 Review
Review

Affiliation:
1Department of Medicine, Washington University School of Medicine, St. Louis, MO 63110, USA
ORCID: https://orcid.org/0000-0001-9048-7294
Affiliation:
2Casma Therapeutics, Cambridge, MA 02139, USA
Email: dory@casmatx.com
ORCID: https://orcid.org/0000-0003-2188-2818
Explor Neuroprot Ther. 2021;1:146–158 DOI: https://doi.org/10.37349/ent.2021.00012
Received: September 25, 2021 Accepted: November 05, 2021 Published: December 30, 2021
Academic Editor: Ta-Yuan Chang, Geisel School of Medicine at Dartmouth, USA
The article belongs to the special issue Cholesterol Dyshomeostasis in Neurological Diseases
Niemann-Pick C disease is a rare neurodegenerative, lysosomal storage disease caused by accumulation of unesterified cholesterol. Diagnosis of the disease is often delayed due to its rarity, the heterogeneous presentation, and the early non-specific symptoms. The discovery of disease-specific biomarkers—cholestane-3β,5α,6β-triol (C-triol), trihydroxycholanic acid glycinate (TCG) and N-palmitoyl-O-phosphocholineserine [PPCS, initially referred to as lysosphingomyelin-509 (lysoSM-509)]—has led to development of non-invasive, blood-based diagnostics. Dissemination of these rapid, sensitive, and specific clinical assays has accelerated diagnosis. Moreover, the superior receiver operating characteristic of the TCG bile acid biomarker and its detection in dried blood spots has also facilitated development of a newborn screen for NPC, which is currently being piloted in New York state. The C-triol, TCG and PPCS biomarkers have also been proved useful for monitoring treatment response in peripheral tissues, but are uninformative with respect to treatment efficacy in the central nervous system (CNS). A major gap for the field is the lack of a validated, non-invasive biomarker to monitor the course of disease and CNS response to therapy.
Niemann-Pick C (NPC) disease is a rare neurovisceral lysosomal storage disorder caused by mutations in either the NPC1 gene (95% cases) [1] or the NPC2 gene (approximately 5% cases) [2]. The disorder has a clinical incidence of 1/100,000–1/120,000 live births [3–5]. Analysis of whole exome sequence databases predicted an incidence rate of approximately 1/90,000 in close agreement with the clinical data [4, 6]. These analyses also identified high frequency NPC1 variants with predicted incidence of 1/19,000–1/39,000, suggesting that there may be a significant under ascertainment of late onset phenotypes [6].
The NPC1 and NPC2 genes encode proteins that are central for regulation of cellular cholesterol homeostasis. NPC1 is a large transmembrane protein that resides in the limiting membrane of the lysosome [7], whereas NPC2 is a small soluble lysosomal protein that binds cholesterol within the lysosomal lumen [2]. Biochemical and structural studies support a hydrophobic handoff model in which NPC2 transfers cholesterol to NPC1 for lysosomal egress [8–10]. Recent structural characterization of the NPC1-NPC2 complex has elucidated the molecular basis of the cholesterol handoff and identified a central tunnel for cholesterol transit from the N-terminal domain to the sterol-sensing domain [11]. Deficiency in either NPC1 or NPC2 proteins impairs cholesterol movement from the lysosome. The consequent lysosomal accumulation of unesterified cholesterol, as well as a broad range of sphingolipids, is the hallmark of the NPC cellular defect and is accompanied by dysregulation of cholesterol homeostatic pathways [12, 13]. The link between cholesterol dyshomeostasis and neurodegeneration is not fully understood, but appears to underlie the neuronal dysfunction and pathology that is central to NPC disease [14].
The clinical presentation of NPC is highly heterogeneous and can generally be classified into four categories based on age at onset of neurological manifestations and disease progression: early-infantile, late-infantile, juvenile, and adolescent/adult-onset [15, 16]. More than half of NPC patients present in the neonatal period with visceral signs such as hepatosplenomegaly and cholestatic jaundice. The liver disease typically resolves in the majority of patients. In a small subset, however, liver disease is fatal during the neonatal period. Although patients usually present during childhood with neurological signs, in a subset of patients the disease is primarily systemic and neurological signs may not manifest for years or even decades. Therefore, NPC should be suspected not only in the neonates with cholestatic jaundice and organomegaly, but also in children of any age with isolated splenomegaly or hepatosplenomegaly. For patients that progress to neurological signs, many early signs are non-specific (e.g., delayed developmental milestones and clumsiness), and typically advance to more specific neurological manifestations, such as cerebellar ataxia, vertical supranuclear gaze palsy, cataplexy, and dementia [15]. These manifestations are progressive and patients often succumb to the disease within the first two decades of life.
Diagnosis of NPC is challenging due to the rarity of the disorder, the heterogeneity of disease onset and presentation, its non-specific early symptoms, and the complexity of the laboratory testing [17]. Consequently, diagnosis is typically delayed 4–5 years for the late-infantile and juvenile forms of the disease, during which time opportunities for early intervention are lost [5, 18]. The discovery of blood-based diagnostics and dissemination of rapid, sensitive and specific clinical assays have lowered the barrier for testing and accelerated diagnosis [19–22]. In addition, the availability of molecular diagnostics (e.g., next generation sequencing) and phenotype-specific gene panels has further increased recognition of NPC disease in at-risk patient populations (e.g., neonates with cholestasis) [4, 23–25].
At present drug treatment options for NPC are limited. The only approved drug for treatment of NPC is the glycosphingolipid synthesis inhibitor miglustat (approved in the EU and > 40 countries worldwide but not the US). The relatively modest disease-modifying effects of miglustat have prompted further drug development to address the unmet clinical needs. Two Phase 2b/3 trials in NPC have been completed over the past five years, though to date neither has led to drug approvals. These trials have underscored the significant challenges in developing and gaining regulatory approval for new treatments in a rare, slowly progressive neurodegenerative disease, as well as the urgent need for biomarkers that could serve as surrogate endpoints and accelerate drug approval.
Here, the development and current status of biomarkers for the diagnosis and treatment of NPC disease are reviewed. This is not intended to be an exhaustive review of NPC biomarkers; rather, it is focused on the most widely accepted biomarkers that have the greatest clinical applicability. We discuss the application of these biomarkers to NPC diagnosis, newborn screening, and clinical development of new treatments, specifically highlighting our laboratory’s contribution to NPC biomarker discovery. For detailed discussion of the use of NPC biomarkers in diagnostic algorithms, the reader is referred to recent consensus statements and reviews [4, 17].
Until recently, the standard for diagnosis of NPC disease was filipin staining of unesterified cholesterol in cultured skin fibroblasts obtained from patients. The filipin test identifies approximately 80–85% “classical” NPC patients; however, the test does not provide a clear result in patients with variant phenotypes [17]. The test is also susceptible to false positives among NPC carriers and other rare disorders that similarly store cholesterol—e.g., acid sphingomyelinase deficiency (ASMD), lysosomal acid lipase deficiency (LAL-D), mucolipidosis II/III, MEGDEL syndrome, Smith-Lemli-Opitz syndrome (SLOS), Tangier disease, and a rare congenital disorder of glycosylation due to Nogo-B receptor [17]. Other liabilities of the filipin assay are its invasiveness, high cost, and long turnaround times (> 7 weeks) [17, 22]. For these reasons, filipin is no longer considered a first-line test for NPC diagnosis. However, it may still have utility in situations where biomarkers yield inconclusive results or to assess pathogenicity of variants of unknown significance identified through molecular diagnostics [26].
Over the past decade, the discovery of disease-specific, blood-based biomarkers has transformed the diagnosis of NPC, supplanting filipin as first-line diagnostics. The first generation of these markers were non-enzymatic oxidation products of cholesterol known as oxysterols. We hypothesized that the intersection of lysosomal cholesterol storage and oxidative stress in NPC disease [27–29] would lead to an elevation in circulating oxysterols. Through targeted metabolomic screening of oxysterols first in Npc1-deficient mice and then in the plasma of NPC1 patients, cholestane-3β,5α,6β-triol (C-triol) and 7-ketocholesterol (7-KC) were discovered as disease-specific biomarkers (Figure 1) [20]. This work led to the development of the first rapid, blood-based assay for diagnosis of NPC1 disease [19]. Although both C-triol and 7-KC have excellent receiver operating characteristics (ROCs) with respect to diagnosing NPC1 patients (0.9958 for C-triol and 0.9907 for 7-KC) [19], the high correlation between the plasma oxysterol values (r2 = 0.86), indicating a common pathway for oxysterol generation, and the higher specificity of C-triol have made it the oxysterol biomarker of choice in the clinical laboratory [17, 19, 26, 30]. An early study showed that C-triol retrospectively identified 100% of NPC patients, whereas filipin staining identified only 65% of known NPC1 patients [31]. Clinical application of the C-triol biomarker for diagnosis of NPC1 patients has been demonstrated on different analytic platforms in multiple clinical laboratories [19, 21, 23, 30, 32–42]. The C-triol assay similarly detects NPC2 patients [38, 43]. The C-triol biomarker is also elevated in other rare genetic disorders including ASMD, cerebrotendinous xanthomatosis (CTX), and LAL-D [30, 36, 37, 40, 42, 44, 45], which may be relevant for the differential diagnosis depending upon the age of the patient.
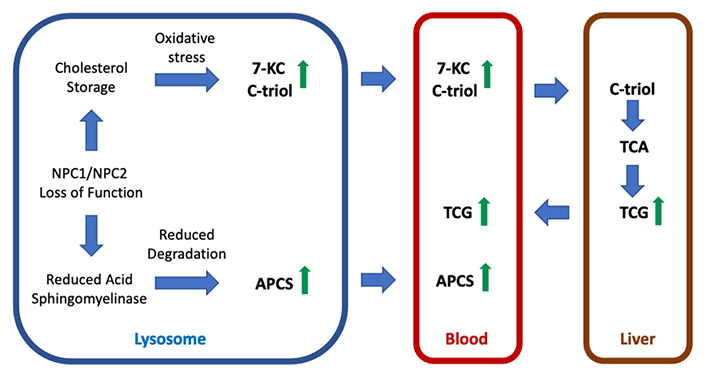
Pathways leading to generation of disease biomarkers in NPC. Either NPC1 or NPC2 loss of function leads to storage of unesterified cholesterol in lysosomes. The consequent oxidative stress promotes non-enzymatic oxidation of cholesterol leading to production of oxysterols (e.g., 7-KC and C-triol) that enter the circulation. C-triol is taken up by the liver and metabolized to trihydroxycholanic acid (TCA) and to its glycinate derivative, TCA glycinate (TCG), which may enter the blood following bile acid secretion and uptake into the enterohepatic circulation. The biosynthetic pathway for N-acyl-O-phosphocholineserines (APCS) species is unknown but catabolism may involve lysosomal acid sphingomyelinase. NPC1 or NPC2 deficiency creates an acid sphingomyelinase-deficient state allowing accumulation of APCS and eventual release into the circulation
To date, the oxysterol assay has been implemented in > 50 clinical laboratories worldwide and widely adopted in NPC diagnostic algorithms [4, 17]. The widespread application of the biomarker has also highlighted some of the assay’s limitations. To achieve sufficient sensitivity for mass spectrometric detection, C-triol requires derivatization that can result in artifactual generation of the oxysterol in situations where cholesterol and 5,6-epoxycholesterol have not been completely removed [40, 46]. C-triol may also show elevation due to autooxidation of cholesterol when the plasma sample is stored at room temperature for more than 24 h [19, 21]. Perhaps the most significant liability of the biomarker is the overlap between NPC1 patients and the upper quartile of NPC1 carriers [19], which has precluded application of oxysterols for purposes of population screening.
The infeasibility of using C-triol or other oxysterol species for newborn screening in NPC disease prompted us to pursue further biomarker discovery. The known perturbations of sterol metabolism, coupled with reports of unusual urinary bile acids in NPC1 disease [47, 48] and the favorable chemical properties of bile acids for ionization and mass spectrometric detection, led us to pursue semi-targeted metabolomic profiling of bile acids in plasma from NPC1 subjects in order to identify a more suitable biomarker [49]. The profiling resulted in discovery of three unknown peaks specifically elevated in the plasma of NPC1 patients. Two of these structures were elucidated and identified as bile acid metabolites 3β,5α,6β-trihydroxycholan-24-oic acid (trihydroxycholanic acid, TCA) and its glycinate derivative, N-(3β,5α,6β-trihydroxy-cholan-24-oyl)glycine (trihydroxycholanic acid glycinate, TCG). The low abundance third peak was not fully elucidated but provisionally assigned as the taurine derivative of TCA. The TCG bile acid was also reported by Clayton and colleagues [50] who identified the biomarker through targeted mass spectrometric profiling of NPC1 patient plasma for presumptive bile acid metabolites of C-triol. These bile acids are unusual in that they have a 3β-hydroxyl-5α-hydroxyl-cholanic acid rather than the 3α-hydroxyl-5β-hydroxyl-cholanic acid configuration typically found in circulating bile acids in human plasma. We further demonstrated that the bile acid biomarkers were derived from C-triol in stable isotopic labeling experiments (Figure 1) [49], thus providing an explanation as to the origin of their unusual A and B steroid ring stereochemistry.
In comparison to oxysterols, the TCA and TCG bile acids exhibit robust ionization, do not require chemical derivatization, and are readily detected and stable at room temperature in plasma and dried blood spots. TCG can be separated from interferences within a total liquid chromatography run time of only 2.2 min. The latter property was crucial to enable development of a fully validated mass spectrometry-based method for detection of the glycine conjugated bile acid biomarker in dried blood spots with throughput of 500 samples per day [49], which is a prerequisite of newborn screening. In contrast to C-triol, the TCG biomarker measured in dried blood spots allowed complete separation between NPC1 carriers and patients, which afforded sensitivity and specificity of 100% (ROC 1.0) and provided the basis for a newborn screen for NPC1. An important limitation of the study, however, was that the dried blood spots were obtained from previously diagnosed NPC1 subjects. To address the key question as to whether the assay would have been able to detect NPC1 subjects at birth, newborn dried blood spots were obtained from 15 NPC1 subjects residing in California, Michigan and New York—states that archive residual dried blood spots in biorepositories—and TCA and TCG bile acids were quantified [51]. Whereas TCG was undetectable in blood spots stored > 13 years, suggesting reduced long-term stability for the bile acid glycinate, it was still able to identify all the NPC1 patients from dried blood spots that have been stored < 10.5 years. The TCA precursor was detected in all the subjects—even those stored > 20 years and discriminated NPC1 patients from carriers and controls completely [51]. These data conclusively demonstrated the feasibility of newborn screening for NPC1 using the bile acid biomarkers.
There is a compelling rationale for newborn screening in NPC disease. Implementation of universal screening has the potential to shift diagnosis to the neonatal period prior to the onset of neurological symptoms. This would not only greatly diminish the diagnostic delay but also allow for early pharmacological intervention that could delay disease progression and extend life. Newborn screening in the US and decisions as to which genetic disorders to include in screening panels are determined at the state level. However, most states adopt Health and Human Services recommendations for disorders included in the Recommended Universal Screening Panel (RUSP). To support a nomination for NPC disease to the RUSP, a requirement is to prospectively obtain data to demonstrate that the disorder can be detected using the screening assay. Working in close partnership with NPC patient advocacy groups (Firefly Fund and Ara Parseghian Medical Research Fund), we partnered with the NIH-funded ScreenPlus program to incorporate the TCG bile acid assay in a panel of 14 rare disorders that are being piloted at the New York State Department of Health [52]. Screening for NPC was initiated in May 2021 with the expectation that it may require up to five years to obtain sufficient experience to support an RUSP nomination.
The strategy of differential liquid chromatography-mass spectrometry profiling of plasma samples from control and NPC1 patients led to discovery of another clinically useful biomarker N-palmitoyl-O-phosphocholineserine [PPCS, initially referred to as lysosphingomyelin-509 (lysoSM-509)] [53]. This biomarker was dramatically elevated in NPC1 plasma and showed even higher levels in ASMD [53–58]. Determination of its structure, however, proved elusive and in the original publication it was misassigned as an isomer of lysosphingomyelin. Following discovery of lysoSM-509, assays were developed for the biomarker using lysosphingomyelin in place of authentic compound for standard curves. Thus, assay accuracy was suspect and accurate reference ranges for control, heterozygotes, and NPC1 patients could not be obtained [53–58]. Nonetheless, application of these assays for relative quantification of the biomarker, despite concerns related to their accuracy, has been useful for discriminating NPC from ASMD [17].
To further explore the potential of lysoSM-509 as an NPC biomarker, accurate mass measurements, tandem mass spectra, and chemical derivatization were used to elucidate its structure. Unexpectedly, it was determined that lysoSM-509 was not a lysosphingomyelin species, as originally proposed by Rolfs and colleagues [53], but PPCS, the most abundant member of an entirely novel lipid class termed APCS [59]. The structure of PPCS was corroborated in a subsequent report by Maekawa and colleagues [60]. All APCS isoforms were dramatically elevated in NPC1 subjects, consistent with the earlier reports. The biosynthetic pathway for generation of the APCS metabolites is poorly understood. It is notable that biomarkers are elevated in both ASMD and NPC subjects [53, 61], the former caused by genetic deficiency of acid sphingomyelinase and the latter a result of a secondary, post-translational defect in the activity of the lysosomal enzyme [62–65]. That these lipids contain a phosphorylcholine moiety implies a possible role for acid sphingomyelinase in the catabolism of these metabolites (Figure 1).
The discovery of the PPCS structure and synthesis of an authentic standard enabled development of a more reliable plasma assay [61]. The plasma PPCS test offers a sensitivity of 100% and specificity of 96.6% for identifying NPC1 subjects from control and NPC1 carriers, with 10% NPC1 carriers above the cutoff for NPC1. Only ASMD overlaps with NPC1 in the PPCS assay [53–56, 61]. PPCS was artifactually produced and elevated 8-fold when plasma, red blood cells, white blood cells, and whole blood were dried on newborn screening cards, suggesting that most PPCS generation might be triggered by unknown substances in cotton fiber card [59]. Taken together, these findings indicate that the PPCS biomarker would appear to have limited utility for population screening.
Lysosphingolipids are sphingolipids lacking an N-acyl chain and have proven to be robust biomarkers for several lysosomal storage disorders, including Fabry disease (lyso-globotriaosylceramide, lyso-Gb3), Gaucher disease (glucosylsphingosine, lyso-GL1) and Krabbe disease (galactosylsphingosine, psychosine). Their accumulation and tissue profile in sphingolipidoses led to the hypothesis that lysosphingolipids may be elevated in the plasma of NPC patients [17]. In two retrospective studies, plasma lysosphingomyelin showed a modest (2.8-fold) elevation in NPC subjects and provided a sensitivity of 100% and specificity of 97–98% to distinguish NPC1 from controls [56, 66]. However, in subsequent studies no increase was seen in patients with NPC and there was a large overlap between NPC and controls [55, 67]. Although plasma lysosphingomyelin has little role in primary diagnosis of NPC, it may have greater utility in ASMD diagnostics, where it has shown to be elevated more than 10-fold and when used in combination with PPCS could discriminate between ASMD and NPC [54, 55, 67, 68]. Simultaneous assay of lysosphingolipids (hexosylsphingosine, lysosphingomyelin, lyso-Gb3) and PPCS was developed to diagnose Fabry disease (lyso-Gb3), Gaucher and Krabbe diseases (hexosylsphingosine), prosaposin deficiency (lyso-Gb3 and hexosylsphingosine), and ASMD and NPC (lysosphingomyelin and PPCS) [55, 56]. Lysosphingomyelin was also used for monitoring treatment with olipudase alfa in patients with ASMD [69].
Lysosomal lipid storage leads to downstream events in the pathogenic cascade, including neuroinflammation, neuroaxonal dystrophy, and neuronal cell death, in particular cerebellar Purkinje cells. Studies of these disease processes using proteomics and genomics have identified numerous plasma and cerebrospinal fluid (CSF) protein biomarkers, including galectin-3 [70, 71], cathepsin D [70], fatty acid binding protein 3 [72], heat shock protein 70 [73], chemokine ligand 3 [74], cathepsin S [75], lysozyme [75], calbindin D [76], and glycoprotein nonmetastatic melanoma protein B [77, 78]. At present none of these biomarkers has a role in primary NPC diagnostics, although some (e.g., calbindin D and fatty acid binding protein 3) have been examined in CSF as potential pharmacodynamic markers for monitoring treatment efficacy [79].
We established in our laboratory fully-validated, FDA-compliant assays for quantification of plasma C-triol, TCG and PPCS, allowing us to directly compare the clinical performance the assays for NPC1 diagnosis in 301 consecutive, unknown patient samples submitted to the Washington University Metabolomics Core (Figure 2) [46]. As anticipated, the C-triol and TCG markers were more highly correlated than TCG and PPCS because C-triol and TCG are in the same metabolic pathway. All NPC1 patients were correctly identified by these biomarkers, but the C-triol and PPCS biomarkers led to more mis-diagnosed non-NPC1 patients. These results indicated that all three biomarkers were equally sensitive in detection of NPC1 patients, but that TCG was more specific [46]. As TCG is a metabolite of C-triol, it may also detect NPC2 patients similar to C-triol. The TCG assay does not require derivatization and is less prone to artefacts than oxysterol measurements; thus, the measurement of TCG is more accurate than C-triol, which may be of particular importance for diagnosing patients with milder phenotypes. TCG is stable in plasma at room temperature and even 37°C for 5 days; therefore, the shipment of plasma sample for TCG assay with dry ice is not necessary and the handling of patient samples in a clinical lab is more forgiving. Based on these results, we expect that TCG will replace C-triol as first-line NPC diagnostic biomarker in the future.
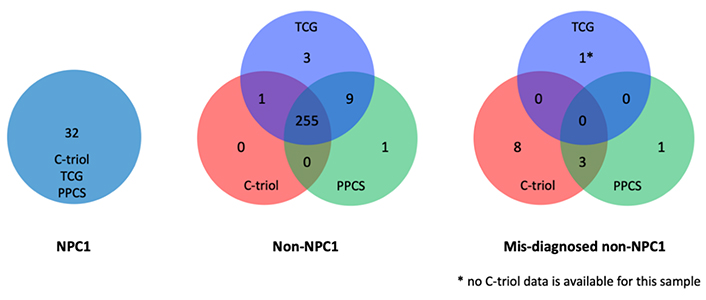
Comparison biomarkers for diagnosis of NPC1 disease. The performance of plasma C-triol, TCG and PPCS assays were directly compared in 301 consecutive, unknown patient samples submitted to the Washington University Metabolomics Core for NPC1 diagnosis. All three biomarkers correctly identified NPC1 patients (left). C-triol and PPCS assays were less specific, mis-identifying 11 and 4 normal subjects, respectively, as NPC1 patients, whereas TCG mis-diagnosed only 1 patient (right). Data reprinted from Sidhu and colleagues [46] with permission of the journal
The slowly progressive nature and clinical heterogeneity in NPC are daunting with respect to development of new treatments. The disappointing regulatory outcomes for the recent Phase 2b/3 trials for arimoclomol (NCT02612129) and for 2-hydroxypropyl-β-cyclodextrin (HPβCD, NCT02534844) underscore the challenges in conducting an interventional clinical trial for this disorder. The insistence by regulatory bodies on a well-controlled trial, the lack of a validated, clinical endpoint accepted by regulators, and the rarity of the disease all contribute to the infeasibility of conducting a trial of shorter than 4–5 years duration. For this reason, it would be highly desirable to use reliable, non-invasive biomarkers to assess response to interventions. Ideally, a panel of biomarkers that correspond to different aspects of the pathogenic cascade or relate to the biological pathway targeted by a therapeutic would be most useful for monitoring the course of disease and response to therapy.
Plasma C-triol, TCG, and PPCS biomarkers have been shown to be useful, but limited, tools for monitoring treatment responses in NPC1 patients. C-triol was significantly reduced in two siblings administered IV HPβCD through an expanded access protocol [80]. Likewise, plasma TCG and PPCS levels were rapidly reduced by IV HPβCD in three expanded access patients [46]. PPCS levels also decreased in a neonate receiving IV HPβCD [81]. However, in the Phase 1/2 trial of intrathecal HPβCD, neither plasma nor CSF levels correlated with the demonstrated clinical benefit in slowing neurodegeneration [79]. This highlights a major caveat for use of these biomarkers, that they reflect almost exclusively metabolism in peripheral tissues and are uninformative with respect to treatment efficacy in the central nervous system (CNS). This is because the production of these metabolites is much lower in brain than liver tissue. As a result, biomarker levels in CSF primarily reflect transcytosis of plasma-derived metabolites down a steep concentration gradient across the blood-brain barrier rather than metabolites that are generated in situ in the brain [61].
By contrast, there is increasing evidence that proteomic rather than metabolomic biomarkers may be better suited to monitoring CNS treatment efficacy. Calbindin D is elevated in the CSF of NPC1 patients and is thought to be a marker for Purkinje cell loss [76]. Significant reduction (33%) was observed in CSF calbindin D in NPC1 patients following initiation of miglustat therapy [76] and CSF calbindin D was also lowered 28% in NPC1 patients treated with intrathecal HPβCD [79]. Similarly, levels of fatty acid-binding protein 3 (FABP3), which is considered as a marker of neuronal inflammation and cell damage, were significantly decreased in miglustat-treated patients relative to untreated patients and in patients receiving intrathecal HPβCD [72, 79]. More recently, neurofilament light chain, a non-specific marker of neurodegeneration, has been reported to be elevated in CSF and plasma of NPC1 patients and may prove useful as a pharmacodynamic marker in future clinical trials.
The discovery of NPC-specific blood-based biomarkers and their rapid dissemination over the past decade into clinical laboratories have transformed the approach to disease diagnosis. The circulating biomarkers most widely used in clinical laboratories—C-triol, TCG and PPCS—all have high sensitivity for identifying NPC disease. However, in contrast to C-triol and PPCS, TCG has significant advantages with respect to sample handling and stability and offers higher specificity for discrimination between carriers and NPC patients. For these reasons, the plasma TCG assay is recommended as the first-line diagnostic. TCG is also the only blood-based biomarker suitable for NPC newborn screening. A pilot screen based on quantification of TCG in dried blood spots is underway, the results of which should provide a more accurate assessment of the true disease incidence and facilitate therapeutic intervention in pre-symptomatic patients. Finally, the drug development landscape for NPC remains challenging. At present, the field lacks a validated, non-invasive biomarker to monitor the course of disease and CNS response to therapy. Thus, there is an urgent need for new biomarkers that could serve as surrogate endpoints to accelerate drug approval.
7-KC: 7-ketocholesterol
APCS: N-acyl-O-phosphocholineserines
ASMD: acid sphingomyelinase deficiency
CNS: central nervous system
CSF: cerebrospinal fluid
C-triol: cholestane-3β,5α,6β-triol
HPβCD: 2-hydroxypropyl-β-cyclodextrin
lyso-Gb3: lyso-globotriaosylceramide
lysoSM-509: lysosphingomyelin-509
NPC: Niemann-Pick C
PPCS: N-palmitoyl-O-phosphocholineserine
RUSP: Recommended Universal Screening Panel
TCA: trihydroxycholanic acid
TCG: trihydroxycholanic acid glycinate
We are grateful to the families and patients who participated in studies that led to discovery and validation of the disease biomarkers.
XJ and DSO conceived of and wrote the manuscript.
XJ and DSO are named as co-inventors on a patent for use of bile acid biomarkers in NPC. DSO is a co-inventor on a patent for use of oxysterols as biomarkers in NPC.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The development of oxysterols, bile acids, and PPCS as NPC biomarkers in authors’ lab was supported by grants from the National Niemann-Pick Disease Foundation (XJ), NIH (R01 NS081985 and U01 HD090845, DSO; UL1 TR000448, XJ), Together Strong NPC Foundation (XJ), the University of Pennsylvania Orphan Disease Center (MDBR-17-124-NPC, XJ), Dana’s Angels Research Trust (DSO), Ara Parseghian Medical Research Foundation (DSO and XJ), and Support of Accelerated Research for NPC Disease (DSO).
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Rafael Franco ... Irene Reyes-Resina
Kamran Tariq, Bryan W. Luikart
Jian Xiao ... Jie Luo
Irina A. Pikuleva
Ta Yuan Chang ... James G. Gow
Lydia Qian ... Andrew J. Brown
