this may not translate to protein expression or enzyme activity.
The ability of neurons to produce cholesterol is dependent on the life stage of the vertebrate. Developing neurons actively and locally synthesize cholesterol with the endogenous sterol pool increasing over time [28]. A comparison of cholesterol synthesis in neurons between adult versus embryonic mice reveals striking differences. Adult mice with a neuron-specific cholesterol synthesis ablation due to a deletion of SQS, are phenotypically similar to wild-type mice, indicating that neurons in adults do not contribute to the overall cholesterol synthesis in the brain [31]. However, ablation of cholesterol synthesis via conditional SQS knockout in murine neuronal precursor cells is perinatally lethal, with embryos developing smaller brains [30]. Neurons compensate by promoting angiogenesis to increase lipoprotein uptake [30]. Together, these studies indicate that cholesterol synthesis predominantly occurs in neuron precursor cells or neurons during early development.
Astrocytes are thought to be the main hubs for cholesterol synthesis in the brain, with most of the sterol supplied to neurons [45]. These glial cells support neuronal growth and development during childhood, and neuronal maintenance throughout adulthood [32]. Cholesterol is synthesized in the endoplasmic reticulum and rapidly transported to the plasma membrane in vesicles and via apolipoprotein-mediated systems [32]. During development, astrocytes maintain strict control of their endogenous cholesterol homeostasis [28].
Several lines of evidence support the idea that astrocytes are the main cholesterol manufacturers in the brain. Murine studies have shown that transcriptional control of cholesterol biosynthesis is more prevalent in astrocytes as SREBP-1 is more highly expressed in the astrocytes compared to neurons [26]. In addition, between murine astrocytes, oligodendrocytes, and microglia, astrocytes express the highest levels of transcripts of the cholesterol synthesis enzymes throughout the entire biosynthetic pathway [42].
The importance of astrocyte cholesterol synthesis in the brain is underscored by the dramatic phenotypic changes observed when the process is disrupted in murine models. The complete inactivation of the murine equivalents of HMG-CoA reductase and SQS, HMGCR, and SQS, via mutational gene disruptions is embryonically lethal [46, 47]. Conditional mutations of Sqs are not lethal to mice but result in many neurological defects. Mice with astrocyte-specific SQS mutations have a limited ability to secrete cholesterol from astrocytes. In addition, neuronal synapses and synaptic vesicles are unable to mature, suggesting that neurons are dependent on astrocyte-derived cholesterol to support the formation of synapses [26]. Evidently, cholesterol sourced from astrocytes is critical for neuronal function.
Oligodendrocytes wrap axons with cholesterol-rich myelin, providing the electrical insulation necessary for a process called saltatory conduction. Electrical impulses can also skip between nodes of the axon not insulated by myelin, speeding up the arrival of the signal to the synaptic terminal [36]. Oligodendrocytes synthesize substantial amounts of cholesterol for myelin formation and maintenance. Like astrocytes, murine oligodendrocytes express high levels of cholesterogenic transcripts, especially those for the post-lanosterol enzymes, LDM, and DHCR24 [42]. Cholesterol in oligodendrocytes is synthesized de novo via a feed-forward process involving phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) signaling [48].
Myelin contains a large proportion of the brain’s cholesterol store, with estimates from murine models indicating that 80% of the brain’s cholesterol resides within myelin [35]. Rodent studies indicate that the timing of myelination correlates with total brain cholesterol accumulation during the first three weeks of life [9, 35]. Nevertheless, myelination and oligodendrocyte maturation continue during the adult life stages of a mammal [37].
A disruption in oligodendrocyte cholesterol synthesis reduces the rate of CNS myelination and perturbs motor function and coordination [38]. Cholesterol-deficient oligodendrocytes can compensate through cholesterol uptake from neighboring cells [38]. Cholesterol availability is a limiting factor of myelin growth during development, with cholesterol synthesis in oligodendrocytes especially crucial during early life stages. Myelination continues throughout adulthood for the growth and repair of white matter tissue [37]. The regulation of this process is dynamic and cholesterol crosstalk occurs between astrocytes and oligodendrocytes. An in vitro study indicates that astrocytes from grey matter (an area of the brain rich in neuronal bodies) rather than white matter (an area in the brain rich in myelinated axons) are able to secrete cholesterol and support myelination [39]. Clearly, when cholesterol supply is disrupted, astrocytes are a critical source of cholesterol necessary for myelination. Further studies are warranted to explore how cells can re-route neural circuits during myelin repair and what signaling processes are involved. Such studies may offer novel insights into the pathophysiology of neurological diseases such as multiple sclerosis, which is characterized by immune-mediated demyelination and loss of axons [49].
Microglia are phagocytic immune cells in the CNS that engulf lipid debris and trigger apoptosis. Microglia survey and respond to the state of synapses, in a process known as synaptic pruning, which is particularly active in the developing brain [41]. A sub-population of microglia is responsible for the removal of myelin debris and remyelination [40]. Microglia adapt and respond to many environmental cues, such as neuroinflammation in diseases such as Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis. Cellular lipid composition influences microglial function [33, 40]; while cholesterol is essential for microglial survival [43], excessive intracellular cholesterol accumulation in aged or pro-inflammatory microglia in multiple sclerosis is toxic and impairs the ability for microglia to phagocytose myelin debris and repair myelin [50, 51].
Microglia do not synthesize large amounts of cholesterol since cholesterogenic enzymes in murine microglia exhibit a low transcription profile compared to other glial cells [42]. Instead, microglia derive most of their cholesterol from neighboring astrocytes [43]. However, the synthesis of cholesterol intermediates is more critical in microglia. A recent study found that desmosterol, the immediate cholesterol precursor in the Bloch pathway, is critical for the repair of demyelinated lesions [42]. Desmosterol activates LXR signaling for cholesterol efflux to resolve inflammation and promote oligodendrocyte differentiation [42]. The synthesis of other sterol intermediates could also play a role in microglia function; however, this requires further investigation.
Transport of cholesterol in the brain is largely contained within the organ itself due to limitations in cholesterol trafficking across the blood-brain barrier. Brain cell types have various mechanisms available to export and/or accept cholesterol from other cells (summarized in Figure 2). Astrocytes are thought to be the primary cholesterol exporters, with the sterol bound to apolipoproteins in high-density lipoprotein (HDL)- like particles [52]. The two main apolipoproteins for cholesterol transport in the brain are apolipoprotein E (ApoE) and ApoJ, also known as clusterin [53]. The apolipoproteins can bind to a variety of receptors for cholesterol uptake into cells, including low-density lipoprotein (LDL) receptor (LDLR), LDLR-related protein 1 (LRP1), and triggering receptor expressed on myeloid cells 2 (TREM2) [20, 53]. As aforementioned, other brain cells often require the utilization of astrocyte-derived cholesterol. Receipt of cholesterol from astrocytes by cholesterol-deficient oligodendrocytes in mice is likely mediated by ApoE and its receptor LRP1 [36, 54]. Microglia cultured in vitro requires ApoE and clusterin-containing lipoproteins for survival and phagocytosis [43]. Lipidated ApoE and/or clusterin efficiently bind to the TREM2 receptor [55] expressed by microglia. Murine neurons in culture require ApoE-bound cholesterol sourced from glial cells to induce synaptogenesis [52, 56]. Astrocytes are proposed to release lipids such as cholesterol for direct insertion into neuronal membranes to influence synaptic transmission, although further studies into the lipid-metabolizing enzymes and lipid trafficking mechanisms in glial cells are needed [33]. Neurons can release cholesterol bound to apolipoproteins [52] or in the oxidized form as 24(S)-hydroxycholesterol (24S-HC) [57].
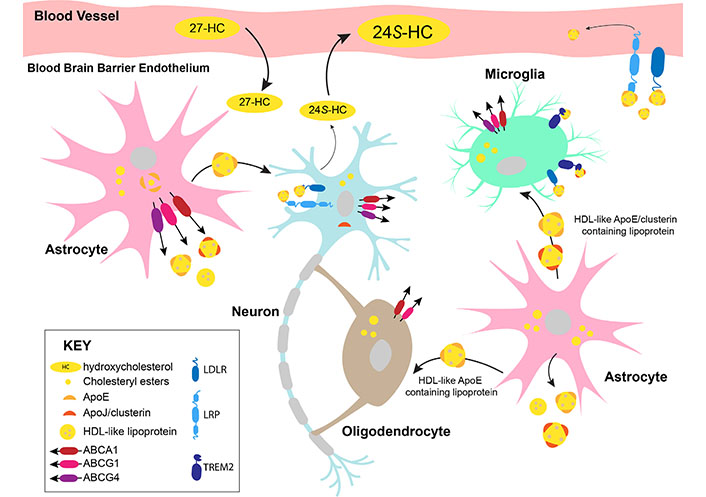
Cholesterol trafficking in the brain. 27-HC: 27-hydroxycholesterol; ABCA1: ABC transporter subfamily A member 1; ABCG1: ABC transporter subfamily G member 1
The variety and brain-cell type-specific distribution of cholesterol trafficking-related proteins, and the difficulty of in situ observations, complicate the in vivo characterization of physiological brain cholesterol transport. ApoE and clusterin have been studied to varying extents and their structure, function, cell-type-specific expressions, and roles in the context of Alzheimer’s disease are discussed below.
Cholesterol is trafficked in the brain in association with ApoE or clusterin in HDL-like particles. Astrocytes are the primary source of cholesterol synthesis and transport in the brain. Astrocytes transport lipidated ApoE-containing lipoproteins to neurons. Neurons express clusterin and excess cholesterol from neurons is converted into 24S-HC by cytochrome P450 (CYP) 46A1 (CYP46A1) for efflux. Oligodendrocytes synthesize large amounts of cholesterol for myelination and may take up additional cholesterol from astrocytes. Microglia encounter HDL-like lipoproteins from astrocytes and take up the cholesterol via the receptors, TREM2 and LDLR. A minor proportion of cholesterol can be exported out of the brain in ApoE-containing lipoproteins. However, most of the excess cholesterol is converted into 24S-HC, which diffuses across the blood-brain barrier into the periphery. 27-HC synthesized by peripheral cells may cross the blood-brain barrier into the brain and act as an indicator of plasma cholesterol levels in the brain. The well-studied cholesterol exporters, ABCA1, ABCG1, and ABCG4 are expressed by astrocytes, neurons, and microglia. ABCA1 and ABCG1 are expressed by oligodendrocytes.
ApoE is the most abundant lipoprotein in the CNS and is synthesized locally chiefly by astrocytes, partially by microglia, and minimally by neurons [58]. ApoE oligomers cannot be complex with lipids. However, upon monomerization, the lipid-binding region is exposed and the protein is able to bind lipids [59–62]. Lipidation of ApoE by cholesterol and phospholipids is mediated by ABC transporters. The resultant HDL-like particles are then secreted by astrocytes into the interstitial fluid [15]. Fully lipidated ApoE undergoes a conformational change to enable interaction with its receptors for cellular uptake [63].
Clusterin is the second major apolipoprotein present in the brain [64]. It is a multifunctional glycoprotein that is distributed in lipoproteins and ubiquitously expressed in the extracellular matrix of many tissues throughout the body and in the CNS [53, 65]. The protein consists of two linked polypeptide chains, which can be configured into different proteoforms by the presence or absence of glycosylation [66]. The proteoform structure confers specificity in the protein’s subcellular localization and function [66]. Intracellular clusterin regulates apoptosis [67, 68] and mitochondrial function [69, 70] and is not known to transport cholesterol. Only the most mature and glycosylated form of clusterin is secreted from the cell and is a known component of circulating peripheral lipoproteins and HDL-like particles in the brain [71]. Clusterin is expressed by both astrocytes and neurons [72], as well as the ependymal cells lining the ventricle [72, 73]. Clusterin is required for lipid trafficking [74] and is able to bind to the LRP2 receptor in vitro [75], with lipidated clusterin enhancing the binding affinity [65]. The interplay between the role of clusterin as a cholesterol carrier and its intracellular functions is an avenue for further exploration.
Other lipoproteins present in the brain, albeit in smaller quantities, include the peripherally-derived apolipoproteins ApoA-I and ApoA-II, and ApoD, ApoC-I, ApoA-IV, and ApoH which are synthesized within the brain. Their roles in cholesterol transport within the CNS are poorly understood [76, 77].
Three major isoforms exist for ApoE in humans, ApoE2, ApoE3, and ApoE4. The ApoE4 isoform, as well as a single nucleotide polymorphism (SNP) in the clusterin gene (CLU) are associated with elevated risk for Alzheimer’s disease [64, 78, 79], suggesting that cholesterol trafficking may be dysregulated in patients with the disease. Indeed, cholesterol abnormalities have been observed in these patients; plasma membranes of brain cells are enriched with cholesterol, and these cholesterol levels persistently increase throughout the course of the disease [80, 81].
The ApoE4 isoform correlates with impaired clearance of amyloid-beta (Aβ) peptides [82], which are a characteristic feature of the Alzheimer’s brain and have been shown to contribute to neurotoxicity and neurodegeneration [79]. The ApoE4 isoform does not operate as efficiently in the delivery of cholesterol to neurons as compared with the ApoE3 isoform [72]. ApoE isoform-dependent processing of Aβ has been recently reviewed by Chai et al [82]. In addition to its role as a cholesterol carrier, secreted clusterin may serve as a protein chaperone outside the cell, in a similar manner to heat shock proteins [83]. Clusterin can be complex with Aβ to mediate endocytosis and transcytosis [84] and is thought to promote cell survival [64]. An Alzheimer’s disease-associated SNP in clusterin affects the alternative splicing of the gene, decreases its expression, and likely reduces Aβ clearance [64]. Cholesterol and Aβ aggregate carried by ApoE and/or clusterin bind with the microglial receptor, TREM2, in human cells cultured in vitro [55], suggesting that cholesterol and Aβ aggregates can be cleared by microglia.
The precise molecular mechanisms of how the different ApoE isoforms and clusterin transport cholesterol in healthy brains versus in Alzheimer’s disease still require further examination. The lipid and apolipoprotein composition of extracellular vesicles from healthy and diseased brains could be analyzed by comprehensive ‘omic’ studies [85]. These vesicles are reflective of intracellular contents, providing a non-invasive method of study. Proteomic and lipidomic studies can be coupled with reductionist approaches with in vivo models to understand the nuances of apolipoprotein-mediated cholesterol transport in healthy versus diseased brains. Further elucidation of structural and conformational changes of ApoE and clusterin upon cholesterol-binding is not only important for understanding cholesterol transport in the brain but may also facilitate future drug target design for Alzheimer’s disease.
The distribution of cholesterol and other lipids within the brain is facilitated by the actions of ABC transporters. These transporters are widely expressed throughout the body and utilize the energy generated from ATP hydrolysis to translocate a broad variety of endogenous and exogenous substrates across cell membranes. In humans, the ABC transporter superfamily encompasses 48 structurally similar proteins categorized into seven families designated ABCA to ABCG. The majority of these are full transporters, exhibiting a four-domain composition consisting of two cytoplasmic nucleotide-binding domains (which house the signature LSGGQ amino acid motif) and two transmembrane domains (which dictate the ligand specificity) [86]. Half transporters, such as ABCDs and ABCGs, contain a single nucleotide-binding domain and a single transmembrane domain and must form homo- or hetero-dimers to be fully functional. Several ABC transporters that are expressed in the brain are also known to transport lipid substrates (Table 2). While many of these have only been investigated with respect to their lipid-transporting capacity in the periphery and thus whether corresponding functions exist for them in the brain remain unexplored, important roles have been identified for ABCA1, ABCG1, and ABCG4 in brain cholesterol transport.
ABC lipid transporters expressed in the brain
| ABC subfamily | Transporter | Cellular location within the brain | Lipid substrates (including brain and periphery) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Neurons | Astrocytes | Oligodendrocytes | Microglia | Choroid plexus epithelial cells | Capillary endothelial cells | References | |||
| ABCA | ABCA1 | • | • | • | • | • | • | [87–89, 90–92] | Cholesterol, phospholipids (PC, PS), oxysterols (24S-HC) [93–95] |
| ABCA2 | • | • | • | - | - | • | [87–89, 91, 92, 96] | Cholesterol, sphingomyelin, glycosphingolipids [97–99] | |
| ABCA3 | • | • | • | • | - | • | [87, 89, 92, 96] | Possibly cholesterol, phospholipids (PC, PG) [89, 100–102] | |
| ABCA7 | • | - | • | • | - | - | [87, 89] | Possibly cholesterol, phospholipids (PC, sphingomyelin) [103–105] | |
| ABCA8 | • | • | • | • | • | - | [87, 89, 106] | Cholesterol [107] | |
| ABCB | ABCB1 | • | • | - | • | • | • | [89, 91, 96, 108–110] | Possibly cholesterol, phospholipids (PC, PE, PS, sphingomyelin), glucosylceramide, glycosphingolipids [111–115] |
| ABCC | ABCC6 | • | - | - | - | - | • | [91, 116] | Associated with plasma lipoprotein and HDL levels [117] |
| ABCD | ABCD1 | - | • | • | • | - | • | [89, 91] | Very long-chain fatty acids (preference for saturated species), fatty acyl-CoAs [118, 119] |
| ABCD2 | • | - | - | - | - | - | [89, 118] | Very long-chain fatty acids (preference for polyunsaturated species), fatty acyl-CoAs [118, 119] | |
| ABCD3 | - | - | - | - | - | • | [96] | Very long-chain fatty acids, long and branched-chain acyl-CoA [118, 119] | |
| ABCG | ABCG1 | • | • | • | • | • | • | [88–91, 120, 121] | Cholesterol, phospholipids (PC), oxysterols, desmosterol [88, 93, 120, 122, 123] |
| ABCG4 | • | • | - | • | - | • | [89, 91, 120, 124] | Cholesterol, cholesterol synthesis intermediates (desmosterol, lathosterol, lanosterol), oxysterols [120, 125–127] | |
Note that the substrates listed here are not limited to the brain and may reflect data from systemic studies since investigations regarding the cholesterol/lipid-transporting activity of ABC transporters within the brain are still limited. “•” denotes expression has been observed in the cell line, “-” indicates no expression or no data. PC: phosphatidylcholine; PE: phosphatidylethanolamine; PG: phosphatidylglycerol; PS: phosphatidylserine; ABCB: ABC transporter subfamily B; ABCC: ABC transporter subfamily C
The two most extensively studied ABC transporters involved in lipid transport in the CNS are ABCA1 and ABCG1. Both transporters are highly expressed in the brain, including in neurons, microglia, astrocytes, and oligodendrocytes [87]. ABCA1 mediates the initial transfer of cholesterol and phospholipids to lipid-free or poorly-lipidated ApoE and ApoA-I, to form nascent discoidal HDL-like particles. This is followed by further lipidation by ABCG1 (or ABCG4), which interacts preferentially with partially lipidated species [93, 128]. This process has been demonstrated to occur in neurons [88], as well as astrocytes and microglia [129, 130]. The resultant lipoproteins may undergo further remodeling by enzymes and lipid transfer proteins to yield mature HDL-like particles that distribute cholesterol around the CNS [131].
Whilst the absence of ABCG1 in mice has minimal effect on brain cholesterol levels [120], ABCA1 deficiency leads to significant cholesterol dyshomeostasis in the murine brain, resulting in structural and functional disturbances to neurons. In the absence of ABCA1, brain cholesterol content is reduced, and cholesterol export from glial cells is impaired, limiting its availability for utilization by neurons. These alterations to brain cholesterol metabolism elicit compensational enhancement of brain uptake of esterified cholesterol from plasma HDL [132]. Abca1-null mice also have lower levels of ApoE in the CNS, with the ApoE particles tending to be smaller and poorly lipidated [129, 133].
Expression of ABCA2 is higher in the brain compared to other tissues and is predominantly found intracellularly, rather than on the cell surface, within lysosomes of oligodendrocytes and to a lesser extent in neurons [89]. Corresponding with the role of oligodendrocytes in myelin formation, ABCA2 has been implicated in the metabolism of myelin lipids, including sphingomyelin and gangliosides [97, 134]. The effect of ABCA2 on cholesterol homeostasis is controversial. ABCA2 deficiency leads to cholesterol accumulation in macrophages [98], and overexpression of ABCA2 in N2a neuroblastoma cells is associated with reduced total, free and esterified cholesterol levels and reduced cholesterol efflux to ApoE3 [99]. However, overexpression of ABCA2 in HEK293 cells had no effect on cholesterol efflux to ApoA-I, ApoE, or ApoE discs [88].
ABCA3 is primarily expressed on the limiting membrane of lamellar bodies of lung alveolar type II (ATII) cells, where it serves a critical role in the formation of pulmonary surfactant by trafficking PC and PG into the lamellar bodies for storage [89, 100]. Its role in cholesterol and sphingomyelin transport is controversial. The levels of these lipids were unaffected by deletion of the transporter in the murine lung [101], whereas the silencing of ABCA3 expression in human ATII cells reduced sphingomyelin and cholesterol uptake into lamellar bodies [102]. The function of ABCA3 in the brain, including oligodendrocytes where expression is highest [89], remains unexplored.
ABCA7 exhibits the greatest sequence homology with ABCA1 (54%). Its expression in the brain is highest in microglia [89]. Human ABCA7 exhibits two isoforms, type I and type II, which arise from alternative gene splicing. These isoforms have consequently been shown to exhibit varying capabilities in transporting cholesterol and phospholipid to ApoA-I [103]. A separate study found that ABCA7 could mediate phospholipid efflux (including PC and sphingomyelin) to ApoA-I, but that its role in cholesterol efflux was limited [104]. ABCA7-mediated cholesterol efflux has been demonstrated to occur to ApoE discs, but not lipid-free ApoE [105]. The ABCA7 gene has been identified in genome-wide association studies as a risk factor for Alzheimer’s disease [135]. Although its exact contribution to pathogenesis remains to be determined, it has been proposed that lipid transport by ABCA7 serves a protective effect [136], with low ABCA7 expression correlating with increased disease risk and poorer prognosis [135, 137, 138].
ABCA8 promotes cholesterol efflux to ApoA-I in the periphery and increases plasma HDL-cholesterol levels, therefore exhibiting some functional overlap with ABCA1 [107]. Although it does not transport sphingomyelin, ABCA8 has been shown to promote sphingomyelin synthesis in oligodendrocytes, thereby implicating a role in myelin formation and maintenance [139].
ABCB1 exports a large profile of structurally and chemically unrelated substrates, the most notable category of which includes therapeutic drugs. ABCB1 has additionally been recognized as a lipid transporter, with in vitro studies utilizing LLC-PK1 pig kidney epithelial cells transfected with human ABCB1 and human gastric carcinoma cells overexpressing ABCB1 demonstrating that the transporter promotes the efflux of phospholipids, glucosylceramide and sphingomyelin [111, 112]. The ability of ABCB1 to transport cholesterol, however, remains controversial [113, 114]. The role of CNS-expressed ABCB1 in transporting lipid substrates has not yet been assessed, but it appears plausible based on existing data that have focused on its activity in the periphery.
Despite its expression in neurons and capillary endothelial cells in the brain [116], the present understanding of the physiological function of ABCC6 remains limited to the periphery. Mutations to the gene encoding ABCC6 may give rise to the autosomal recessive disorder, pseudoxanthoma elasticum (PXE), which is characterized by atherogenesis and calcification of connective tissues in the body. PXE patients and Abcc6-deficient mice exhibit reduced ApoE, ApoA-I, ApoA-II, and HDL-cholesterol levels, and enhanced cholesterol biosynthesis activity [140, 141], implicating a role for ABCC6 in maintaining cholesterol homeostasis.
ABCD1–3 is expressed at the peroxisomal membranes of glial cells, neurons, and capillary endothelial cells, where they transport fatty acids into the peroxisome for metabolism [89]. The importance of ABCD1 activity for lipid homeostasis is highlighted by the recognition that mutations to the ABCD1 gene result in X-linked adrenoleukodystrophy, a severe neurological disorder characterized by progressive demyelination of the CNS. These patients exhibit accumulation of very long-chain fatty acids in the brain due to impaired ABCD1-mediated peroxisomal uptake for subsequent oxidation [118].
ABCG4 expression in humans is limited to the brain. ABCG4 exhibits 74% amino acid homology with ABCG1, thus exhibiting overlapping functionality in promoting cholesterol efflux to HDL [89, 93, 125]. ABCG1 and ABCG4 may also be co-expressed and form heterodimers, to facilitate the efflux of oxysterols (24S-HC, 27-HC) and sterol intermediates (desmosterol, lathosterol, lanosterol) from astrocytes to HDL [120, 126, 142, 143].
Cholesterol metabolism is regulated by two transcriptional axes; the SREBP transcription factors and the LXR and retinoid X receptor (RXR) transcription factors. The genes responsible for cholesterol synthesis are upregulated by SREBP-1a and SREBP-2 [144]. Components of the SREBP pathway are widely expressed across all tissues, including in the brain [17]. SREBP transcription factors reside within the endoplasmic reticulum, where it is bound to SREBP cleavage-activating protein (SCAP). When cholesterol levels fall, the complex moves into the Golgi, where SREBP undergoes successive proteolytic cleavages, releasing the domain for transcription of genes responsible for cholesterol biosynthesis. Conversely, in the presence of cholesterol or its derivatives, insulin-induced gene (Insig) binds to the SCAP-SREBP complex to inhibit SREBP transcriptional activity.
The LXR-RXR transcriptional axis upregulates genes responsible for cholesterol efflux [15]. LXRs are nuclear receptors that form obligate heterodimers with RXRs, which remain bound to DNA elements of target genes regardless of sterol levels. Both isoforms of LXR (α and β) are expressed in the brain. To modulate their transcriptional activity in the absence of cholesterol derivatives, a corepressor complex interacts with the heterodimer to prevent transcription. However, in the presence of oxysterol ligands, LXR undergoes a conformational change, dissociates from the corepressor complex, and recruits a coactivator complex to promote target gene expression [15].
The ‘canonical’ roles of SREBP and LXR in cholesterol synthesis and efflux in the brain are well established in the literature, but these factors are also involved in the cholesterol-intensive processes of cell proliferation, neuron migration, and myelination.
SREBP-mediated transcription is required for regulating normal levels of cholesterol synthesis and is critical for the survival of embryonic cells [145, 146]. As one of the main cholesterol synthesizers in the brain, astrocytes require SREBP to upregulate cholesterol biosynthetic enzymes, and its activity is modulated by SCAP [17, 147, 148]. The cholesterol generated in astrocytes can then be taken up by neurons for neuronal growth and synaptic maintenance. Mice with SREBP-2 specific knockouts in CNS astrocytes survive after birth, but display microcephaly, impaired brain development, and behavioral and motor defects [34]. Similarly, neurological deficits and early mortality are seen in mice with a loss of astrocytic SCAP [26, 149]. In neuron-astrocyte co-cultures, SREBP-2-regulated transcription in astrocytes is critical for neurite outgrowth [34]. Curiously, mice with a loss of astrocytic SREBP2 display metabolic defects including adiposity reduction and a shift in their metabolism towards carbohydrate oxidation, driven by increased glucose oxidation by the brain [34]. A complex crosstalk between carbohydrate and cholesterol pathways may be present in the brain, and extricating the underlying mechanisms involved is important for understanding the potential relationships between brain health, neurodegenerative conditions, and metabolic disorders.
To date, the importance of SREBP transcription factors in other brain cell types has not been examined closely. SREBP is likely to be critical for oligodendrocytes and developing neurons, which are other major sites for cholesterol synthesis in the brain. Mice with a whole-body disruption of one Insig isoform were viable and showed close to normal lipid metabolism, suggesting potential compensation by the other Insig isoform or other mechanisms [150–152]. Nevertheless, an animal model with CNS-specific perturbations to both Insig-1 and Insig-2 isoforms may provide further insights into the regulation of the SREBP pathway in the brain.
In the brain, LXR activation directly upregulates the transcription of cholesterol transporters, and indirectly downregulates cellular cholesterol uptake. LXR controls the transcription of ApoE and ABC transporters including ABCA1 and ABCG1 [153], which lipidated apolipoproteins for intercellular cholesterol distribution [15]. Meanwhile, LXR also transcriptionally upregulates the gene, inducible degrader of the LDLR (Idol) [154]. Idol is an E3 ubiquitin ligase that specifically targets LDLR for proteasomal degradation and thus decreases cholesterol uptake into the cell. Disruption of both LXRα and β isoforms in mice leads to pathological changes in the brain, including lipid accumulation and neurodegeneration, with significant downregulations in the mRNA levels of direct LXR targets, including ABCA1 and ABCG1 gene expression, as well as indirect LXR targets, including LDLR, SREBP-1c, and SREBP-2 genes and cholesterol synthesis genes [155].
The LXR pathway itself is regulated by an endoplasmic reticulum-bound transcription factor, nuclear factor erythroid 2 related factor 1 (NRF1). Under low cholesterol conditions, NRF1 translocates into the nucleus and represses the LXR pathway, preventing cholesterol removal. However, in situations of cholesterol excess, cholesterol-binding retains the transcription factor in the endoplasmic reticulum, resulting in a de-repression of LXR activity and activation of cholesterol export [156]. Thus, NRF1 is a master regulator of the LXR pathway, and, indeed in the mouse liver, it exerts a protective effect by preventing cholesterol accumulation and suppressing inflammation. Although, NRF1-mediated control of cholesterol levels has not been well investigated in the brain, disrupting its activity in mouse neurons leads to neurodegeneration [157], suggesting the importance of NRF1 in this organ.
During brain development, LXR signaling influences neuronal migration, where cells arrange themselves in appropriate spatio-temporal patterns to optimize neuronal circuit function [158]. When LXRβ is knocked out in mice, neuronal migration is unsuccessful and disorganized [159]. This could occur via LXR-mediated modulation of the Reelin signaling pathway, which is responsible for controlling neuronal migration and the formation of cellular layers [160]. The LXR target, Idol, has been shown to regulate expression of the Reelin and lipoprotein receptors, very-LDLR (VLDLR), and ApoE receptor 2, in vitro [18, 161]. Correspondingly, in vivo pharmacological activation of the LXR pathway in mice increases Idol expression and decreases VLDLR levels [18]. Evidently, neuron migration in developing brains and cholesterol transport is coordinated by the same cellular machinery. Consequently, the crosstalk between LXR-mediated cholesterol transport and Reelin signaling is a potential avenue for research.
Myelin production and repair are also regulated by LXR signaling. Thinner myelin sheaths, and altered motor coordination and spatial learning linked to cerebellar deficits, are seen in LXRα/β-double knockout mice [162]. Primary murine oligodendrocytes in culture require LXR for maturation [162], and during differentiation, LXRβ expression is increased at both neonatal and adult life stages. Treating differentiating primary murine oligodendrocytes with a synthetic LXR ligand-induced cholesterol efflux [19]. Oligodendrocyte differentiation is required for myelin repair and the LXR signaling pathways could be a potential therapeutic target for treating demyelination in multiple sclerosis [49].
Despite highly efficient reutilization of cholesterol in the brain, circumstances may arise in which cholesterol levels exceed cellular requirements, necessitating clearance mechanisms in order to maintain homeostasis. Three metabolic pathways are possible, including esterification followed by intracellular storage, efflux of apolipoprotein-cholesterol complexes from the brain via CSF or the blood-brain barrier, and conversion into 24S-HC.
Excess cholesterol can be esterified and stored within intracellular organelles known as lipid droplets. Within the brain, cholesteryl esters only constitute ~1% of total cholesterol content. Esterification occurs through the action of acyl-CoA: cholesterol acyltransferase 1 (ACAT1), which is localized in the endoplasmic reticulum [25]. ACAT1 expression and activity have been detected in neurons, but not in glial cells [163]. However, cholesterol esterification by ACAT1 may be induced in astrocytes under conditions of cholesterol loading or absence of ApoE [130]. Not only do lipid droplets protect cells from toxic accumulation of excess lipids, but they also serve as reservoirs of cholesteryl esters and triglycerides, which can be hydrolyzed into free cholesterol and fatty acids respectively, to fulfill cellular needs for cell signaling, membrane formation, and energy production [164, 165].
Cholesterol may be eliminated directly from the brain complexed with apolipoproteins. Cholesterol bound to ApoA-I or ApoE is released by glial cells into the CSF, and subsequently cleared from the brain via CSF bulk flow [166]. However, this only constitutes a minor elimination pathway as it is estimated to remove 2 mg of cholesterol each day [166].
Although it is widely recognized that cholesterol is unable to pass through the blood-brain barrier owing to the presence of tight junctions [167], interestingly, one study has reported direct transport of free cholesterol across the blood-brain barrier via ABCA1 in mice [168]. It also remains possible that physiological and morphological changes to the blood-brain barrier as a result of age or neurodegenerative disease may compromise its permeability, rendering a greater likelihood of leakage of substances, including cholesterol, into and out of the brain [169]. Indeed, increased leakage of oxysterols out of the brain at the blood-brain barrier has been reported in pericyte-deficient mice [170].
The third and most quantitatively significant route of elimination of cholesterol from the brain involves conversion into the oxysterol, 24S-HC. Excess cholesterol undergoes 24S-hydroxylation by the CYP enzyme, CYP46A1 (also known as cholesterol 24-hydroxylase), producing a less hydrophobic molecule that traverses lipophilic membranes much more readily than cholesterol [25, 171, 172]. Deletion of CYP46A1 in mice reduces cholesterol efflux from the brain by 64%, highlighting the importance of this elimination pathway [172]. CYP46A1 is localized to neurons of the brain. Immunohistochemical analyses have identified its expression in the endoplasmic reticulum of cortical pyramidal cells, Purkinje cells of the cerebellum, and hippocampal and thalamic neurons [173, 174], thus implicating neurons as the primary site for the initiation of cholesterol turnover in the brain. Despite this, some studies have demonstrated that CYP46A1 expression may occur in microglia and astrocytes in circumstances of brain injury or disease, however, its functionality at these sites has not yet been confirmed [175–178]. Although it is generally accepted that 24S-HC can passively diffuse across the neuronal membrane, Matsuda et al [94] demonstrated that 24S-HC may be actively exported from SH-SY5Y neuronal cells by ABCA1 in the presence of HDL.
Approximately 1% of 24S-HC flux from the brain occurs via the CSF, while the remaining 99% occurs via diffusion through the blood-brain barrier [179]. Active transportation by organic anion transporter proteins can also facilitate the removal of 24S-HC at the blood-brain barrier [180]. 24S-HC is released into circulation at a rate of 6–7 mg/day and delivered to the liver via LDL and HDL for biliary excretion [77, 167, 181]. In healthy individuals, the magnitude of brain flux closely reflects that of hepatic uptake, indicating that 24S-HC production is exclusive to the brain [181]. However, circulating levels of 24S-HC can be affected by age and the brain-to-liver size ratio [182].
In addition to serving as the major form by which excess cholesterol is removed from the brain, oxysterols can also influence cholesterol homeostasis at the transcriptional and post-translational levels. For instance, 24S-HC and 27-HC are ligands for LXR in neurons and initiate transcription of cholesterol transport genes [183–185]. Other oxysterols, such as 24,25-EC, can activate the Hedgehog pathway, which is critical for embryogenesis [5, 186, 187].
Recently, oxysterols have been implicated in many neurodegenerative diseases including multiple sclerosis and Alzheimer’s disease [49], where increased oxysterol levels are associated with adverse diagnostic and clinical outcomes [188], suggesting the clinical potential for oxysterols as biomarkers and therapeutic targets.
24S-HC is one of the most abundant oxysterols in the brain [15]. The CYP46A1 enzyme serves a dedicated function in producing the brain-specific oxysterol to remove excess cholesterol from the brain [186]. 24S-HC can also facilitate cholesterol homeostasis via transcriptional regulation [45]. In situations of cholesterol excess, 24S-HC inhibits SREBP-2 to suppress cholesterol biosynthesis in glial cells [189, 190]. 24S-HC also serves as a ligand for LXR, mediating the expression of target genes encoding cholesterol transporters, ApoE, ABCA1, and ABCG1, thus promoting the distribution of cholesterol from astrocytes to neurons [191].
Other functions of 24S-HC unrelated to cholesterol metabolism include modulation of Aβ production, prevention of tau hyperphosphorylation, and induction of N-methyl-D-aspartate receptor-mediated excitotoxicity. Excess accumulation of 24S-HC may promote oxidative stress and cytotoxicity [191, 192]. 24S-HC may also be involved in the pathogenesis of neurodegenerative disorders and could be a suitable biomarker for such conditions, as its level reflects the number of metabolically active neurons in the brain [193, 194].
While 24S-HC is the predominant oxysterol generated in the brain, the analogous oxysterol generated in the periphery is 27-HC. Excess cholesterol is converted to 27-HC by the CYP 27A1 enzyme in the first step of the alternative bile acid synthesis pathway, which can then diffuse across the blood-brain barrier into the brain at a rate of 5 mg/day [77, 195, 196]. Decreased levels of oxysterol are observed in Alzheimer’s, Parkinson’s, and Huntington’s diseases [5]. Correspondingly, treatment of primary human neurons with 27-HC has been shown to downregulate Aβ production, potentially in an LXR-mediated manner [183], suggesting that the oxysterol is a possible therapeutic target for Alzheimer’s disease. 27-HC may promote LXR activation, as the peripheral injection of the oxysterol into rats has been shown to upregulate LXRα and ABCA1 in the brain, and downregulate genes for cholesterol synthesis, and uptake [197]. However, high doses of 27-HC impair spatial memory performance [197] and further studies are required to understand how 27-HC is involved in brain function.
24,25-EC is produced differently from other oxysterols; it is not derived from cholesterol but generated from sterol intermediates. Partial inhibition of the LSS enzyme channels sterol intermediates into the epoxycholesterol Shunt pathway, where epoxylanosterol is metabolized into 24,25-EC (Figure 1) [198]. In addition, desmosterol can be converted into epoxycholesterol by the action of CYP46A1 [199]. 24,25-EC is produced in human brains [200], and in rat neonate brains it is more abundant than 24S-HC [201].
24,25-EC is involved in the development and maintenance of brain function. Increasing flux in the Shunt pathway via LSS inhibition enhances oligodendrocyte formation and remyelination [202]. Overexpression of human CYP46A1 in mice increases levels of 24S-HC and 24,25-EC, and this is associated with enhanced midbrain dopamine neurogenesis [203], a process important for controlling voluntary movement, reward processing, and working memory [204]. It is uncertain whether CYP46A1 activity or the epoxycholesterol Shunt pathway is more important for 24,25-EC production in the brain, but an interplay between the two mechanisms is likely [186].
Our understanding of cholesterol synthesis and disposal in the brain has progressed significantly over the past few decades, however many questions remain unanswered. Which cell type synthesizes the most cholesterol during development and maturation? Do cells in the brain exhibit preference for the usage of the Kandutsch-Russell or Bloch pathways, and does this change over the course of an individual’s life? Is there an interplay between the roles of ApoE and clusterin as cholesterol carriers with their non-lipid trafficking duties, and how do they confer pathological risk in Alzheimer’s disease? To what degree do ABC lipid transporters share substrates and functionalities between the periphery and CNS? What functions do oxysterols serve in the human brain and could they be used as biomarkers and therapeutic targets for neurodegenerative disorders?
The precise mechanisms for cholesterol regulation are still yet to be elucidated for the brain, due to the predominance of studies focusing on peripheral tissues [205]. Nonetheless, transcriptional and post-translational machinery necessary for homeostatic control of cholesterol expression are present in the brain [17–19], and it is likely that the regulation of brain cholesterol is similar to that which occurs in peripheral tissues. While we have a general understanding of how cholesterol is regulated within the main cholesterol synthesizing cells, astrocytes, and oligodendrocytes, a particular focus on other cell types such as neurons, is warranted. Further studies on the roles of ABC transporters in mediating cholesterol distribution within the brain will also facilitate our understanding of how cholesterol homeostasis is maintained within the brain, and how disruptions to their activity may elicit pathogenic consequences.
Cholesterol serves crucial structural, functional, and supportive roles in the brain. Cholesterol deficiency and excess cholesterol in the brain can contribute to the pathogenesis of diseases alike [20, 22, 49, 206, 207]. Thus, elucidating the nuances of cholesterol regulation, from its synthesis, to distribution, and to disposal, from the brain, provides a critical foundation upon which developments in the understanding of the pathophysiology of neurodegenerative and metabolic disorders can be made.
24,25-EC: 24(S),25-epoxycholesterol
24S-HC: 24(S)-hydroxycholesterol
27-HC: 27-hydroxycholesterol
ABC: ATP-binding cassette
ABCA1: ATP-binding cassette transporter subfamily A member 1
ABCB: ATP-binding cassette transporter subfamily B
ABCC: ATP-binding cassette transporter subfamily C
ABCG1: ATP-binding cassette transporter subfamily G member 1
ACAT1: acyl-coenzyme A: cholesterol acyltransferase 1
ApoE: apolipoprotein E
Aβ: amyloid-beta
CNS: central nervous system
CoA: coenzyme A
CSF: cerebrospinal fluid
CYP46A1: cytochrome P450 46A1
DHCR14: 14-dehydrocholesterol reductase
DOS: dioxidosqualene
HDL: high-density lipoprotein
HMG: 3-hydroxy-3-methylglutaryl
HMGCR: 3-hydroxy-3-methylglutaryl-coenzyme A reductase
Idol: inducible degrader of low-density lipoprotein receptor
Insig: insulin-induced gene
K-R: Kandutsch-Russell pathway
LDL: low-density lipoprotein
LDLR: low-density lipoprotein receptor
LDM: lanosterol 14α-demethylase
LRP1: low-density lipoprotein receptor-related protein 1
LSS: lanosterol synthase
LXR: liver X receptor
MOS: monooxidosqualene
NRF1: nuclear factor erythroid 2 related factor 1
NSDHL: NAD(P) dependent steroid dehydrogenase-like
PC: phosphatidylcholine
PG: phosphatidylglycerol
RXR: retinoid X receptor
SCAP: sterol regulatory element-binding protein cleavage-activating protein
SM: squalene monooxygenase
SQS: squalene synthase
SREBP: sterol regulatory element-binding protein
TREM2: triggering receptor expressed on myeloid cells 2
The authors thank members of the Brown laboratory for their intellectural assistance to this review.
AJB and ICG: funding acquisition and conceptualization. AJB, ICG, ABC, LQ: writing-original draft and editing. LQ and ABC: visualization and preparation of figures and tables. All authors approved the final version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by the Australian Research Council Discovery Project Grant [DP170101178]. Amanda B. Chai and Lydia Qian are supported by an Australian Research Training Program scholarship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Rafael Franco ... Irene Reyes-Resina
Kamran Tariq, Bryan W. Luikart
Jian Xiao ... Jie Luo
Xuntian Jiang, Daniel S. Ory
Irina A. Pikuleva
Ta Yuan Chang ... James G. Gow
