 Review
Review

Affiliation:
1Lahore Grammar School, Lahore 54792, Pakistan
ORCID: https://orcid.org/0009-0005-2295-0810
Affiliation:
2Stanford Neurosciences Interdepartmental Program, Stanford University School of Medicine, Stanford, CA 94305, USA
Email: jungyoj@stanford.edu
ORCID: https://orcid.org/0000-0002-9015-5529
Explor Neuroprot Ther. 2023;3:281–298 DOI: https://doi.org/10.37349/ent.2023.00052
Received: May 08, 2023 Accepted: August 01, 2023 Published: October 26, 2023
Academic Editor: Mahesh Narayan, University of Texas at El Paso, USA
Post-translational modifications (PTMs) of alpha-synuclein (α-syn) can alter protein aggregation propensity to affect α-syn oligomer and fibril formation. The inflammatory response in Parkinson’s disease (PD) is mediated by microglia, astrocytes, T cells, B cells, macrophages, and neutrophils, which respond to α-syn aggregates in an attempt to clear synucleinopathy and restore brain homeostasis. This review focuses on the effects of PTMs on α-syn aggregation and cell-specific immune responses to α-syn aggregates in the context of PD.
Parkinson’s disease (PD) is a neurodegenerative disease mainly characterized by the death of dopaminergic neurons in the substantia nigra pars compacta, which results in motor symptoms such as tremors, bradykinesia, and rigidity, as well as non-motor symptoms such as constipation, hypotension, anxiety, and sleep disorders. In PD, a neuronal protein called alpha-synuclein (α-syn) aggregates in neurons to form Lewy bodies, though it is generally expressed in the presynaptic membrane at the axon terminal [1, 2].
In healthy conditions, α-syn plays a role in lipid packaging and transport, vesicle trafficking, dopamine synthesis and secretion, and neurotransmitter release, though the entirety of its functions is not yet understood. Homeostatic α-syn exists in its monomeric form and does not form aggregates. The misfolding of α-syn leads to oligomer and fibril formation, which ultimately leads to α-syn aggregation. The propensity of α-syn to aggregate can change as a result of genetic modifications; for example, the A53T mutation in the SNCA gene increases α-syn aggregation. α-syn aggregation is also influenced by post-translational modifications (PTMs)—the effects of which will be expanded upon in this review [1, 3].
α-syn aggregates have been shown to induce neuroinflammation, which is another central feature of PD pathology. While inflammation is a protective mechanism meant to regenerate and repair tissue or to remove damaged cells, toxins, and pathogens from the body [4], in PD, chronic inflammation in response to neurodegeneration leads to the progression of the disease [5, 6]. It is mediated by microglia, astrocytes, T cells, B cells, macrophages, and neutrophils, which interact with each other to generate an immune response to α-syn [7]. This review describes how various forms of α-syn activate immune cells, which work synergistically to generate an inflammatory response in PD patients.
PTMs are changes to a protein which occur after amino acid translation and synthesis of the primary protein structure. PTMs are divided into subtypes based on the functional groups that are added to the protein, with common functional groups including polypeptide chains, amino acids, or complex molecules [8]. The propensity of α-syn to aggregate is affected by the category and site of modification [8, 9].
Phosphorylation is a common PTM, characterized by the addition of a phosphate group to an amino acid in a polypeptide chain, and is highly implicated in α-syn aggregation. Phosphorylation at Tyr125, Tyr133, Tyr136, and Ser87 has been shown to reduce the aggregation of α-syn, while phosphorylation at Tyr39 has been shown to enhance α-syn aggregation [9]. Interestingly, Tyr125 phosphorylation has been shown to decrease with aging in humans [10]. Phosphorylation at Ser87 and Ser129 are both upregulated in PD brains [11], but phospho-Ser87 (pSer87) is found in significantly lower amounts than α-syn phosphorylated at Ser129 [12] which is consistently upregulated in PD patients—with 90% of α-syn containing phospho-Ser129 (pSer129) in PD patient brains compared to less than 5% in non-patient brains. Studies have failed to reach a consensus on its consequences, as there is conflicting evidence for Ser129 phosphorylation to both reduce and enhance the propensity for α-syn to aggregate [9, 13]. The mechanisms underlying these effects are yet to be understood.
Another common PTM is ubiquitination, which plays a role in protein degradation. Ubiquitinated α-syn is primarily found on phosphorylated α-syn in Lewy bodies [11]. This pathway has three steps in its catalysis and the last step which involves E3 ubiquitin ligases, also has varied results in whether ubiquitination leads to an increase or decrease in aggregation. One ubiquitin ligase for α-syn, neuronally-expressed developmentally down-regulated gene 4 (NEDD4), reduces aggregation of α-syn, promoting its degradation [14]. The involvement of NEDD4 extends to its interactions with p21-activated kinase 4 (PAK4) where NEDD4 presence is necessary for upregulation of PAK4 to reduce α-syn aggregation [15]. Carboxyl terminus of Hsp70-interacting protein (CHIP) is another E3 ligase which reduces α-syn oligomerization [16–18]. It further reduces α-syn aggregation through its interactions with ubiquitin A-52 residue ribosomal protein fusion product 1 (UBA52) [19]. On the other hand, some E3 ligases like seven in absentia homolog (SIAH) can increase α-syn aggregation [20, 21]. Overall, the effect of ubiquitination on α-syn aggregation varies.
Similar to ubiquitination, truncation, the shortening of a polypeptide chain, does not have a consistent effect on α-syn aggregation. In α-syn, it has been shown that truncation at the C-terminal promotes aggregation in most cases [8, 9], with amino acids 1–87, 1–97, 1–102, 1–103, 1–110, 1–114, 1–119, 1–120, and 1–122 making up the most common occurrences of truncated polypeptide chains in aggregated α-syn proteins [9]. In Lewy bodies, α-syn truncated at amino acids 115, 119, or 122 were found to be present. However, α-syn truncated at Asp119 is also found outside Lewy bodies, indicating that it is a normal metabolite of α-syn [11]. Although C-terminally truncated α-syn itself is more prone to aggregation, some studies also found that it can reduce aggregation by preventing the fibrillization of full-length α-syn [22]. N-terminally truncated α-syn exhibits reduced aggregation, showing that the section of α-syn which causes aggregation is found near the N-terminal [8, 22].
Acetylation, the binding of an acetyl group onto amino acid residues [8], catalyzed by N-acetyltransferase enzymatic complexes [23], is also a PTM which reduces aggregation and is found in Lewy bodies [11]. Acetylation of the N-terminal, which most commonly occurs at Lys6 and Lys10 [8], does not change the properties of monomeric α-syn, retaining its ability to bind with lipid membranes and its solubility [24, 25]. The presence of lipids has been shown to induce the aggregation of α-syn [26, 27]. Acetylation reduces α-syn aggregation and reduces the rate of fibril elongation in the presence of lipids [18] but does cause the elongation of fibrils in other conditions [28]. However, acetylation increases the formation of toxic α-syn oligomers [25, 27, 28]. Furthermore, although the α-syn fibrils formed from acetylated monomers modified the secondary structure of the α-syn fibrils, it did not affect their stability or their cytotoxicity [25, 28].
Glycosylation, or more specifically O-linked β-N-acetylglucosaminylation in the case of α-syn, most commonly affects Ser and Thr residues [9]. Glycosylation does not affect the function of monomeric α-syn, similarly to acetylation, and inhibits α-syn aggregation [29–32]. When mixed with unmodified α-syn in a one-to-one ratio, glycosylated α-syn slowed the formation of fibrils the most, also preventing the elongation of fibrils which were already present. This reduction in fibril formation causes an increase in formation of toxic oligomers at Thr81 and Ser87 [29] but not at Thr72 [31]. Though glycosylation is associated more with Alzheimer’s disease than PD, its role in preventing aggregation of α-syn is being investigated as a possible treatment for Lewy body disorders [31].
Nitration is the binding of a nitro group onto a polypeptide chain and is another PTM which is upregulated in PD through the upregulation of inducible nitric oxide synthase [33]. In α-syn, Tyr39, Tyr125, Tyr133, and Tyr136 can all be subject to nitration [23]. The effect of nitration on α-syn aggregation, rather than being dependent on modification site, is dependent on factors such as protein concentration and molecular weight. For example, nitrated α-syn is unable to aggregate on its own, which may be why oligomers of high molecular mass inhibit aggregation. However, when present in low concentrations, nitrated monomers and dimers are incorporated into fibrils of unmodified α-syn and increase the rate of fibril formation [34].
The covalent attachment of a small ubiquitin-like modifier (SUMO) onto a Lys residue is called SUMOylation and is also upregulated in PD patient brains [35]. SUMOylation occurs mainly on Lys96 and Lys102 in wildtype α-syn [23] but can affect many other Lys residues of α-syn as well in diseased conditions. While SUMOylation has been reported to promote the formation of α-syn aggregates through prevention of ubiquitin linkage by competing for Lys residues and by increasing α-syn hydrophobicity [35, 36], SUMOylation has also been shown to reduce aggregation in some circumstances, with Lys96 and Lys102 SUMOylation increasing α-syn solubility to limit protein fibrillation [37, 38]. A summary of the amino acids in the α-syn protein which commonly undergo the PTMs discussed above is provided as Figure 1.
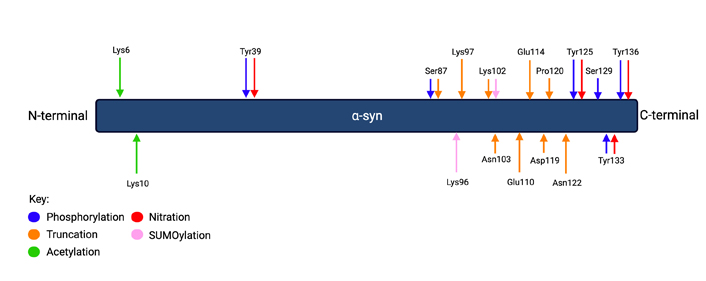
Common PTMs of α-syn. The key shows which PTM corresponds with the color-coded arrows which show the location of the labeled amino acids. The double arrows of different colors with the same label show where two PTMs occur on the same amino acid. Phosphorylation occurs at Tyr39, Tyr125, Tyr133, Tyr136, and Ser87. Truncation occurs at Ser87, Lys97, Lys102, Asn103, Glu110, Glu114, Asp119, Pro120, and Asn122. Acetylation occurs at Lys6 and Lys10. Nitration occurs at Tyr39, Tyr125, Tyr133, and Tyr136. SUMOylation occurs at Lys96 and Lys102. Figure created with Biorender.com
Microglia are resident immune cells in the central nervous system (CNS) which exist in a homeostatic state, patrolling for pathogens, dying neurons, and inflammatory signals under healthy conditions. When exposed to acute danger signals, they are activated and produce pro-inflammatory cytokines. Activated microglia can become overwhelmed under chronic pathological states, contributing to disease progression [39].
Microglial activation can be triggered by monomeric, oligomeric, and fibrillar α-syn binding to cell membrane receptors, with misfolded, oligomeric, and fibrillar α-syn yielding greater pro-inflammatory responses than monomeric forms [7, 40]. The extent of microglial activation and its downstream functions vary based on α-syn form. For instance, monomeric α-syn does not activate microglia but does promote microglia-mediated phagocytosis [41].
Toll-like receptors (TLRs) are transmembrane receptors found in innate immune cells (as well as some non-immune cells) that bind to extracellular ligands and recognize pathogen-associated molecular patterns (PAMPs) [42]. TLRs are sensitive to protein confirmation, with some TLRs binding to some, but not all conformations of α-syn. For example, TLR2 cannot be activated by fibrillar α-syn but can be activated by α-syn monomers and oligomers. TLR2 knockout studies have shown that TLR2 is necessary for the activation of the nucleotide-binding oligomerization domain-like receptor pyrin domain containing 3 (NLRP3) inflammasome [43], which leads to increased cytokine production, impaired clearance of intracellular α-syn, and can exacerbate neurodegeneration [43–47]. The role of TLR2 in synucleinopathy is further emphasized by its upregulation in PD-affected regions of the brain and its correlation with α-syn levels, as well as its involvement in Lewy body formation [44].
TLR4 is more sensitive to α-syn oligomers than TLR2 at low oligomer concentrations, which suggests that it may be the more significant receptor in physiological conditions. It has a role in the response of TLR2 as activation of TLR4 causes an upregulation of TLR2. TLR4 has also been found to be necessary for oligomer-induced cytotoxicity in microglia [48–50]. TLR4 is upregulated in microglia in response to neuronal α-syn, which is released and engulfed by microglia. This leads to downstream activation of the nuclear factor-kappaB (NF-κB) pathway and upregulation of autophagy receptor p62, ultimately allowing for microglial autophagy of α-syn released from neurons [51]. TLR4 is also necessary for astrogliosis, which contributes to a neuroinflammatory environment [49, 52].
Cell membrane receptors are responsible for the internalization of α-syn as well. Lymphocyte activation gene 3 (LAG3) is a part of the immunoglobin superfamily and binds preferentially with α-syn fibrils and the C-terminal of α-syn. LAG3 binding of α-syn, which is strengthened by PD-related PTMs such as pSer129, can lead to α-syn internalization and seeding. LAG3 depletion has been shown to reduce α-syn aggregation and pSer129 and subsequent glial activation in human α-syn A53T transgenic (hA53T) mice [53, 54]. However, the pathological role of LAG3 is not completely supported as other studies have also shown that overexpression or knockout of LAG3 showed no effect on the uptake of α-syn in murine cells [53], suggesting that LAG3 does not affect α-syn-related inflammation [55].
Another cell membrane receptor implicated in synucleinopathy is metabotropic Glu receptor 5 (mGluR5), a G-protein coupled receptor usually found in neurons but also present in microglia. Activation of mGluR5 has been shown to significantly inhibit the microglial inflammatory response through limiting inflammatory signaling caused by α-syn. mGluR5 activation reduced cytokine secretion and inhibited neurotoxicity. However, only N-terminally truncated α-syn binds with mGluR5 [56], and its expression does not change with the progression of PD [57].
N-methyl-d-aspartate receptors (NMDARs) are membrane receptors which respond to Glu to allow the influx of calcium ions (Ca2+). Ca2+-mediated events are important for both neurotransmission and neural cell function but can induce excitotoxicity in excess [58]. Although α-syn oligomers do not directly activate NMDARs, α-syn oligomers induce the release of Glu from astrocytes, which promotes NMDAR activation. Activation of NMDARs has been shown to lead to synaptic loss, highlighting the potential influence of α-syn oligomers on neuronal excitotoxicity and synaptic integrity [59].
Triggering receptor expressed on myeloid cells 2 (TREM2) is a transmembrane receptor found on the surface of microglia and other myeloid cells. TREM2 binds to extracellular ligands to trigger downstream signals that regulate inflammation, microglial activation, phagocytosis, and cell survival [60]. TREM2 has been shown to reduce inflammation and neurodegeneration in PD through negative regulation of the mitogen-activated protein kinase (MAPK) and NF-κB signaling pathways, which are upregulated with TLR4 signaling [61]. TREM2 inhibition aggravates neurodegeneration and the inflammatory responses of microglia and astrocytes, while TREM2 overexpression causes a downregulation in the expression of inflammatory TLRs [60, 62].
The receptor for advanced glycation end products (RAGE) is a pattern recognition receptor (PRR) of microglia which, like LAG3, is part of the immunoglobin superfamily. RAGE has been found to bind the C-terminal of α-syn with high affinity, more so with fibrillar α-syn than monomeric α-syn. Although RAGE has a role in the α-syn fibril-induced activation of microglia, its absence does not completely block microglial activation [63].
Proinflammatory microglia can release proinflammatory cytokines to activate astrocytes and induce a secondary inflammatory response. These astrocytes are neural cells that aid in the inflammatory response and help remove and degrade debris, as well as damaged organelles and toxic proteinaceous accumulations at the synapse. Microglia induce reactive astrocytes by producing cytokines such as interleukin (IL)-1α, tumor necrosis factor-alpha (TNFα), and complement 1q (C1q) [46, 64–66]. After activation, astrocytes surround, take up, and degrade α-syn as microglia do, engaging in phagocytic activity in cases of microglial dysfunction or exhaustion [40, 67–70].
α-syn aggregates are stored in multiple astrocytic organelles, shown by Rostami et al. [69] to be the trans-Golgi network (TGN), endoplasmic reticulum (ER), and mitochondria (Figure 2). In mitochondria, α-syn aggregates cause an increase in the production of reactive oxygen species (ROS) through the inhibition of complex-I dependent respiration, the impairment of ATP production, and depolarization of the mitochondrial membrane [71]. In the ER, α-syn accumulation leads to ER stress [72] and can also cause ROS production [73]. This oxidative environment causes further oligomerization of α-syn monomers, which induces a positive feedback loop where the oligomer invokes ROS production to further increase aggregation [71]. In the TGN, accumulation of α-syn can lead to Golgi fragmentation and apoptosis [72, 74].
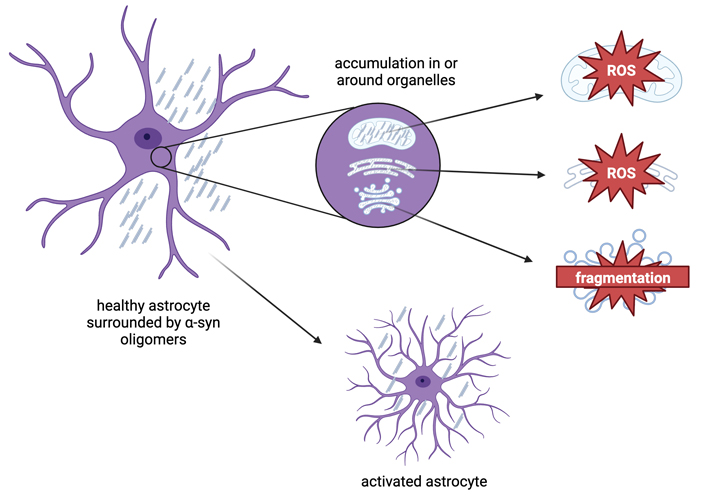
Effects of α-syn accumulation in astrocytes. When α-syn accumulates in an astrocyte, it aggregates in or around organelles within the cytoplasm, such as mitochondria, the Golgi apparatus, and the endoplasmic reticulum. Aggregation of α-syn in mitochondria and the ER leads to the production of ROS, while α-syn aggregation in the TGN leads to Golgi fragmentation. These byproducts lead to greater activation of astrocytes. Figure created with Biorender.com
As mentioned above, ROS production and mitochondrial damage resulting from α-syn aggregation, as well as aggregated, fibrillar, or monomeric α-syn can activate the NLRP3 inflammasome through activation of TLRs [75–79]. The NLRP3 inflammasome regulates inflammatory signaling and the innate immune response to α-syn by initiating microglial production and secretion of proinflammatory cytokines IL-1β and IL-18 [77, 80].
Activation of NLRP3 has been shown to have a neurotoxic effect: Fyn kinase which regulates α-syn uptake and NLRP3 activation in microglia has been shown to contribute to microgliosis [75]; the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-mediated NLRP3 activation results in neuronal cell death [81]; and inhibition of NLRP3 has neuroprotective effects [82–84]. NLRP3 activation compromises the ability of microglia to clear α-syn, further contributing to its neurotoxic effects [43].
Although the resident immune cells in the CNS play a significant role in neuroinflammation in PD, non-resident immune cells like lymphocytes, neutrophils, and monocytes can infiltrate into the CNS through the blood-brain barrier (BBB) and react to synucleinopathy as well.
T cells have been shown to infiltrate into the CNS in response to α-syn expression [85, 86] and recognize α-syn peptides, specifically at the Y39 amino acid [87]. Lymphocytes that recognize and respond to α-syn primarily produce interferon-gamma (IFN-γ) [85]. Compared to wildtype mice, immunocompromised mice exhibited an 8-fold increase in phosphorylated α-syn in response to intrastriatal α-syn injection. This increased synucleinopathy was rescued with T cell reconstitution, suggesting the role of T cells in α-syn clearance, possibly through sustained activation of microglia [88]. However, T cell reactivity to α-syn may also be detrimental to PD pathology; for example, the drainage of nitrated α-syn into the lymphatic system can activate and recruit T cells and macrophages to the CNS, further contributing to the pathogenesis of PD by increasing death of dopaminergic neurons [86].
CD4 (helper) T cells are known to worsen neurodegeneration as their absence in CD4-deficient mice ameliorates α-syn-induced neurodegeneration [85]. The T cell response to α-syn also depends on the form of α-syn the T cells are exposed to. In a study of mouse inoculation with human α-syn, Olesen et al. [89] found that nitrated α-syn increased the total number of T cells, while monomeric α-syn and fibrillar α-syn caused a change in the ratio between helper and non-helper T cells [89]. Fibrillar α-syn caused a compromise in the survival and activation of helper T cells and led to the production of antibodies while monomeric α-syn, fibrillar α-syn, and nitrated α-syn all led to infiltration of peripheral CD4 T cells into the brain parenchyma [90]. Monomeric α-syn and nitrated α-syn promote inflammation by increasing expression of dopamine receptor (DR)-D3 and decreasing expression of DR-D2, which alters the availability of dopamine [90, 91]. The effect of α-syn on CD4 T cells is further shown by the vaccination of mice with α-syn. Nitrated α-syn upregulated retinoic acid receptor-related orphan nuclear receptor gamma t (RORγt) protein [91], which protects against autoimmune diseases [89].
The role of CD4 cells in α-syn-mediated neurodegeneration is further corroborated through studies of Lewy body dementia (LBD) patients. C-X-C motif chemokine receptor 4 (CXCR4) is upregulated on T cells circulating through LBD patient cerebrospinal fluid (CSF). Increased CXCR4 expression was associated with greater CD4 T cell activation and cytokine signaling, as well as a greater degree of neurodegeneration. pSer129 α-syn can also activate and induce clonal expansion of human T cells, further endorsing the likelihood of an antigen-dependent helper T cell response to synucleinopathy [92].
CD8 cells, however, are not integral to neurodegeneration induced by α-syn [85]. When exposed to common blood antigens from PD patients, CD8 T cells from PD patients and healthy controls showed no difference in inflammatory cytokine expression [93]. In a longitudinal case study [94], α-syn T cell reactivity was detected in patients prior to PD diagnosis and the onset of motor symptoms, followed by a decline in T cell reactivity. Lindestam Arlehamn et al. [95] speculated that T cell reactivity may have initiated the spread of inflammation and confirmed an association between α-syn specific T cells and PD.
Although little evidence has been found of the involvement of B cells compared to that of T cells, B cell presence has been confirmed in the CNS of PD patients [96]. Studies have also found that PD patients have reduced B cell populations [97] and leukocytes of women with PD express lower levels of B cell-related genes [94]. Autoantibodies, which can aid in the clearance of pathological α-syn, have also been shown to be diminished in PD, indicating a protective role for B cells in the context of PD [96, 98].
In addition to B cell presence in the brains of PD patients, immunoglobulin G (IgG) expression has also been observed near dopaminergic neurons and Lewy bodies in PD patients [99] along with a general upregulation of IgA and IgG when compared to healthy controls [100]. However, there is a lack of consistent findings for differences in plasma concentrations of naturally occurring anti-α-syn autoantibodies between PD patients and healthy controls [98]. Additionally, PD patients have a higher proportion of TNFα-producing B cells, suggesting that B cells may play more of a regulatory role in PD as opposed to conferring an adaptive immune response via antibody production [101].
Major histocompatibility complex of class II (MHCII) is responsible for antigen presentation to CD4 T cells and has been shown to have a protective role in the spread of α-syn. Their absence in mice seeded with α-syn preformed fibrils (PFFs) led to a reduced lifespan and accelerated seeding of α-syn [102]. However, although MHCII molecules have been shown to have a significant role in the spread of α-syn, it has also been shown that absence of mature lymphocytes does not affect survival of mice seeded with PFFs [102], putting in question the protective role of T cell immunity against synucleinopathy.
Monocyte derived macrophages are also recruited to the brain in response to α-syn aggregation. In both murine and human macrophages, α-syn fibrils have been shown to induce leucine-rich repeat kinase 2 (LRRK2) expression as well as Rab10 phosphorylation, which contrasts with the limited effect of α-syn on LRRK2 expression in microglia. These LRRK2 monocytes infiltrate into the brain following intracranial injection of α-syn fibrils. Injections of monomeric α-syn did not lead to the recruitment of peripheral immune cells into the brain. LRRK2 inhibition prevented macrophage responses to α-syn fibrils, showing that LRRK2 is necessary for macrophage recruitment in synucleinopathy [103].
Macrophages take up α-syn in different ways depending on protein form. Fibrillar α-syn was shown to be taken up by endocytosis while monomeric α-syn was shown to be able to pass through the cell membrane without an actin rearranging process. After taking up α-syn, macrophages were able to degrade α-syn via both the lysosomal and proteasomal pathways [104], but phagocytosis was compromised by α-syn inside the cells [104, 105]. Additionally, monocytic activation increases in the late stages of PD, further supporting the role of monocytes and macrophages in the immune response and PD progression [106].
Although the mechanisms of neutrophil contributions to synucleinopathy are not entirely understood, neutrophils play a significant role in the progression of PD, as shown by the increase in neutrophils across PD patients when compared to healthy controls [107]. When activated, neutrophils release neutrophil extracellular traps (NETs), which entangle pathogens. Neutrophils have been shown to release NETs in response to α-syn and engulf α-syn aggregates for phagocytosis [108].
Additionally, BBB permeability has been shown to be increased in PD patients [109], which may lead to further immune cell infiltration into the brain and increased neuroinflammation. Inflammation may be exacerbated by the potential damage to the BBB caused by pathological α-syn, which activates endothelial cells and astrocytes that line the BBB [110]. Peripheral immune cells may cross the BBB more easily due to the damage caused by pathological α-syn accumulation, further contributing to neuroinflammatory processes in PD pathology.
In both microglia and astrocytes, the lysosomal machinery fails to degrade α-syn oligomers, causing mitochondrial damage and reduced cell viability [40, 67–69], as α-syn oligomers are toxic [111, 112]. This cytotoxicity can be attributed to their ability to affect membrane permeability. The presence of these oligomers reduces cell viability significantly [113, 114], especially in dopaminergic neurons [115]. Therefore, the accumulation of this neurotoxic species leads to astrocytes and microglia leads them to form tunneling nanotubes (TNTs) to uniformly distribute α-syn and reduce aggregate burden. TNTs are thin connections of plasma membranes between remote cells and aid the intercellular transfer of α-syn aggregates [40, 116, 117].
In microglia, the accumulation of α-syn fibrils can lead to the formation of TNTs with short and thick membranes or longer, thinner membranes, the latter of which transfers α-syn at a faster rate [116]. In astrocytes, α-syn accumulates upon failure to degrade ingested oligomers. Astrocytes show similar variation in the membrane protrusions to microglia, with long TNTs transferring smaller aggregates and shorter protrusions directly connecting cells for faster α-syn transfer [40].
In both astrocytes and microglia, the formation of TNTs facilitates the transfer of α-syn between acceptor and donor cells [40, 116, 118]. Although vesicular α-syn transportation causes aggregation of the soluble α-syn present in acceptor cells, the exchange of α-syn between astrocytes and microglia is especially effective in reducing inflammation, as microglia are able to degrade α-syn more efficiently through even distribution of protein aggregations across cells [116–119]. Glial cells also interact with peripheral immune cells in response to α-syn. Accumulation of α-syn in glia leads to the release of proinflammatory cytokines and chemokines that attract peripheral immune cells [120, 121]. For example, fibrillar α-syn causes the activation of stimulator of interferon genes (STING) in microglia and astrocytes, which triggers the release of interferons and proinflammatory cytokines [122, 123]. This inflammatory response is accompanied by an upregulation of chemokines such as C-X-C motif chemokine ligand 10 (CXCL10) and C-C motif chemokine ligand 5 (CCL5), which increase peripheral immune cell infiltration into the brain parenchyma. STING expression is positively correlated with levels of pSer129 α-syn, indicating its role in synucleinopathy [122]. α-syn fibrils also induce upregulation of MHCII molecules, which recruit peripheral monocytes and macrophages to parts of the brain where α-syn fibrils are present [121].
Recruitment of CD4 and CD8 T cells has been shown to exacerbate α-syn aggregation [85, 121]. The detrimental outcomes of T cell presence may be due to their influence on microglial function, as microglia showed greater phagocytosis of α-syn in the absence of lymphocytes and reduced phagocytosis in the presence of lymphocytes [85, 124].
An alternative explanation for this exacerbation of α-syn aggregation may be attributed to its function in the immune system. Viral infections, bacterial infections, and general immune responses have been shown to induce production of α-syn [125–128]. α-syn production leads to an immune response which then inversely induces α-syn production leading to a cyclic relationship with would exacerbate both aspects of the cycle and is a suspect trigger of PD [129].
The progression of PD is affected by a wide variety of factors—some of which involve immune responses to α-syn summarized in Figure 3. Immune cell activation and downstream functions depend on α-syn protein modifications and structure, with phosphorylated, nitrated, oligomeric, and fibrillar α-syn inducing the most inflammatory responses.
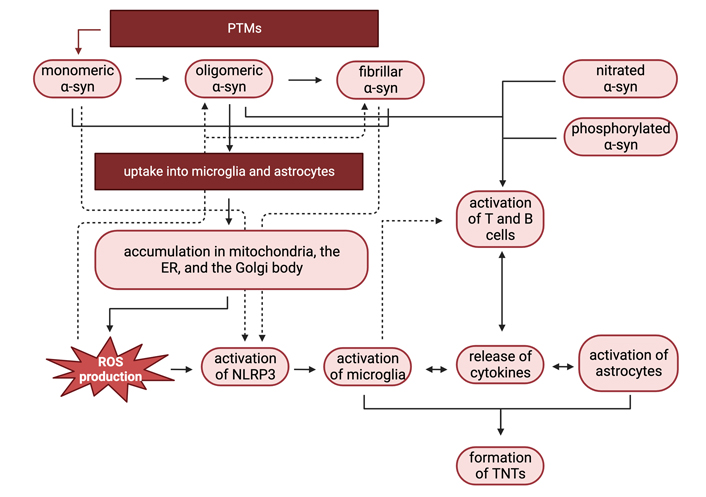
Interactions between immune cells in response to pathological α-syn. PTMs of monomeric α-syn can enhance α-syn aggregation, promoting α-syn oligomer and fibril formation. Aggregated α-syn is then taken up into astrocytes and microglia and accumulates in the mitochondria, ER, and the Golgi body. Accumulation in the mitochondria and ER causes ROS production, which can lead to microglial activation. Oligomeric, nitrated, and phosphorylated α-syn, along with inflammatory cytokine release by activated glia, activate T and B cells, which also contribute to inflammation and glial activation. In response to α-syn aggregate accumulation, astrocytes and microglia form TNTs, which help propagate the immune response to α-syn aggregates. Figure created with Biorender.com
PTMs can change the propensity of α-syn to aggregate and form oligomers. PTMs such as phosphorylation at Tyr39 or SUMOylation at Lys96 and Lys102 can increase α-syn aggregation, while other PTMs such as phosphorylation near the C-terminal and glycosylation can reduce α-syn aggregation. These post-translationally modified forms of α-syn can activate immune cells in the CNS such as microglia, which are triggered by pathological forms of α-syn. TLR signaling and NLRP3 inflammasome activation in microglia can propagate an innate immune response in the CNS through the release of inflammatory cytokines, ROS production, and α-syn intercellular transfer. Microglia can also upregulate MHCII molecules in response to α-syn exposure, resulting in recruitment and activation of peripheral monocytes, macrophages, and adaptive immune cells such as T cells, which can further exacerbate neuroinflammation.
There are currently no treatments capable of preventing, slowing, or stopping PD onset or progression. Although PD diagnosis has been historically difficult due to a lack of reliable biomarkers, CSF levels and aggregation propensity of α-syn have recently been shown to accurately diagnose 87–89% of PD [130], encouraging the development of drugs that can reduce α-syn aggregation to prevent or slow PD progression. Research studying PTMs of α-syn that increase monomer fibrillation and are commonly observed in human Lewy bodies is especially useful, as it pinpoints PTMs and regions of α-syn to target to prevent α-syn aggregation. Mechanistic studies of α-syn activation of immune cells in the CNS and periphery also shed light on inflammatory pathways that are activated in response to α-syn aggregates, highlighting ways to alter the immune response to increase α-syn clearance or reduce excessive neuroinflammation in PD [131–133].
Additionally, antibody-based therapies seem promising despite the lack of antibody involvement in the PD immune response. This is especially true for treatments making use of passive immunization against α-syn, as active immunization may not be entirely effective in elderly PD patients with diminished immune responses [132]. The use of monoclonal antibodies to target α-syn has already been shown to be effective in reducing α-syn aggregation [133] and could increase the involvement of phagocytic immune cells in the inflammatory response to PD, aiding the clearance of α-syn. Another promising option to prevent α-syn aggregation is using antibodies to target protofibrils, the oligomeric form of α-syn which is on its way to forming a fibril [131]. This alleviates the toxicity of α-syn oligomers as opposed to the less toxic fibrils [111] while also preventing the protofibrils from developing into fibrils.
At this time, most research on the effects of PTMs on α-syn aggregation is focused on singular PTMs in vitro. The effects of α-syn PTMs in vivo and the interplay between multiple PTMs present in the same environment or even on the same protein have yet to be elucidated. PTMs such as SUMOylation and phosphorylation have been shown to interact synergistically to protect against the formation of α-syn inclusions in in vitro systems [134], while SUMOylation has also been reported to decrease ubiquitination to enhance α-syn aggregation [35]. Nitration can also prevent phosphorylation at Ser129 which enhances α-syn aggregation [135]. Whether these interactions hold true under physiological conditions has yet to be validated.
There is also a lack of research on the effects of specific α-syn PTMs on cell-specific immune responses to monomeric α-syn. Although there is some existing data that suggests nitrated α-syn correlated with increased T lymphocytes in comparison to monomeric and fibrillar α-syn [86, 89, 136, 137], very little is known about the effects of other α-syn PTMs on α-syn immunoreactivity and PD pathology.
In conclusion, the tendency of α-syn to be inflammatory is dependent on its form. Aggregated α-syn fibrils are the most inflammatory, and certain PTMs can increase the aggregation propensity of α-syn. The activation of immune cells in response to α-syn is dependent on protein structure and exposure to proinflammatory cytokines secreted from other immune cells. The elucidation of α-syn PTMs that increase α-syn aggregation and immune activation will allow for the development of novel PD therapeutics that target aggregation-prone α-syn forms and chronic neuroinflammation commonly observed in PD patients makes directing α-syn (PTMs) and the use of antibodies seems promising in treatment.
α-syn: alpha-synuclein
BBB: blood-brain barrier
CNS: central nervous system
ER: endoplasmic reticulum
IgG: immunoglobulin G
IL: interleukin
LRRK2: leucine-rich repeat kinase 2
mGluR5: metabotropic Glu receptor 5
MHCII: major histocompatibility complex of class II
NEDD4: neuronally-expressed developmentally down-regulated gene 4
NLRP3: nucleotide-binding oligomerization domain-like receptor pyrin domain containing 3
NMDARs: N-methyl-d-aspartate receptors
PD: Parkinson’s disease
pSer129: phospho-Ser129
PTMs: post-translational modifications
RAGE: receptor for advanced glycation end products
ROS: reactive oxygen species
SUMOylation: the covalent attachment of a small ubiquitin-like modifier onto a Lys residue
TGN: trans-Golgi network
TLRs: toll-like receptors
TNTs: tunneling nanotubes
TREM2: triggering receptor expressed on myeloid cells 2
ZK: Conceptualization, Writing—original draft, Writing—review & editing, Visualization. YJ: Supervision, Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

