 Review
Review

Affiliation:
Department of Ophthalmology and Visual Sciences, Case Western Reserve University, Cleveland, OH 44106, USA
Email: iap8@case.edu
ORCID: https://orcid.org/0000-0001-9742-6232
Explor Neuroprot Ther. 2021;1:159–172 DOI: https://doi.org/10.37349/ent.2021.00013
Received: October 12, 2021 Accepted: November 19, 2021 Published: December 30, 2021
Academic Editor: Ta-Yuan Chang, Geisel School of Medicine at Dartmouth, USA
The article belongs to the special issue Cholesterol Dyshomeostasis in Neurological Diseases
The brain cholesterol content is determined by the balance between the pathways of in situ biosynthesis and cholesterol elimination via 24-hydroxylation catalyzed by cytochrome P450 46A1 (CYP46A1). Both pathways are tightly coupled and determine the rate of brain cholesterol turnover. Evidence is accumulating that modulation of CYP46A1 activity by gene therapy or pharmacologic means could be beneficial in the case of neurodegenerative and other brain diseases and affect brain processes other than cholesterol biosynthesis and elimination. This minireview summarizes these other processes, most common of which include abnormal protein accumulation, memory, and cognition, motor behavior, gene transcription, protein phosphorylation as well as autophagy and lysosomal processing. The unifying mechanisms, by which these processes could be affected by CYP46A targeting are also discussed.
The brain is separated from the systemic circulation by the blood-brain barrier, which is impermeable for cholesterol. Therefore, the major source of cholesterol for the brain is in situ biosynthesis, and cholesterol elimination is mainly realized via enzymatic hydroxylation at the carbon atom 24 to convert cholesterol to 24-hydroxycholesterol (24HC) [1–3]. 24HC is more soluble than cholesterol and can enter the systemic circulation by crossing the blood-brain barrier [4], after which this oxysterol is delivered to the liver for further biotransformations [2, 5, 6]. Cholesterol 24-hydroxylation is catalyzed by cytochrome P450 46A1 (CYP46A1), a microsomal enzyme specific for the brain and retina. In the brain, CYP46A1 is normally expressed in the pyramidal neurons of the hippocampus and cortex, in Purkinje cells of the cerebellum as well as in hippocampal and cerebellar interneurons [7, 8]. In the retina, CYP46A1 is mostly found in the neurons of the inner nuclear layer and ganglion cell layer [9]. Studies in mice with genetic ablation of Cyp46a1 or pharmacologic activation of CYP46A1 revealed that there is a tight coupling between cholesterol production and elimination from the brain, i.e., when there is an increase or decrease in the cholesterol 24-hydroxylation rate, there is a compensatory increase or decrease, respectively, in the cholesterol biosynthesis rate. This coupling keeps the steady-state cholesterol levels unchanged and thus maintains cholesterol homeostasis [3, 10–14].
Mutations in the genes involved in the biosynthesis and intracellular trafficking of cholesterol underlie Smith-Lemli-Opitz syndrome and Niemann-Pick disease type C (NPC), respectively, developmental disorders with a broad spectrum of manifestations including neurodegeneration [15, 16]. In addition, data point to cholesterol dyshomeostasis in the brain as a contributing factor to such neurodegenerative disorders as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), spinocerebellar ataxia (SCA), and prion disease [17–26]. Consistent with this contribution, the levels of CYP46A1 protein or its metabolite 24HC are decreased in some or all the affected brain regions in AD, HD, SCA3 (Machado-Joseph disease) as well as in sporadic Creutzfeldt-Jakob disease (a human prion disease) and status epilepticus [27–33]. Also, ectopic CYP46A1 expression was found in non-neuronal brain cells (astrocytes) in AD [34, 35], possibly to compensate for a decreased enzyme content in the brain due to the loss of neurons. Accordingly, CYP46A1 is intensively studied by different laboratories as a target for the treatment of neurodegenerative and other brain disorders. Different mouse models are utilized, which are either subjected to gene therapy with CYP46A1 to restore the enzyme expression or a pharmacologic treatment with a small dose of efavirenz, an anti-human immunodeficiency virus (HIV) drug, to activate CYP46A1. The effects of a decreased CYP46A1 expression and activity are also investigated [13, 14, 28, 29, 36–42]. This minireview will first summarize the major common processes affected by modulation of CYP46A1 expression or activity in vivo and then will discuss several unifying mechanisms that can explain the CYP46A1 expression or activity effects.
Accumulation of specific proteins that form toxic aggregates is a common feature of many neurodegenerative diseases. These proteins include amyloid β peptides (Aβ) and hyperphosphorylated tau protein in AD; mutant forms of huntingtin protein in HD, mutant forms of ataxin protein in SCA, the prion protein in prion disease, and α-synuclein in PD.
The Aβ burden was significantly reduced in the brain of APP23 and APP/PS mice, a model of amyloidogenesis in AD after the CYP46A1-containing adeno-associated vector (AAV; AAV-CYP46A1) was injected into the cortex and hippocampus of these animals before or after the onset of amyloid plaques [36]. Similarly, pharmacologic activation of CYP46A1 with efavirenz also had the Aβ decreasing effect when tested on 5XFAD mice, another AD model of amyloidogenesis. The effect was either on the whole brain levels of insoluble Aβ peptides (when efavirenz treatment started before the Aβ deposition) or on the number and area of the dense core amyloid plaques in the mouse cortex and hippocampus (when efavirenz treatment started after the Aβ plaque appearance) [13, 14].
The opposite effect, i.e., increased abundance of the Aβ peptides, was documented in normal or APP23 mice, when their CYP46A1 expression was decreased via hippocampal injections with the AAV containing the short hairpin RNA for Cyp46a1 (AAV-shCyp46a1). Similarly, the Aβ load was increased in Tg2576 mice, when they were treated with efavirenz at the CYP46A1 inhibiting drug dose [37, 38].
Tau hyperphosphorylation was not affected after the AAV-CYP46A1 injections into the hippocampus of THY-Tau22 mice, a model of AD-like tau pathology, despite the CYP46A1 expression being increased and cognitive deficits, impairments in long-term depression (LTD) as well as spine defects that characterize this model were completely rescued [39]. Yet, abnormal tau phosphorylation was increased (with no evidence of tangles) in normal mice after their CYP46A1 expression was decreased as a result of the hippocampal AAV-shCyp46a1 delivery [37].
Aberrant accumulation of phosphorylated tau was reduced in neurons and astrocytes derived from induced pluripotent stem cells (a human model of AD) after the former were treated with the CYP46A1 activating concentration of efavirenz [43, 44].
The number and volume of the aggregates of the mutant forms (with polyglutamine expansion) of huntingtin protein were decreased in the R6/2 HD mouse model following the striatal AAV-CYP46A1 injection [28]. Similarly, a decreased aggregate load was observed in the zQ175 mice (a knock-in mouse model of HD) injected striatally with AAV-CYP46A1 [40].
A reduction in the number and size of the mutant (with polyglutamine expansion) ataxin-3 aggregates were observed in the lentiviral-based model of SCA3, the most prevalent form of ataxia, after mice were injected into the cerebellum with the AAV-CYP46A1 [29].
Oral administration of efavirenz after intracerebral prion inoculation significantly prolonged the lifespan of mice, which was consistent with preliminary studies in cell culture showing that efavirenz treatment significantly mitigated the propagation of the misfolded prion protein [32].
Studies of the therapeutic value of CYP46A1 for PD have not yet been carried out but are warranted by immunolocalization of the two isoforms of isopentenyl diphosphate isomerase involved in the biosynthesis of isoprenoids and ultimately cholesterol. Both isomerase isoforms were found to be strongly expressed in Lewy bodies in patients with PD and were co-localized with phosphorylated α-synuclein [45]. It is thus possible that cholesterol biosynthesis could play a role in the α-synuclein aggregation. Hence, enhancement of cholesterol elimination by CYP46A1 could be beneficial.
Neurodegenerative diseases vary in their clinical manifestations with some mainly affecting memory and other impairing motor function.
Improved performance in behavioral tests was documented in two mouse models of AD (APP23 mice, Morris water maze (MWM) test, and THY-Tau22 mice, Y-maze and MWM tests) after intracerebral injections with the AAV-CYP46A1 [36, 39]. Spatial memory was improved in the MWM test in old CYP46A1 transgenic female mice, i.e., animals which had increased CYP46A1 expression [10]. Similarly, improved performance in the MWM test was observed in 5XFAD mice treated with the CYP46A1 activator efavirenz [13, 14]. Also, pharmacologic CYP46A1 activation with efavirenz improved the performance of NPC1nmfl64 mice (a model of NPC1) in the object placement recognition test, the Y-maze test, and fear conditioning tests [46].
In contrast, a complete lack of CYP46A1 activity in Cyp46a1−/− mice led to severe deficiencies in spatial, associative, and motor learning [47]. Memory deficits (the open-field task and MWM test) were also triggered in wild-type mice by a partial decrease in Cyp46a1 expression as a result of the hippocampal AAV-shCyp46a1 injection [37].
Long-term potentiation (LTP) and LTD are the two primary forms of synaptic plasticity, or efficacy, i.e., strengthening and weakening, respectively, of synaptic transmission in response to electrical stimulation. Synaptic plasticity is the cellular basis of learning and memory [48].
Pharmacologic activation of CYP46A1 with efavirenz restored basal synaptic transmission and LTP in hippocampal slices from NPC1nmf164 mice [46]. Impairments in LTD were completely rescued by gene therapy of THY-Tau22 mice with AAV-CYP46A1 [39].
Hippocampal LTP was impaired in Cyp46a1−/− mice [47], and LTD was inhibited in hippocampal slices of rats treated with the CYP46A1 inhibitor voriconazole as well as the two newly developed enzyme inhibitors [49].
Activation of N-methyl-D-aspartate receptors (NMDARs) is a key mediator of the LTP induction, and 24HC was found to be a potent positive allosteric modulator of the GluN2B subunit of NMDARs [50–54]. Also, CYP46A1 transgenic mice were shown to have increased expression of some of the synaptic proteins, including one of the NMDAR subunits [10]. Cyp46a1−/− mice were found to have reduced activity and function of these receptors [55].
The motor deficits and impairments were improved in R6/2 mice (an HD model, the rotarod and clasping tests) and Q69 mice (a SCA3 model, the rotarod and footprinting pattern tests) after the brain injections with AAV-CYP46A1 [28, 29]. Similarly, pharmacologic CYP46A1 activation with efavirenz improved motor abilities of NPC1nmfl64 mice in the rotarod test [46].
A decreased CYP46A1 brain expression after the AAV-shCyp46a1 injection into wild-type mice led to motor deficits (the rotarod test) [28]. Pharmacologic CYP46A1 inhibition with soticlestat reduced seizure frequency in 14 human subjects with developmental and/or epileptic encephalopathies associated with NMDAR overactivation [56].
Studies by different omics approaches provide unbiased and sometimes unanticipated insights that then can be followed up in more detail and expand our understanding of the significance of the protein in question. Only in vivo effects as assessed by RNA-seq will be considered here as such an approach reveals global changes in the brain transcriptome.
Gene therapy with CYP46A1 significantly modified the striatal transcriptome of zQ175 mice. The affected genes play a role in neurotransmission, synaptic activity, RNA processing, Toll-like receptor signaling, cytoskeleton dynamics, immune response, phagosome, proteasome, small RNA metabolism, neuronal apoptosis, and other processes [40]. Gene expression was also affected in the brain of 5XFAD mice treated with the CYP46A1-activating dose of efavirenz. These were the genes involved in amyloid precursor protein processing, neurotransmission, neurogenesis, inflammation, immune response, autophagy, ubiquitin-proteasome systems, hypoxia, apoptosis, and glucocorticoid-related stress [13, 57, 58].
Like with the gene expression, only a global effect (as assessed by a phosphoproteomic approach) and only on animal samples will be presented here.
Changes in protein phosphorylation were detected in the brain of both 5XFAD mice treated with efavirenz and Cyp46a1−/− mice [57, 59, 60]. In Cyp46a1−/− mice, differentially phosphorylated proteins were involved in brain processes related to cognition, cytoskeletal organization, and genetic information transfer [59]. In efavirenz-treated 5XFAD mice, differentially phosphorylated proteins encompassed similar processes (synaptic function, cytoskeletal organization, and genetic information transfer) plus cell differentiation and proliferation [57, 60]. Notably, there were differentially phosphorylated proteins common between Cyp46a1−/− mice and efavirenz-treated 5XFAD mice: seven of them [centrosomal protein of 170 kDa protein B (CEP170B), microtubule-associated protein 2 (MAP2), MAP1B, dihydropyrimidinase-like 2 (DPYSL2), catenin delta 2 (CTNND2), growth associated protein 43 (GAP43), and neurofilament heavy chain (NEFH)] pertained to the cytoskeletal organization and one protein [eukaryotic initiation factor 4B (EIF4B)] was involved in the initiation of protein synthesis [60]. The differentially phosphorylated protein kinases [e.g., brain-specific kinase 2 (BRSK2), cyclin-dependent kinase 5 (CDK5), casein kinase 1 or 2 (CK1/2), glycogen synthase kinase 3 α/β (GSK-3α/β), mitogen-activated protein kinase kinase kinase kinase-4 (MAP4K4), misshapen-like kinase 1 (MINK1), p38 mitogen-activated protein kinase (MAPK), protein kinase C epsilon type (PRKCE), and Traf2 and NCK-interacting protein kinase (TNIK) and protein phosphatases serine/threonine-protein phosphatases 1/2A and 2B (PP1/2A and PP2B), tumor-promoting effect of tyrosine phosphatase receptor type N (PTPRN), and protein tyrosine phosphatase receptor delta (PTPRD)] affected by the CYP46A1 activity modulation were identified as well [57, 60, 61].
In neurodegenerative and other diseases, these mechanisms play important roles in the clearance of small molecules as well as aberrantly processed and misfolded proteins [62, 63].
Striatal gene therapy with CYP46A1 induced autophagy and led to a decreased deposition of mutant ataxin-3 in a transgenic Q69 SCA3 model [29].
Restoration of CYP46A1 expression in the striatum of zQ175 mice increased the formation of autophagosomes and improved the clearance of the aggregated mutant huntingtin protein [40].
Endosomal trafficking was disturbed in the CA3a pyramidal cells of wild-type mice injected hippocampally with the AAV-shCYP46A1 vector [37]. In addition, the AAV-shCyp46a1-mediated Cyp46a1 downregulation led to lysosomal swelling and an increase in their number as well as the appearance of abnormal lysosomes and the accumulation of autophagolysosomes. Collectively, these data suggested that endosomal-lysosomal membrane trafficking was impaired in mice with decreased hippocampal Cyp46a1 expression [64]. Also, the endosome and lysosome accumulation was observed after the striatal AAV-shCyp46a1 -injection to wild-type mice [29].
NPC1 is the classical lysosome storage disorder characterized by cholesterol accumulation in the lysosomes in nearly all tissues [65]. CYP46A1 activation with efavirenz improved the lysosomal phenotype in the hippocampus of NPC1nmf164 mice, i.e., significantly reduced the lysosomal cholesterol accumulation, and extended the animal life span [46].
The effects of genetic or pharmacologic CYP46A1 activity modulation extend beyond the processes presented in this minireview and include, but not limited to, apoptosis, the stress of the endoplasmic reticulum, transcriptional regulation via liver X receptors, expression of important synaptic proteins, stimulation of dopaminergic neurogenesis, the function of lipid rafts and other, often disease-specific mechanisms [13, 14, 28, 29, 32, 36–42, 46, 66–68].
In vivo data keep accumulating and demonstrate that CYP46A1 targeting by gene therapy or pharmacologic treatments affect multiple brain processes and pathways and can serve as a potential treatment for many brain disorders, both neurodegenerative and non-neurodegenerative (e.g., glioblastoma and depression [66, 67]). Hence, the existence of the mechanisms that integrate multiple CYP46A1 expression or activity effects (called the unifying mechanisms) was proposed [25, 61]. Two of these unifying mechanisms, which involve the cholesterol biosynthesis and cholesterol elimination pathways, were discussed in detail elsewhere [21, 25, 69, 70]. Hence, herein, we will present these pathways only briefly and mainly focus on a third potential unifying mechanism that was discovered recently and is mediated by the brain acetyl-coenzyme A (acetyl-CoA) production [60].
The cholesterol biosynthesis pathway, or the first unifying mechanism, starts from acetyl-CoA as the first substrate and includes more than 20 enzymatic reactions [71]. Cholesterol is synthesized from acetyl-CoA in three stages, which end in the formation of 3-isopentenyl pyrophosphate (PP; stage 1), squalene (stage 2), and cholesterol (stage 3) (Figure 1). The production of mevalonate during stage 1 is a committed (or irreversible) step in cholesterol biosynthesis, hence stage 1 as well as stage 2, which involves the condensation of the phosphorylated form of mevalonate (3-isopentenyl PP), is often called the mevalonate pathway [71–73]. Many non-sterol intermediates produced in the mevalonate pathway [isopentyl transfer RNAs (tRNA), dolichol, heme A, ubiquinone, farnesyl PP, and geranylgeranyl PP] represent essential compounds with key roles in a variety of brain or cellular processes such as normal brain development [74, 75], LTP and higher-order brain functions [47, 76], motor function [77], lysosomal function [75], autophagy [78], malignant cell growth and survival [79, 80], DNA replication [81], protein N-glycosylation [74, 82–84], protein prenylation [69, 70], and mitochondrial electron transport [85]. Since the mevalonate pathway is tightly coupled to cholesterol elimination by CYP46A1 [3], modulation of CYP46A1 expression or activity can lead to changes in the rate of cholesterol biosynthesis and hence the rate, at which important non-sterol cholesterol intermediates are produced. The latter in turn may affect biological processes and pathways, in which these cholesterol intermediates are utilized, and thus explain a variety of biological manifestations associated with CYP46A1 targeting and the CYP46A1 beneficial role in a variety of brain disorders.
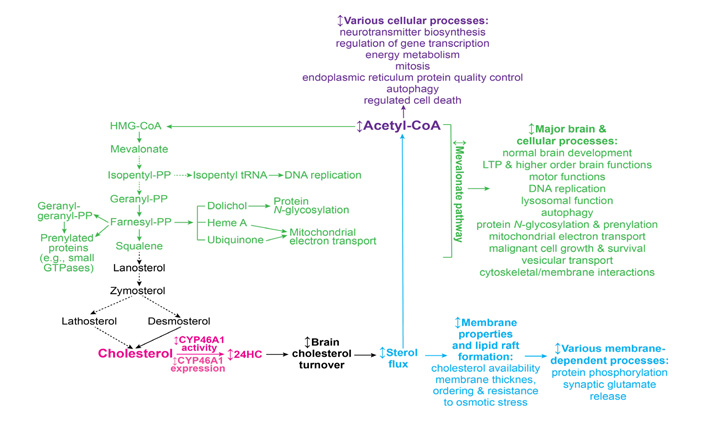
Schematic representation of the three potential mechanisms (mevalonate pathway, sterol flux, and acetyl-CoA production, colored in green, blue, and violet, respectively) that can integrate a variety of CYP46A1 targeting effects. The initial event, CYP46A1-mediated cholesterol 24-hydroxylation, is colored in magenta. See main text for details. Dashed arrows indicate multiple steps; ↕: the up-down arrow indicates modulation (increase or decrease); HMG: 3-hydroxy-3-methylglutaryl
This mevalonate pathway mechanism is supported by a requirement for constant production of geranylgeranyl PP in neurons for LTP and learning as revealed by studies of Cyp46a1−/− mice [47, 76]. Also, increased prenylation of small GTPases, which regulate diverse cellular activities (e.g., intracellular vesicle transport, cell adhesion, endocytosis, cytoskeletal organization, cell cycle progression, receptor signaling, vesicle trafficking, and gene expression), was documented in neurons with increased CYP46A1 expression [70, 86, 87].
Cholesterol elimination by CYP46A1, or the second unifying mechanism (Figure 1), determines the rate of brain cholesterol turnover [3], and hence the rate of sterol flux through the plasma and subcellular membranes. The sterol flux rate can in turn affect the physico-chemical properties of plasma membranes along with the properties of lipid rafts that are present in these membranes. As a result, membrane-dependent events can also become affected including synaptic transmission and protein phosphorylation [61].
The sterol flux mechanism is supported by changes in the lipid raft cholesterol content elicited by modulations in CYP46A1 expression in wild type mice and animal models of AD and HD [36, 37, 88]; altered protein phosphorylation in the brain of animals with decreased (Cyp46a1−/− mice) and increased (efavirenz-treated 5XFAD mice) sterol fluxes [57, 59, 60]; as well as altered properties of synaptosomal fractions isolated from the brain of animals with modulated sterol flux [61]. The latter include changes in cholesterol accessibility, thickness, and ordering of synaptosomal membranes, synaptosome resistance to osmotic stress, their total glutamate content, and the ability to release glutamate in response to mild stimulation. In addition, the sterol flux rate was found to affect the ability of several protein kinases (GSK-3 and CDK5) and protein phosphatases (PP1/2A and PP2B) to regulate glutamate release from brain synaptosomes [61].
Finally, the third unifying mechanism (Figure 1) can rely on the production of acetyl-CoA as the levels of this compound were found to depend on the rate of sterol flux [60]. Indeed, relative to the corresponding wild-type mice, the total and mitochondrial acetyl-CoA pools were decreased in the brain of Cyp46a1−/− and control 5XDAD mice. Yet, the acetyl-CoA content was either restored to normal levels (in the total pool) or became even higher (in the mitochondrial pool) in efavirenz-treated 5XFAD mice. Also, the abundance of several enzymes involved in the acetyl-CoA biosynthesis and the pathways that provide substrates for the synthesis of acetyl-CoA was increased, whereases the brain glucose levels were decreased in 5XFAD mice with increased sterol flux (the efavirenz-treated cohort) vs. decreased sterol flux (the control, vehicle-treated cohort). Collectively, these data suggested that an increase in the total acetyl-CoA pool in efavirenz-treated vs. control 5XFAD mice could be due to an increase in acetyl-CoA production as a result of increased glucose utilization, normally the principal source of acetyl-CoA and energy in the brain [89].
Acetyl-CoA is a central molecule in a myriad of biological processes including energy metabolism, mitosis, endoplasmic reticulum protein quality control, autophagy, regulated cell death, epigenetic regulation of gene transcription, neurotransmitter biosynthesis, and many others [90]. Hence, the CYP46A1-acetyl-CoA link could certainly represent the third mechanism integrating multiple CYP46A1 activity effects. Of various processes requiring acetyl-CoA, two could be of particular importance for brain disorders. These are the acetylation of choline to generate acetylcholine, a neurotransmitter essential for memory and learning, and acetylation of histones to increase gene transcription [91, 92].
Inhibition of acetylcholine breakdown with donepezil is currently a symptomatic treatment for AD [92]. It is conceivable that an increase in the acetyl-CoA production as a result of CYP46A1 expression or activity increase could lead to an increase in the acetylcholine synthesis and thus improvement in cholinergic neurotransmission as well as memory.
A reduction in histone acetylation has been causally implicated in memory impairment associated with neurodegeneration, aging, and neurodevelopment disorders [93]. The CYP46A1-mediated increase in the brain acetyl-CoA levels could certainly increase acetylation of some of the histones as acetyl-CoA is a sole donor of acetyl groups for protein acetylation, including those of histones [91]. As such, acetyl-CoA could also exert a general control of gene expression via epigenetic regulation as well as alteration of the acetylation state of transcription factors [90, 94, 95]. These functions can explain changes in brain transcriptome and protein abundance observed in mouse models with CYP46A1 activity modulation described in the previous sections, and the data suggesting epigenetic regulation of cholesterol homeostasis [96]. Further studies are required to determine what processes involving acetyl-CoA are affected by CYP46A1 activity modulation.
Cholesterol biosynthesis and elimination are usually tightly coupled in the brain [3, 10–14] and cholesterol biosynthesis is believed to depend directly on the acetyl-CoA levels [89, 90]. Thus, it is conceivable that it is an increase in the acetyl-CoA levels that likely enables a feedback increase in cholesterol biosynthesis rates in mouse models with increased CYP46A1 activity [10, 12–14]. If so, all three unifying mechanisms could be interlinked via acetyl-CoA, thus explaining the CYP46A1 activity effects that cannot be explained only by one mechanism. Also, it is possible the contribution of each of these three mechanisms depends on the biological context and will vary depending on the brain disorder.
This as well as a previous review [25] is an attempt to start building a bigger picture of the CYP46A1 roles in the brain by summarizing the work of different laboratories in the field. Not all the results were mentioned here and perhaps other unifying mechanisms or explanations exist, which are based on the effects of 24HC, a biologically active molecule [53, 97–103]. Nevertheless, this minireview provides a testable concept and arms CYP46A1 researchers as well as neuroscientists with potential general mechanisms that can be further explored and used as a guide for the investigation of the CYP46A1 expression or activity effects and finding the modifying treatments for different brain disorders.
24HC: 24-hydroxycholesterol
AAV: adeno-associated vector
acetyl-CoA: acetyl-coenzyme A
AD: Alzheimer’s disease
Aβ: amyloid β peptides
CYP46A1: cytochrome P450 46A1
HD: Huntington’s disease
LTD: long-term depression
LTP: long-term potentiation
MWM: Morris water maze
NPC: Niemann-Pick disease type C
PD: Parkinson’s disease
PP: pyrophosphate
SCA: spinocerebellar ataxia
The author contributed solely to the work.
The authors declares that she has no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Some of the studies described in this work were supported by the National Institute on Aging grant [AG067552]. The study sponsor did not play a role in the writing of the paper and in the decision to submit the paper for publication.
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Rafael Franco ... Irene Reyes-Resina
Kamran Tariq, Bryan W. Luikart
Jian Xiao ... Jie Luo
Xuntian Jiang, Daniel S. Ory
Ta Yuan Chang ... James G. Gow
Lydia Qian ... Andrew J. Brown
