 Review
Review

Affiliation:
1Department of Medical Biotechnology, Bioresources Development Centre, National Biotechnology Research and Development Agency, Ogbomoso 210211, Nigeria
Email: asufem2017@gmail.com
ORCID: https://orcid.org/0000-0003-0071-0531
Affiliation:
2Memorial University of Newfoundland, Newfoundland and Labrador, St John’s, NL A1C5S7, Canada
ORCID: https://orcid.org/0009-0004-7645-5957
Affiliation:
3Department of Physiology, Faculty of Basic Medical Sciences, Redeemer’s University, Ede 232102, Nigeria
ORCID: https://orcid.org/0000-0002-9587-9307
Explor Neuroprot Ther. 2025;5:1004119 DOI: https://doi.org/10.37349/ent.2025.1004119
Received: July 05, 2025 Accepted: October 14, 2025 Published: October 30, 2025
Academic Editor: Mahesh Narayan, University of Texas at El Paso, USA
Parkinson’s disease (PD) is a devastating neurodegenerative condition characterized primarily by the degeneration of the dopaminergic neurons in the substantia nigra, causing motor dysfunction and many non-motor symptoms. Available pharmacological treatments and therapies provide symptomatic relief but do not halt the progression of PD. Gene therapy has been recognized as a valuable therapeutic frontier, providing the possibility of disease modification by targeting the underlying molecular and cellular mechanisms of PD. The parts of the methodology used for gene therapy entail the delivery of genetic material into particular regions of the brain with the aid of viral vectors to improve the synthesis of dopamine, maintain the integrity of neurons, or control pathological pathways. Recent clinical trials have shown promising efficacy and safety profiles for many gene therapy methods, consisting of those targeting enzymes in the biosynthesis of dopamine [e.g., L-amino acid decarboxylase (AADC)], synuclein alpha pathology, and neurotrophic factors [e.g., growth-derived neurotrophic factor (GDNF)]. However, in spite of these developments, there are limitations in vector delivery and prolonged expression of genes, as well as patient-specific responses. This review highlights the present landscape of gene therapy in PD, discussing the latest successes, ongoing clinical trials, and future perspectives that could shape therapeutic paradigms for PD.
Parkinson’s disease (PD) is the second most prevalent neurological condition after dementia [1, 2]. According to an analysis by the Global Burden of Disease Study (2016), which was published in Lancet Neurology two years later, the probability of having PD increases with aging, with rates ranging from 41 to 100,000 in those in their 50s and up to 1,900 per 100,000 in those aged 80 and above [3]. Furthermore, it was reported that a prevalence percentage increase of 54.9% to 66.2% occurred from the year 1990 to 2021 [4]. Within this chronic, severe neurodegenerative condition, which is recognized by a rapid degeneration of dopamine (DA)-associated neurons in the pars compacta of the substantia nigra par compacta (SNpc), an intracellular protein called synuclein alpha (SNCA) is broadly dispersed [5, 6].
PD is a complicated condition that is triggered by genetic, environmental, or age variables. Motor manifestations develop whenever DA within the basal ganglia is insufficient [7]. People with PD often exhibit olfactory impairment, rapid eye movement (REM) sleep behavioral disorder, and autonomic nervous system (ANS) problems such as orthostatic hypotension, bladder malfunction, and constipation, coupled with the usual motor warning signs of rigidity, resting tremor, bradykinesia, and gait abnormalities, as well as lack of postural stability [8].
PD normally progresses until it leaves its patients completely debilitated. The rate of progression and its course vary among patients. The course is relatively benign in some patients with little disability after twenty years, and may be more aggressive among others who may be severely disabled after ten years. Untreated PD worsens over the years, and it may lead to the deterioration of all brain functions and premature death.
According to [9], the breakdown of DA pathways that connect the SNpc with the striatum is thought to be caused by an aberrant buildup of fibrillar SNCA. This leads to a lack of DA and poor motor activity.
The gold standard for treating PD motor symptoms is levodopa (L-DOPA) [10, 11]. In order to increase L-DOPA availability in the brain, carbidopa functions as a decarboxylase inhibitor [12]. PD drugs have a lot of adverse effects (AEs). The most frequent AEs of the combination of L-DOPA and carbidopa are nausea, compulsive gambling, depression, low blood pressure, hallucinations, and irregular sleep patterns.
L-DOPA treatment-induced off symptoms and dyskinesia are still unsolved; according to one study, off symptoms and dyskinesia affect 55.9% and 13.5% of the study group, respectively [13]. It’s unclear how long-term L-DOPA medication causes dyskinesia [14]. It underscores the essential need to investigate novel therapeutic options for the medical management of PD. By suppressing or delaying DA-producing cell death within the brain, disease alteration and neural protection are key therapy options to reduce PD symptoms related to movement [15]. L-DOPA is one of the only short-term PD drugs currently on the market [16].
This review aims to explore the advancing role of gene therapy in PD treatment, with an emphasis on its ability to modify PD progression, tackle underlying molecular and genetic mechanisms, and surmount the challenges of current symptomatic treatments. The objective of this review is to highlight recent advancements, current methods and strategies, and future perspectives, coupled with the evaluation of the limitations, ethical concerns, and clinical outcomes associated with its uses in the management of PD.
The selection of literature was done systematically to identify relevant reviews, studies, and reports relating to the title. Major databases like Web of Science, Scopus, PubMed, and Google Scholar were searched to ensure comprehensive coverage of the existing literature. Other sources, including conference proceedings and institutional repositories, were also examined to minimize publication bias. The strategy used for searching involves the combination of keywords, like gene therapy, PD, preclinical, and clinical trials, which are relevant to the main concepts of the review. In order to optimize retrieval specificity and sensitivity, terms were modified for every database. The primary time frame for inclusion spanned from 2015 to 2025; however, earlier studies were also considered in an appropriate situation. Inclusion criteria were studies published in English, peer-reviewed articles, and literature on gene therapy and PD. Exclusion criteria were duplicate records and non-English publications.
The etiology of PD constitutes a cellular buildup of the SNCA gene in insoluble particles, which damages cells [17]. Depletion of dopaminergic neurons leads to SNCA assembly, disrupting several pathways in the SNpc that rely on the dopaminergic tone. Because of this imbalance, the striatal projections in the neural circuits that contribute to the Parkinsonian phenotype diverge. PD is mostly brought on by a mix of both external and genetic variables. Subsequently, it is considered that PD is multidimensional rather than the outcome of a single, unambiguous cause.
Genetic involvement in the development of PD is currently established by data from several genes and genetic risk factors [18]. Multiple research projects have suggested that genetics is the primary cause of PD risk, and several loci and related markers have been identified based on data from association studies and linkage analyses, respectively [19]. Many different kinds of disease-causing genes were found [18]. The phenotype of the important form of genes is remarkably comparable to that of sporadic PD. Conversely, recessive variants demonstrate a younger age of onset and thus are predictive of pure Parkinsonism in addition to other clinical characteristics.
More than 20 genes have been identified to cause autosomal dominant PD (AD-PD), autosomal recessive PD (AR-PD), and X-linked Parkinsonism, in addition to a variety of symptoms ranging from nonspecific PD-like to early indications of Parkinsonism. These traits may be simple or complex in terms of both non-motor and motor medical aspects [20].
As shown in Figure 1, AD-PD is caused by transmutations in the SNCA and leucine-rich repeat kinase 2 (LRRK2) genes, while AR-PD is caused by transmutations in the PTEN-induced kinase 1 (PINK1), Parkin PBRE 3 ubiquitin protein ligase (PRKN), and Parkinson’s disease protein 7 (DJ-1) genes.
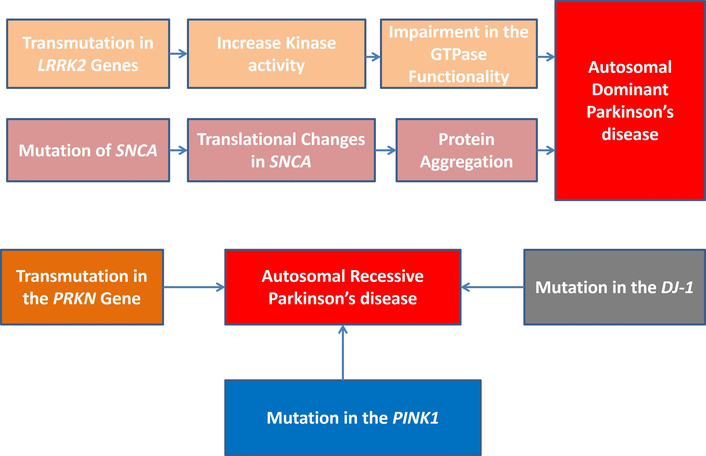
Gene mutations and autosomal dominant and autosomal recessive PD. PD: Parkinson’s disease; LRRK2: leucine-rich repeat kinase 2; SNCA: synuclein alpha; PRKN: Parkin PBRE 3 ubiquitin protein ligase; PINK1: PTEN-induced kinase 1; DJ-1: Parkinson’s disease protein 7.
Pharmacotherapy and functional neurosurgery are the two categories of treatment for PD. There are two forms of pharmacotherapy: dopaminergic and non-dopaminergic methods. Varieties of dopaminergic treatments for PD include monoamine oxidase (MAO) inhibitors, DA receptor agonists, and catecholamine-O-methyl transferase (COMT) inhibitors [11, 14] (Table 1). Patients’ motor fluctuations can be minimized by employing COMT and MAO inhibitors. The mainstay of modern therapy for PD consists of L-DOPA-based medications. L-DOPA-based medications work by replacing the DA that has been depleted in the striatum. Since L-DOPA, the precursor of DA, can cross the blood-brain barrier (BBB), it is employed for PD treatment. Following its passage through the BBB, 3,4-dihydroxyphenylalanine (DOPA) decarboxylase transforms L-DOPA into DA. Serious motor impairments could arise from peripheral, extra-central conversion by the enzyme. MAO-B inhibitors selectively raise DA within synaptic clefts, block catabolism of DA, increase DA signaling, and decrease MAO-B activity in the brain. These drugs have inverse correlations with striatal dopaminergic activity. Dopaminergic activity rises as the MAO-B inhibiting enzyme diminishes. This family of drugs can occasionally be appropriate to ease initial symptoms of PD.
Currently approved Parkinson’s disease treatments.
| Classes/Types | Examples of drugs | Mechanisms of action |
|---|---|---|
| Levodopa-based therapy | Levodopa + carbidopa, levodopa + benserazide | Levodopa is converted to dopamine in the brain; carbidopa/benserazide prevents peripheral disintegration |
| Dopamine agonists | Pramipexole, ropinirole, and apomorphine | They mimic dopamine by direct activation of the dopamine receptor |
| COMT inhibitors | Opicapone, tolcapone, and entacapone | They block COMT, prolonging the levodopa effect |
| MAO-B inhibitor | Rasagiline, selegiline, and safinamide | They inhibit MAO-B, decreasing dopamine breakdown |
| Amantadine | Amantadine | NMDA receptor antagonist; enhances dopamine release and decreases reuptake |
| Anticholinergics | Benztropine and trihexyphenidyl | They block ACh receptors, restoring the balance of dopamine-ACh |
| Surgical therapy | Deep brain stimulation | Electrical stimulation of the basal ganglia |
COMT: catecholamine-O-methyl transferase; MAO-B: monoamine oxidase-B; NMDA: N-methyl-D-aspartate; ACh: acetylcholine.
In PD, anticholinergic medications are also given. They function via non-dopaminergic pathways. Acetylcholine activity is decreased by anticholinergic medications. The main purpose of these medications is to alleviate moderate motor symptoms, particularly muscle stiffness and tremors. Also, amantadine, a non-dopaminergic medication that blocks the N-methyl-D-aspartate (NMDA) receptor, has some therapeutic advantages [21]. It was first developed mainly to treat flu, but it has also been used to treat a number of PD conditions with symptoms that include resting tremors and stiffness in particular. Amantadine is titrated up from a lower beginning dose, just like L-DOPA [22]. This medication causes headaches, sweating, nightmares, and insomnia as side effects. Clinical trials and animal experiments are still being conducted on other non-dopaminergic drugs, such as antagonists of metabotropic glutamate receptors.
Two surgical options for treating PD are subthalamotomy and subthalamic nucleus deep brain stimulation (DBS), as well as pallidotomy and globus pallidus internus DBS [23]. Functional neurosurgery was utilized for DBS and lesions in severe PD [24]. While globus pallidus internus surgery directly reduces dyskinesia, subthalamic nucleus DBS has the benefit of reducing the dosage of dopaminergic drugs [25].
Numerous drugs or therapies have exhibited clear neuroprotection in DA-producing neurons when tested in vitro as well as in vivo; yet, a number of them did not pass clinical trials [26]. Novel therapies have been developed as a result of the previously outlined flaws of the drugs that are already available. Gene therapy has been utilized in clinical investigations for ameliorating a range of disorders in the human brain, and it might provide an option for PD treatment. It is a very innovative technique for treating PD. Gene therapy is highly versatile, and its numerous approaches are being tested for curing PD. A lot of research studies were either finished or are currently in progress.
In order to restore normal cellular function and provide therapeutic benefits, gene therapy attempts to replace or repair a damaged gene by injecting genetic material into specific cells [27]. By protecting and repairing dopaminergic neurons, gene therapy for PD seeks to slow or even reverse the course of the disease. The primary treatment for the motor symptoms of PD has been L-DOPA for about six decades. Despite being effective, this treatment may have a number of AEs, which may only be somewhat controlled by changing the drug’s formulation or taking it with other drugs that increase L-DOPA’s effectiveness and lessen its AEs.
DBS and continuous enteral or subcutaneous dopaminergic infusions are two alternate therapies that have been developed for the relief of symptoms of PD, although their efficacy is limited by their non-physiological mechanisms of action. Furthermore, when neuron loss worsens, these treatments become less effective since they don’t address the underlying neurodegeneration. To considerably enhance people’s well-being, a treatment that slows down the progression of the condition and/or gives a robust, long-lasting functional benefit is still required.
Adeno-associated viruses (AAVs) are small viruses with single-stranded DNA, and they are members of the family Parvoviridae [28]. Despite their small size, they are the most promising gene therapy vehicle due to their ability to generate long-term transgene expression and their clinical safety and effectiveness in transduction of quiescent and proliferative cells [28]. Almost all PD gene therapy clinical trials have employed AAV-based vectors. Effective AAV-based gene therapy requires tissue tropism, biodistribution, and sensitivity to neutralizing antibodies, all of which are largely determined by AAV serotypes [29]. AAV2 has also been used in a number of clinical trials. Due to its sustained expression in neurons and relative safety profile, it is presently considered a viable vector for gene treatment of neurodegenerative diseases like PD.
Targeting SNCA is considered one of the vital approaches in reducing the progression of PD. Furthermore, proteins in the DA synthesis pathway have been an obvious target for gene therapy. A gene therapy approach that enhances DA synthesis is promising for the treatment of PD. Also, targeting of trophic factor genes has been reported to result in the continuous production of trophic factors and increased DA turnover [30].
Gene therapy holds promise for a long-lasting impact that could not only enhance existing dopaminergic treatment but also change the progression of PD. Gene therapy’s main goal is to restore the synaptic function of the nigrostriatal dopaminergic pathway, which could help prevent long-term issues brought on by maladaptive brain remodeling and side effects from stimulation or medication. Figure 2 shows the different methods of gene delivery for PD.
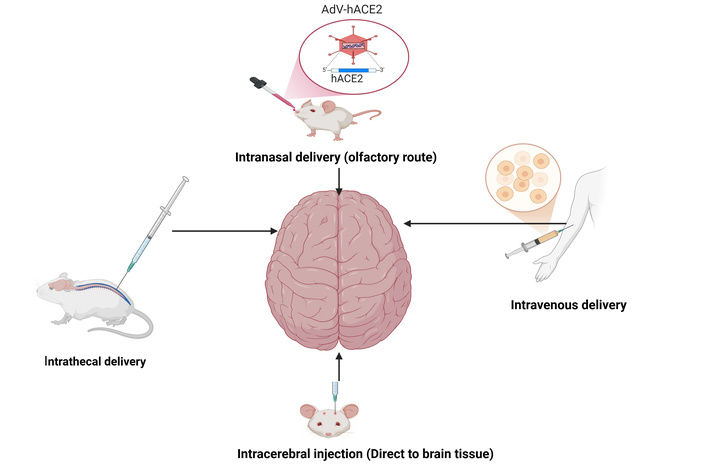
Different gene delivery methods for Parkinson’s disease. AdV-hACE2: adenovirus-transduced human angiotensin converting enzyme 2. Created in BioRender. Ajibare, A. (2025) https://BioRender.com/r1r1qqt.
AAVs have emerged as the most reliable vectors for central nervous system (CNS) gene therapy due to their ability to achieve long-term transgene expression, coupled with their established clinical safety and efficacy in transducing both quiescent and dividing cells. However, their use is constrained by the limited size of the therapeutic payload they can accommodate. Studies of PD gene therapy have primarily used AAV-based vectors.
Multiple genetic therapy techniques are currently being explored to restore the dopaminergic pathways [31]. AAV-L-amino acid decarboxylase (AADC), AAV-growth-derived neurotrophic factor (GDNF), AAV-glutamic acid decarboxylase (GAD), and striatal viral transduction have all been examined in studies in monkey models of PD [32, 33]. In non-human primates (NHPs), these studies demonstrated enhanced motor behavior, persistent gene transfer, robust expression, and regeneration of DA signaling in PD. AAV vector is the most commonly used vehicle in PD due to its biocompatibility, safety, delivery capability, and non-pathogenic characteristics. Although there are currently no active clinical studies or traditional medical treatments for PD, targeting microRNAs (miRNAs) seems to be a viable therapeutic strategy. One promising strategy appears to be the development of PD therapies using miRNAs that target and inhibit the production of SNCA. It has been demonstrated that in mice with sporadically produced SNCA, AAV-induced SN synthesis of short hairpin RNA (shRNA) targeting SNCA reduces neurological impairments [34].
Additionally, the SN of the adult male Lewis rats receives AAV-shRNA targeting SNCA, which lowers SNCA protein levels by roughly three-quarters and shields the animals from the toxicity of mitochondrial rotenone [35], as indicated in Table 2.
Present developments in Parkinson’s disease preclinical gene therapy.
| Animal models | Targeted genes and vectors | Administration process | Safety/Toxicity | Outcome measures | Follow-up and monitoring | Reference(s) |
|---|---|---|---|---|---|---|
| Adult male Sprague Dawley rats | SNCA, AAV-shRNA | Bilateral injection into the SN | Not safe: loss of striatal projecting dopamine neurons was evident in the vector injection site | Reduced behavioral deficiencies in rats and silenced ectopically expressed SNCA | Followed up on [35] | [36] |
| Adult male Lewis rats | SNCA, AAV-shRNA | Unilateral injection into the SN | No neurodegeneration or cell death | Protected the SN against the neurotoxic mitochondrial rotenone and lowered SNCA protein levels by about 35% | [35] | |
| Rhesus monkeys | GDNF, AAV2-GDNF | Bilateral injection of AAV2-GDNF in the putamen after MPTP-lesioning | There was no adverse effect | Bilaterally increased striatal metabolism, indicating increased dopaminergic activity in the nigrostriatal pathway | [37] | |
| MPTP-lesioned macaque monkey | Aromatic AADC, AAV2-hAADC | Unilateral injection into the striatum | Safe, minimal, or no side effects | Positron emission tomography-verified increase in AADC activity after 2 years. Consistently higher L-DOPA sensitivity in the treatment group | Followed up for 8 years | [38, 39] |
| Mice | GBA1, AAV-GFP-micro RNA-GBA | Intracerebroventricular administration | Safe | Slower progression of neurological complications and an increased lifespan | [40] | |
| Mice | GBA1, AAV-PhP.B-GBA1 | Tail vein injection | It did not lead to evident dysfunction in the integrity and permeability of the BBB | Decreased SNCA pathology and attenuated behavioral deficits | [41] | |
| Macaque monkeys (n = 4) | GDNF, AAV5-GDNF | Unilateral administration into the putamen | No adverse effect | At higher dosages, striatal and SN GDNF expression levels are greater | [32] | |
| Unilaterally MPTP-treated macaques (n = 15) | GDNF, AAV2-GDNF | Bilateral delivery to the putamen following a lesion | The persistent high level of GDNF in the basal ganglia after AAV2-GDNF delivery to the putamen is well tolerated | Motor behavior significantly improved at 24 months. Impact was proportionate to the lesion’s severity. A threefold rise in striatal dopamine | [42] |
SNCA: synuclein alpha; AAV: adeno-associated virus; shRNA: short hairpin RNA; GDNF: growth-derived neurotrophic factor; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; AADC: L-amino acid decarboxylase; L-DOPA: levodopa; hAADC: human AADC; GBA1: glucocerebrosidase 1; GFP: green fluorescent protein; SN: substantia nigra; BBB: blood-brain barrier.
The efficacy of delivering the typical glucocerebrosidase 1 (GBA1) gene via an AAV vector to various animal experimental models of PD or PD-GBA has been shown by multiple independent researchers, as shown in Table 2. These studies include various genetic studies involving models with SNCA or GBA1 gene mutations [39, 40]. In the majority of these in vivo investigations, the AAV-GBA1 vector was injected into the mouse’s CNS. In these several PD models, it has been demonstrated that increasing GCas and its functionality via the administration of a GBA1 vector reduces irritation and aggregated SNCA formation. This research indicated improvements in motor behavior, secure transmission of genes, robust expression, and recovery of DA communication [43]. There aren’t any human trials for disease-modifying compounds that target DJ-1 at the moment. Nonetheless, some intriguing preclinical research could result in future clinical uses.
Several investigations using rat PD models have demonstrated the effectiveness of recombinant wild-type DJ-1 in preserving dopaminergic neurons [44, 45]. It was observed that lower 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity was related to better behavior and less dopaminergic dysfunction as revealed in a hemi-Parkinsonian mouse model [46]. Finding medications that prevent the excessive oxidation of a crucial cysteine (Cys) that is still present at position 106 of the DJ-1 protein (Cys106) is a second crucial tactic. There are presently no known investigations focusing on PRKN or PINK1 mutations or the associated metabolic pathways. Nonetheless, some promising preclinical research could result in future clinical uses. As was previously indicated, two essential hallmarks of PRKN, alongside PINK1-related PD, are mitophagy and mitochondrial dysfunction. Addressing the PINK1/Parkin route to restore normal mitophagy thus seems to be the most promising treatment approach [47]. In cell models, it has been shown that the matrix protein nipsnap homolog 1 (NIPSNAP1) affects this mitophagy pathway, indicating that it may restore PINK1-Parkin-dependent mitophagy in PINK1 phenotypes [48]. Reactive oxygen species (ROS) were shown to be elevated, and brain mitophagy decreased in zebrafish deficient in NIPSNAP1 [48].
Gene therapy has shown promising results in the preclinical models of PD in numerous studies, resulting in several clinical trials. Results from the clinical trials may at first seem somewhat disappointing, but they have shown that safe delivery of viral vectors into the brain is feasible.
AAV2-hAADC (human AADC) and lentiviral-GTP cyclohydrolase 1-tyrosine hydroxylase (LV-GCH1-TH)-AADC are important clinical trials that are presently being conducted to evaluate safety, dose escalation, and efficacy in PD patients.
As indicated in Table 3, there were several enhancements in the off-state Unified Parkinson’s Disease Rating Scale (UPDRS)-III using gene therapy strategies that focused on DA restoration (AAV2-AADC and LV-GCH1-TH-AADC) or basal ganglia network neuromodulation (AAV2-GAD). With the gene therapy approach based on increasing the synthesis of neurotrophic factors (AAV2-GDNF), a varying degree of off-state UPDRS-III score reduction or stability was also noted.
Recent results of clinical studies on PD gene therapy.
| Eligibility criteria | Targeted genes and vector | Administration process | Safety/Toxicity/Efficacy | Outcome measures | Follow-up and monitoring | Clinical trials identifier and reference(s) |
|---|---|---|---|---|---|---|
| PD subjects | AADC, AAV2-hAADC | hAADC-expressing adeno-associated serotype 2 viral vectors were delivered by bilateral intra-putaminal infusions | Safe and tolerable, very few persons reported SAEs. Phase I studies | There was a significant improvement in putaminal uptake and AADC activity, and signs of dopamine signaling restoration. There is currently a clinical hold on the phase 2 VY-AADC02 project | NCT03065192/NCT03562494 [49] | |
| PD patients with mild to moderate, and moderate to severe | AADC, LV-GCH1-TH-AADC | Bilateral administration into the putamen | Safe and tolerable, but there were 3 SAEs: dyskinesia, severe psychosis, and an unidentified nervous system disorder. Every dosing cohort has a comparable safety profile. After four and six years, two deaths were reported; these were deemed to be unconnected to the treatment | In all patients, an improvement in motor score was observed. Specifically, a remarkable improvement in mean UPDRS part III motor scores off medication was recorded in all patients at 6 months compared with baseline | 6-month follow-up by Sio Gene Therapies | NCT00627588/NCT01856439 |
| Early PD patients | Small molecular GBA gene | Oral administration of GZ/SAR402671 | Safe and tolerable | The phase I trial was ended since its primary and secondary objectives were not achieved | NCT02906020 | |
| Idiopathic PD patients | CERE-120 (neurturin gene), AAV2-neurturin | Administered bilaterally into the substantia nigra and putamen | It was well tolerated and safe, but also caused AEs | It failed to improve the patients’ conditions | NCT00985517/NCT00400634 | |
| Advanced-stage PD patients | GAD genes, AAV-GAD | Surgical infusion into the subthalamic nucleus | It was safe and well-tolerated | UPDRS showed significant improvements in motor function | NCT00195143 [50–52] |
AADC: L-amino acid decarboxylase; AAV2-hAADC: adeno-associated virus 2-human AADC; PD: Parkinson’s disease; SAEs: serious adverse effects; LV-GCH1-TH-AADC: lentiviral-GTP cyclohydrolase 1-tyrosine hydroxylase-AADC; UPDRS: Unified Parkinson’s Disease Rating Scale; GBA: glucocerebrosidase; GAD: glutamic acid decarboxylase.
In the AAV2-AADC and AAV2-GDNF studies, an increase in AADC activity or DA storage terminal capacity was validated by the use of neuroimaging biomarkers, primarily fecal microbiota transplantation (FMT) and 18F-DOPA positron emission tomography (PET) [53–56]. Following treatment, metabolic alterations by fludeoxyglucose-18 (FDG) PET demonstrated a functional reconfiguration of the pathways connecting the SN with cortical motor areas with AAV2-GAD [49]. Given its relative improvement, neuroimaging may prove to be a crucial goal for gene therapy research; nevertheless, there has been inconsistency and challenge in the correlation between clinical change and neuroimaging.
A plethora of clinical trials have investigated the preliminary efficacy and safety of gene therapy for PD using viral vectors [37, 49, 53–57]. Four of these investigations [37, 53, 54, 56] have reported their outcomes from similar patient cohorts. In this long-term observational extension experiment, individuals who have previously undergone AAV2-hAADC therapy are asked to take part (NCT03733496). Also, one completed research study and one ongoing investigation have used ProSavin, which expresses GCH1, TH, and AADC (LV-GCH1-TH-AADC) [58, 59]. As indicated in Table 4, there is a significant unmet need to find disease-modifying therapeutics for PD, and some active clinical trials are also looking into alternative viral vectors (AAV2) and recombinant genes. A number of PD gene therapy studies have demonstrated relative safety in a variety of clinical trials intended to improve the production of trophic factors (NCT04167540) or restore DA synthesis [37, 49, 53–57].
Ongoing clinical trials on gene therapy for PD.
| Eligibility criteria | Targeted genes and vectors | Administration process | Safety/Toxicity/Efficacy | Outcome measures | Clinical trials identifier and reference |
|---|---|---|---|---|---|
| Phase 1/2a clinical trial of PR001 (LY3884961) in patients with PD with at least one GBA1 mutation | Small molecular GBA gene, AAV9-GBA1 | Intra-cisterna infusion followed by methylprednisolone, sirolimus optional | Modified owing to SAEs in the first enrolled participant to receive active treatment | It is anticipated to be finished by 2029, but no data is provided | NCT04127578 [60] |
| Mild to moderate, and moderate to severe PD patients | GDNF, AAV2-GDNF | Intra-putaminal injection | A robust safety profile | Restoration of dopamine function. To be completed June 2026 | NCT04167540 |
PD: Parkinson’s disease; GBA: glucocerebrosidase; AAV2-GDNF: adeno-associated virus 2-growth-derived neurotrophic factor; SAEs: serious adverse effects.
Unfortunately, some of the clinical trials on gene therapy for PD have been discontinued due to the inability to achieve the set objectives and the inability to produce the desired patient outcomes. The efficacy of gene therapy for PD can be affected by various factors, with the route and mode of administration being one of the most important. Intraparenchymal administration has been used in many PD trials to deliver the gene vector directly to the target area of the brain [37, 49, 53–57]. This approach significantly lowers the vector dose and the probability of off-target diffusion into peripheral tissues, both of which lower the potential immunogenicity [61]. Intracerebroventricular, intrathecal, subpial, and intracisternal infusions can also be employed when a broader delivery inside the CNS is required. These methods are limited by a more heterogeneous distribution, higher dose requirements, and lower target tissue levels of vector transduction.
Few studies look at the long-term safety profile, and no firm conclusions can be made regarding the long-term therapeutic efficacy, even if the follow-up period in PD gene therapy is sufficient to show the short-term safety profile for all examined medicines. Given that neurotrophic factors are anticipated to exhibit their neuroprotective effect over an extended period of time, this is extremely important.
Investigating the “disease-modifying” effect of gene therapy in PD was hampered by the preferred inclusion of advanced PD patients and the lack of cerebrospinal fluid biomarkers to track the course of PD. Current research is expected to yield more information by including a population of early PD patients, even though the absence of broadly accepted biomarkers of disease progression remains a barrier to evaluating the long-term expected impact of gene therapy in PD. Lastly, the ability to do a meta-analysis and a direct comparison between the various therapy options is limited by the variety of study designs.
The most important developments in gene therapy are expected to come from advancements in molecular techniques for modifying gene expression and target specificity, as well as growing interest in a precision-medicine approach. These developments will likely be crucial for future clinical trials.
By delivering different transgenes, viral vectors like AAV have become a powerful tool for neuroscience since they allow for neural tracing and functional probing. A transgene, regulatory elements like enhancers or promoters that limit expression to particular cells or tissue types, and a capsid, an outer protein shell that encloses the genetic material and controls the vector’s tropism, or capacity to infect various cell types, make up viral vectors [62]. In the end, BBB-crossing AAVs’ enhanced effectiveness might be paired with other technologies to produce localized expression. However, a significant obstacle to effective gene delivery is getting beyond the BBB, which serves as a gatekeeper by keeping toxins and pathogens in the systemic circulation from accessing the CNS.
There are currently 13 different wild-type, or naturally occurring, AAV serotypes (AAV1–13) that have been found in humans and NHPs [63]. The capsid structure and, consequently, tropism of each of these serotypes vary [64]. AAV5 and AAV9 are the serotypes most frequently found in NHPs. Because AAV9 can pass the BBB, it has been extensively researched and used in a number of CNS-targeted gene treatments [65]. An AAV’s capsid is its main point of contact with host cell surface receptors, allowing the virus to be internalized and eventually transport its genetic material to the cell nucleus [66].
Since multiple permissive locations for logical and random amino acid changes and insertions have been identified, one method of changing an AAV’s tropism and efficacy is by capsid modification or engineering [66]. We can find new AAV serotypes with better BBB crossing characteristics and improve and hone AAV tropisms through capsid engineering.
Capsid engineering can be accomplished by controlled evolution or logical design. In order to systematically forecast and improve virus function, rational design makes use of the information of current AAV serotypes [67]. Conversely, directed evolution is a high-throughput method that creates variations with the desired characteristics, like tissue and cell-type tropism and/or antibody neutralization, through a selection process. Researchers have found novel capsid variations with a greater transduction effectiveness at lower titers or concentrations by repeatedly generating a large number of non-naturally occurring viral serotypes and choosing those with the necessary tropism for the subsequent evaluation round [68, 69]. The development of the AAV capsid has been significantly impacted by technological advancements. Viral vector-based gene therapy has upped the bar for what can be accomplished, not least in the CNS.
Genome editing and engineered AAVs are ideal for new clinical treatments. We anticipate that several of the capsids shown here will be moving to the clinic for a variety of diseases, including PD. Although there is still a long way to go, gene therapy has a bright future ahead of it, and many other capsids with much more promise will emerge.
Since the toxic species of SNCA have not yet been identified, the therapeutic benefit of these approaches may be limited. Currently, a number of therapeutic strategies are being researched, such as antibodies and small-molecule approaches that target various forms and conformational states of SNCA [70]. Furthermore, tiny compounds and antibodies frequently exclusively target the protein’s extracellular pools, which may make these strategies less effective. The limitations of these methods may be addressed by antisense oligonucleotide (ASO) therapy, which targets SNCA RNA intracellularly to block SNCA formation and reduce SNCA protein in all its forms [71].
ASO-mediated suppression of SNCA in a primary cortical culture preformed fibril system decreased SNCA mRNA and reversed phospho-Ser129 (pSer129+) pathology and cellular dysfunction [72]. Cole et al. [73] show that ASO-mediated suppression of SNCA prevented and reversed the progression of alpha-mediated pathology in rodent transmission models of PD, indicating the potential of ASOs as a therapy for PD patients. Because ASOs are sequence-specific and can reach CNS targets by i.t. delivery, ASOs represent a therapeutic approach for directly lowering SNCA production. The ASO platform has become a therapeutic strategy for the treatment of CNS diseases, in part because of advancements in ASO design, which have improved stability, affinity, potency, and tolerability [71, 74].
In addition to having a long-lasting effect [75], ASOs have also been demonstrated to disperse extensively throughout the brain and spinal cord in the NHP [76, 77]. The ongoing studies for an LRRK2-targeted ASO for PD (NCT03976349) and an SNCA-targeted ASO for synucleinopathy (NCT04165486) show the approach’s viability.
NBIb-1817, also known as VY-AADC, is an investigational recombinant AAV serotype 2 vector encoding the gene for hAADC, which is intended to help brain cells produce the AADC enzyme that converts L-DOPA to DA. In early 2019, Voyager Therapeutics and Neurocrine Biosciences entered into a strategic collaboration in an effort to combine Neurocrine Biosciences’ expertise in neuroscience, drug development, and commercialization with the creative gene therapy programs targeting severe neurological diseases established by Voyager [78]. Targeted delivery aided by magnetic resonance imaging (MRI) is used in conjunction with intraoperative monitoring.
The Food and Drug Administration (FDA) informed Neurocrine in December 2020 that it was putting a clinical hold on the phase 2 clinical trial of NBIb-1817, called RESTORE-1 (NCT03562494), after the data safety and monitoring board (DSMB) requested that dosing be stopped until it received information about MRI abnormalities seen in trial participants. The agency gave Neurocrine information so that it could respond fully to the FDA regarding the hold, including an assessment of any potential link between the agent and the negative findings, a plan to mitigate and manage those findings, and supporting documentation to demonstrate that there is a favorable benefit/risk profile. Before this, the medication seemed to be headed toward success in patients with advanced PD. According to 3-year data released in September 2020 from two cohorts, VY-AADC01 treatment led to stable or better motor function, quality of life, and a significant reduction in the demand for antiparkinsonian drugs. When compared to a second phase 1 open-label evaluation, the gene therapy and related administration method were generally well-tolerated within the time frame and seemed to support further clinical development in this population [79]. Three cohorts of fifteen patients received bilateral intraoperative MRI-guided putaminal infusions of the drug. A dose of ≤ 7.5 × 1011 vg was given to Cohort 1, ≤ 1.5 × 1012 vg to Cohort 2, and ≤ 4.7 × 1012 vg to Cohort 3. There were no documented severe AEs associated with VY-AADC01. One person experienced atrial fibrillation and pulmonary embolism, while another experienced two small intestinal obstructions six days apart, 29 months after the treatment. These four nondrug-related significant adverse events were all eventually resolved. Headache (Cohort 1: n = 5; Cohort 2: n = 3; Cohort 3: n = 3) and hypoesthesia (Cohort 1: n = 3; Cohort 2: n = 1; Cohort 3: n = 3) were the most frequently reported adverse events.
Through the use of clustered regularly interspaced short palindromic repeats (CRISPR) screening, genes or genetic sequences that control particular morphological or physiological impacts can be found. Understanding the genes linked to PD processes and focusing future therapy on them depends on CRISPR editing.
It has long been known by scientists that certain neural cells are more vulnerable to neurodegeneration and aging-related damage. PD and other neurological disorders, including Alzheimer’s, have been linked to this phenomenon, which is sometimes referred to as selective neuronal vulnerability. Conventionally, gene expression investigations (transcriptome and proteome) have demonstrated the distinctions between vulnerable versus resilient neurons. To target pertinent genes therapeutically and comprehend the molecular pathways at a functional level, genetic editing is necessary.
Gene function in neural cell models may be efficiently studied using CRISPR. The Hoffman lab at the University of Pittsburgh in Georgia provides a relevant example of one such study, which created nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX1, NOX2, and NOX4) knockout cell models for PD by utilizing Synthego’s genome editing capabilities [80]. The study showed how the NOX2 enzyme contributes to oxidative stress-induced degeneration, which includes LRRK2 activation, SNCA buildup, and impaired protein import in mitochondria. Additionally, researchers created a rhesus macaque model that replicated the human Parkinson’s phenotype by using CRISPR-Cas9-mediated PINK1 deletion. Interestingly, scientists found that the SN region of the affected animals’ midbrain had lost neurons, which is a common finding in Parkinson’s patients. Different levels of PINK1 deletion were made possible by CRISPR-Cas9 mosaicism, which helped scientists comprehend the intricacy of the related phenotypes [81].
There are still certain problems despite the promising preclinical and early clinical studies for gene therapy in PD. Accurately targeting and delivering genetic treatments to the impacted regions, such as the SN and striatum, is one of the most pressing issues, as when compared to backward transfer, anterograde delivery of GDNF to the SN is less successful than surgical repair [82]. Neuronal recovery has also been demonstrated by NRTN [83] and GDNF [84] following striatal injection of a rat 6-hydroxydopamine (6-OHDA) model of PD. Furthermore, the BBB and the diverse nature of PD pathophysiology present significant challenges to effective gene delivery [85]. AAV vectors and gene delivery techniques need to be further improved in order to increase their specificity and efficacy. Also, there has been great development in gene editing techniques like CRISPR-Cas9, and an increasing understanding of genes responsible for PD is the key driver of the development of more effective, personalized, and precise therapies. There have been reports of potential efficacy and safety in early clinical trials. Despite encouraging initial findings, it is important to carefully assess the long-term implications of gene therapy on the progression of PD and off-target effects. Other significant safety issues that need to be addressed are the immunological response to AAV vectors and the possibility of insertional mutagenesis. Despite these obstacles, gene therapy for PD treatment is advancing quickly thanks to finished and ongoing research.
Patients with PD can still benefit from gene therapy, which has the potential to significantly improve their quality of life. Despite the lack of approval, encouraging outcomes from completed and continuing clinical trials guide the development of gene therapy. For PD gene therapy trials to be successful, patient selection must take into account the mechanism of action of a specific vector design as well as its broad distribution in the targeted brain region. Longer observation periods and early participation are anticipated to be necessary to assess the growth factor-based gene therapy capacity to alter the course of the disease. Moreover, there is a need for combination strategies like the applications of both gene therapy and neuroprotective drugs, the development of efficient CNS-delivery vectors, better patient stratification (prodromal PD, genetic subtypes), and extensive testing to achieve adequate safety for the treatment of PD.
AADC: L-amino acid decarboxylase
AAVs: adeno-associated viruses
AD-PD: autosomal dominant Parkinson’s disease
AE: adverse effects
AR-PD: autosomal recessive Parkinson’s disease
ASO: antisense oligonucleotide
BBB: blood-brain barrier
CNS: central nervous system
COMT: catecholamine-O-methyl transferase
CRISPR: clustered regularly interspaced short palindromic repeats
Cys: cysteine
DA: dopamine
DBS: deep brain stimulation
DJ-1: Parkinson’s disease protein 7
DOPA: 3,4-dihydroxyphenylalanine
FDA: Food and Drug Administration
GAD: glutamic acid decarboxylase
GBA1: glucocerebrosidase 1
GDNF: growth-derived neurotrophic factor
hAADC: human L-amino acid decarboxylase
L-DOPA: levodopa
LRRK2: leucine-rich repeat kinase 2
LV-GCH1-TH-AADC: lentiviral-GTP cyclohydrolase 1-tyrosine hydroxylase L-amino acid decarboxylase
MAO: monoamine oxidase
miRNAs: microRNAs
MRI: magnetic resonance imaging
NHPs: non-human primates
NIPSNAP1: nipsnap homolog 1
PD: Parkinson’s disease
PET: positron emission tomography
PINK1: PTEN-induced kinase 1
PRKN: Parkin PBRE 3 ubiquitin protein ligase
shRNA: short hairpin RNA
SNCA: synuclein alpha
SNpc: substantia nigra par compacta
UPDRS: Unified Parkinson’s Disease Rating Scale
AOA: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. GOO: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. GAF: Validation, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

