Review
Review

Affiliation:
Head of Research, CRC Scotland & London, Eccleston Square, SW1V 1PG London, UK
Email: anderson.george@rocketmail.com
ORCID: https://orcid.org/0000-0001-7243-0817
Explor Neurosci. 2026;5:1006132 DOI: https://doi.org/10.37349/en.2026.1006132
Received: November 10, 2025 Accepted: March 17, 2026 Published: April 15, 2026
Academic Editor: Dirk M. Hermann, University of Duisburg-Essen, Germany
There is a growing appreciation of the role of mitochondria in determining the interactions of CNS astrocytes, microglia, and neurons. The influence of circadian and systemic processes in regulating these interactions is relatively underexplored. Recent work has indicated the importance of night-time dampening and resetting in the pathoetiology of a diverse array of aging-associated medical conditions, including neurodegenerative disorders. The 10-fold decrease in pineal melatonin at night between childhood and the 9th decade of life is a major determinant of how aging associates with neurodegenerative disorders, cardiovascular disorders, and a wide range of tumors. It is proposed that the beneficial effects of pineal melatonin are mediated via its upregulation of the mitochondria-derived peptides (MDPs), including humanin. Although potentially induced in all mitochondria-containing cells, humanin is primarily produced in the CNS by astrocytes. The capacity of pineal melatonin to increase astrocyte humanin leads to the induction of the local melatonergic pathway in microglia to shift microglia from a pro-inflammatory M1-like to prophagocytic M2-like phenotype. In neurons, astrocyte-derived humanin optimizes mitochondrial function and decreases oxidant production to increase function and survival, possibly also involving mitochondrial melatonergic pathway upregulation. Concurrent effects of pineal melatonin in decreasing gut dysbiosis/permeability and stimulating oxytocin to activate the vagal nerve contribute to more optimized dampening and resetting that influences CNS interactions of glia and neurons. Overall, the conceptualizations of how astrocyte, microglial, and neuronal mitochondria interact require integration with wider circadian and systemic processes. A plethora of novel research implications are highlighted.
Mitochondria are evolutionarily modified bacteria arising from an endosymbiotic alpha-proteobacterium within an archaeal-derived pre-existing single cellular organism to initiate multicellular life on planet Earth [1]. Although classically conceptualized as energy providers via oxidative phosphorylation (OXPHOS) derived adenosine triphosphate (ATP) production, recent work highlights the diverse impacts that mitochondria have on cellular and intercellular interactions [2, 3]. Mitochondria not only provide ATP but are also the major source of cellular oxidants, which seem to arise from their bacterial roots, given that bacteria produce oxidants to exogenous challenges. Mitochondrial oxidant production may therefore be conceptualized as an indicant of suboptimal mitochondrial function within a challenging microenvironment. Almost all current medical conditions show indicants of altered and suboptimal mitochondrial function, including neurodegenerative, neuropsychiatric, and cardiovascular disorders [4–6], as well as in cancer [7]. As most of these conditions have an increased risk over aging [8], understanding the alterations in mitochondrial function over aging is cutting edge research across diverse medical conditions [8].
There is a growing appreciation of the roles of mitochondria in determining homeostatic regulation of cells and their microenvironments [7]. Many of the beneficial effects of the gut microbiome are mediated via the production of the short-chain fatty acid, butyrate, and its impacts on systemic mitochondrial function [9]. This has been conceptualized as a communication between actual bacteria and evolutionarily modified bacteria in the form of mitochondria across CNS and systemic cells, microenvironments, and body systems [9]. More direct intercellular interactions of mitochondria across cells in local microenvironments are changing the nature of how aging-associated neurodegenerative conditions are conceptualized. This is primarily mediated by the regulation of various mitochondrial fluxes that influence the function and interactions of astrocytes, microglia, and neurons by modulating mitochondrial function [10] as well as mitochondrial transfer among cells [11].
Recent work has highlighted the roles of mitochondria-derived peptides (MDPs), such as humanin, mitochondrial open reading frame of the 12S ribosomal RNA type-c (MOTS-c), and small humanin-like peptides (SHLPs), in the regulation of aging and associated neurodegenerative disorders [12, 13]. Humanin has been most extensively investigated and is the focus of the current article, where the interactions of humanin with the mitochondrial melatonergic pathway are highlighted.
This review evaluates the hypothesis that pineal melatonin resets mitochondrial function following daytime stressors/challenges to increase mitochondrial fusion and humanin production, which then acts on neighboring glia and neurons to reset mitochondrial function and upregulate the mitochondrial melatonergic pathway, thereby resolving inflammatory activity. The suppressed capacity to produce melatonin over aging underpins inflammaging and the emergence of neurodegenerative conditions.
The following terms were searched on Medline/PubMed, Scopus, and Google: ‘Glia’, ‘astrocytes’, ‘melatonin’, ‘mitochondria’, ‘humanin’, ‘neurodegenerative disorders’, ‘NF-κB’, ‘STAT3’, ‘aging’, ‘circadian’, ‘vagal’, ‘oxytocin’, ‘gut microbiome’, and ‘butyrate’ to provide the background data for this article.
First, data on humanin are briefly overviewed.
Although all mitochondria-containing cells, including those in the CNS, can produce humanin, astrocytes seem to be the primary source of CNS humanin [14]. Astrocyte-derived humanin is protective of challenged neurons and is decreased over aging and aging-associated conditions, including Alzheimer’s disease and the menopause, as shown primarily in preclinical studies [14–16]. The suppressed capacity of astrocyte mitochondria to produce humanin over the course of aging may therefore contribute to the maintained inflammaging currently proposed to underpin many neurodegenerative conditions, including Alzheimer’s disease [17]. As estrogen and progesterone can increase humanin and have mitochondrial receptors [14], it requires investigation whether other mitochondrial receptors, including brain-derived neurotrophic factor (BDNF) receptor, tropomyosin receptor kinase (Trk) B, and cannabinoid receptors, as well as melatonin receptor 1 (MT1r)/MT2r and glucocorticoid receptors (GRs) [18], modulate humanin production. Currently, it is proposed that mitochondrial transcription factor A (TFAM) induction and the actions of growth hormone/insulin-like growth factor 1 (GH/IGF-1) drive humanin induction [14]. As melatonin can increase GH/IGF-1, partly via hypothalamic somatostatin suppression [19], and increase TFAM in neurodegenerative conditions [20], pineal melatonin and its loss over aging may be a significant contributory factor to the aging-linked decline in humanin levels [21].
Released humanin binds to a cell-surface receptor complex comprised of glycoprotein 130 (gp130), ciliary neurotrophic factor receptor (CNTFR) α, and the IL-27 receptor subunit (WSX-1), which increases the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) 3 pathway. The beneficial effects of humanin are prevented by STAT3 inhibition [22, 23], indicating the importance of STAT3 induction in humanin’s intercellular effects. Humanin is also a ligand for the G protein-coupled formyl peptide receptor-like 1 (FPRL1) and formyl peptide receptor-like 2 (FPRL2) [24, 25]. Intracellular humanin binds Bcl-2-associated X protein (BAX), Bcl-2-interacting mediator of cell death (Bim), and BH3 interacting domain death agonist (Bid), as well as insulin-like growth factor-binding protein (IGFBP) 3, allowing humanin to modulate mitochondrial function, cell apoptosis and survival by diverse routes, both intracellularly in cells of humanin production and via humanin release and paracrine effects [26].
Astrocytes produce and secrete humanin, which can be transferred to other cells in the astrocyte microenvironment, including by mitochondrial transfer to other cells [27]. These authors also showed that astrocyte-derived humanin shifts microglia from an M1-like pro-inflammatory phenotype to an M2-like pro-phagocytic phenotype [27]. This is a phenotypic shift that requires a change in nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) dimer composition (from p65/p50 to c-Rel/p50), which is coupled to microglia/macrophage melatonergic pathway upregulation and release and autocrine effects of melatonin [28, 29]. This suggests that astrocyte mitochondria released humanin acts on other cells to regulate their mitochondrial melatonergic pathway, as indicated in microglia [27]. The melatonergic pathway is regulated across diverse human cell types by the interactions between STAT3 and NF-κB [30]. Humanin’s necessary STAT3 upregulation [22, 23] coupled to changing the NF-κB dimer composition [27] indicates that STAT3 interactions with NF-κB dimer composition contribute to melatonergic pathway upregulation. Data indicating humanin effects on STAT3 and NF-κB interactions are covered next.
As noted above, the mitochondrial melatonergic pathway seems to be available but not necessarily continually active in all body cells [1, 30], including astrocytes [31], macrophages [32], microglia [28], and neurons [33]. Data across diverse human cells indicates that the mitochondrial melatonergic pathway is regulated by the differential interactions of canonical (nuclear) and non-canonical (mitochondrial) STAT3 with NF-κB dimer composition. Nuclear bound STAT3 is phosphorylated at Tyrosine705 (pSTAT3Tyr705), whilst mitochondria-translocating STAT3 is phosphorylated at Serine727 (pSTAT3Ser727). Phosphorylated STAT3 at Serine727 (pSTAT3Ser727) translocation to mitochondria can be to different sites, namely the mitochondrial matrix and/or mitochondria-associated membranes (MAMs). At MAMs, pSTAT3Ser727 (and in some studies, phosphorylated STAT3 at Tyrosine705 (pSTAT3Tyr705) [34]) down-regulates Ca2+ influx into mitochondria, thereby modulating mitochondrial apoptotic susceptibility. However, the mitochondrial translocation of pSTAT3Ser727 can also lead to the translocation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome to the mitochondrial membrane as well as the mitochondrial transfer of NF-κB and one of its dimer components, p65 [35]. Mitochondria-translocating pSTAT3Ser727 can therefore have diverse and dramatic effects, as indicated by data across an array of cell types [36, 37], with pSTAT3Ser727 mitochondrial translocation increasing pro-inflammatory responses and mimicking many aging-associated changes in immune cells [38]. pSTAT3Ser727 effects in astrocyte mitochondria require coordinated investigation, as pSTAT3Ser727 induces a reactive astrocyte phenotype [39].
The mitochondrial translocation of pSTAT3Ser727 requires 14-3-3ζ as a chaperone [40], with 14-3-3ζ bound pSTAT3Ser727 forming a positive feedback loop with leucine zipper EF-hand containing transmembrane protein 1 (LETM1)-domain-containing protein 1 (LETMD1) to modulate mitochondrial Ca2+ and K+ regulation [41, 42]. Whether this increases or limits the availability of 14-3-3ζ, which is necessary to stabilize aralkylamine N-acetyltransferase (AANAT) and therefore the initiation of the mitochondrial melatonergic pathway [43], requires investigation in astrocytes and microglia. The complexity of STAT3 effects may partly arise from its distinct interactions with NF-κB dimer composition.
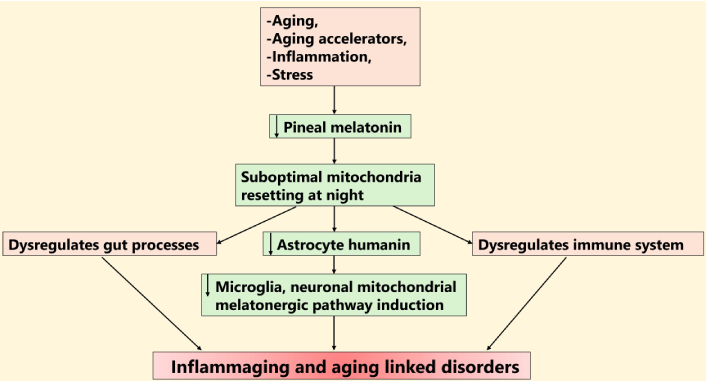
NF-κB can have diverse effects in different cells that are strongly determined by the composition of the NF-κB dimer, with a p65/p50 NF-κB dimer being pro-inflammatory in astrocytes and microglia, whilst the c-Rel/p50 dimer is integral to the resolution of astrocytes from a pro-inflammatory phenotype [44], as in microglia [29]. Nuclear translocating NF-κB dimer composition differentially interacts with pSTAT3Tyr705 to initiate or suppress the melatonergic pathway, mediated by different NF-κB dimer components across different cell types [30]. As indicated above, in astrocytes and microglia, the induction of the melatonergic pathway is highly likely to be mediated by the resolution inducing c-Rel/p50 dimer [29, 44]. Whether STAT3 and NF-κB nuclear interactions drive kinase(s) that regulate the phosphorylation of mitochondria-translocating pSTAT3Ser727 requires further investigation. This allows for a transient inflammatory process to be initiated and quickly shifted to a pro-phagocytic or quiescent phenotype that is dependent upon the regulation of the NF-κB dimer components, as shown in macrophages and microglia [28, 29]. How this interacts with humanin regulation of NF-κB dimer composition [45], and the presence of NF-κB at mitochondria, as well as within the nucleus, also requires clarification in CNS cells. The regulation of the mitochondrial melatonergic pathway and its potential interface with humanin is shown in Figure 1.
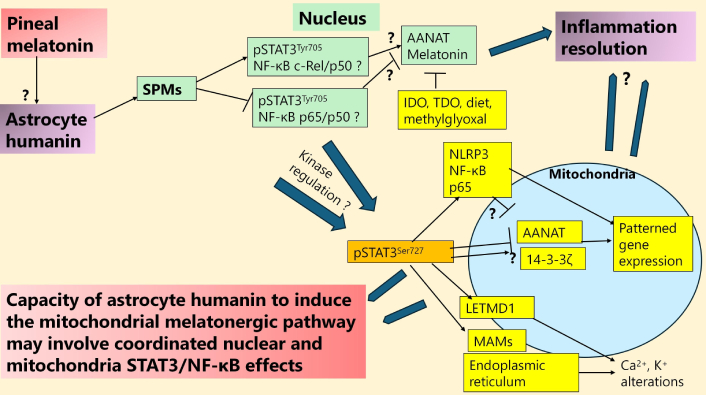
STAT3 interaction with NF-κB dimer composition in humanin melatonergic pathway regulation. The dramatic decrease in pineal melatonin over aging may attenuate its induction of astrocyte humanin and therefore the capacity of released astrocyte humanin to suppress microglia and neuronal inflammation via mitochondrial melatonergic pathway upregulation. Data in macrophages indicates that humanin increases SPMs, which can shift the NF-κB dimer composition to resolution inducing. The raised levels of IDO, TDO, and methylglyoxal over aging, especially when T2DM/hyperglycemia/hypertension is evident, suppress the availability of tryptophan, as does inadequate dietary tryptophan. Suppressed tryptophan availability attenuates the initiation of the tryptophan-melatonin pathway and, therefore, the capacity to attain resolution. The interactions of mitochondrial pSTAT3Ser727 with various processes in mitochondria, including regulating Ca2+ from the endoplasmic reticulum at MAMs, whilst also regulating 14-3-3ζ availability for AANAT stabilization to initiate the mitochondrial melatonergic pathway, as well as interacting with LETMD1 to regulate mitochondrial Ca2+ and K+, whilst also coupled with a capacity to increase the mitochondrial translocation of the NLRP3 inflammasome, NF-κB and p65 indicate potential complex effects dependent upon specific cellular circumstances. Whether pSTAT3Try705 nuclear interactions with NF-κB c-Rel/p50 to drive a resolution seeking phenotype include kinase regulation of pSTAT3Ser727 that suppresses pSTAT3Ser727 ‘detrimental’ effects at mitochondria requires investigation. Thicker arrows indicate an accumulation of processes influencing where the arrows are pointing. SPMs: specialized pro-resolving mediators; pSTAT3Tyr705: phosphorylated signal transducer and activator of transcription 3 at Tyrosine705; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; AANAT: aralkylamine N-acetyltransferase; IDO: indoleamine 2,3-dioxygenase; TDO: tryptophan 2,3-dioxygenase; NLRP3: NLR family pyrin domain containing 3; LETMD1: leucine zipper EF-hand containing transmembrane protein 1-domain-containing protein 1; MAMs: mitochondria-associated membranes; T2DM: type 2 diabetes mellitus.
As shown in Figure 1, pineal melatonin may upregulate humanin, which then acts to upregulate pSTAT3 interactions with NF-κB c-Rel/p50 to induce the local melatonergic pathway, which in microglia shifts these cells from an M1-like pro-inflammatory phenotype to an M2-like pro-phagocytic phenotype in the course of inflammation resolution. Whether pSTAT3Tyr705 interactions with NF-κB c-Rel/p50 induce kinases to regulate the mitochondria-translocating pSTAT3Ser727 and/or the site and effects of its mitochondrial translocation requires future investigation. Data in macrophages show humanin to increase specialized pro-resolving mediators (SPMs) [46], including genes crucial for SPMs synthesis, such as Alox15 and Retnla [46]. SPMs are crucial to shifting microglia to an M2-like, pro-resolving phenotype [47], as is microglia autocrine melatonin [28], indicating that the effects of exogenous humanin may be mediated via the SPMs regulation of the NF-κB dimer composition and the melatonergic pathway, as shown in Figure 1.
It is important to emphasize that pSTAT3Ser727 has diverse effects at mitochondria, including the downregulation of Ca2+ mitochondrial entry via MAMs, as well as having a positive feedback loop with LETMD1, thereby modulating the regulation of Ca2+ and K+. Given the importance of ionic flux to the diverse modes of mitochondrial function, the investigation of pSTAT3Ser727 interactions with different aspects of mitochondrial function and regulation in glia and neurons will be important to clarify. The translocation of pSTAT3Ser727 to mitochondria requires binding with the chaperone 14-3-3ζ [43], which may act to positively or negatively regulate the availability of 14-3-3ζ to stabilize AANAT and thereby initiate the mitochondrial melatonergic pathway. In other cells, pSTAT3Ser727 increases the mitochondrial translocation of the NLRP3 inflammasome, NF-κB, and p65 that contribute to suboptimal mitochondrial function and enhanced pro-inflammatory activity [35]. Whether this occurs in CNS cells and the wider cellular factors regulating it require investigation.
Recent decades have witnessed a shift from a neuron-centric conceptualization of CNS function to one where neuronal activity may be seen as a form of immune-to-immune communication, with astrocytes and microglia being the predominant CNS immune cells [48]. Astrocytes can autonomously regulate circadian rhythms as well as modulate the rhythmicity of the suprachiasmatic nucleus (SCN), the CNS central pacemaker [49–51], highlighting the importance of astrocytes in shaping the diurnal variations in the function of neurons and other CNS cells. The influence of astrocytes on the circadian rhythm and neuronal and microglial function is complicated by aging-associated suppression of night-time pineal melatonin.
Pineal melatonin regulates all systemic cells, including astrocytes, neurons, and other CNS glial cells [52] as well as systemic processes that regulate CNS function, including gut permeability/dysbiosis [53] and systemic immune cells/systems [54]. Pineal melatonin inhibits glial oxidative stress, apoptosis, inflammation, amyloid-β induction, and fibrillogenesis, as well as upregulating the aquaporin (AQP) 4 on astrocyte end-feet to induce a polarization that drives the debris clearing by the glymphatic system during sleep [52, 55]. The dramatic 10-fold loss of melatonin over the course of aging [56] significantly modulates astrocyte function and astrocyte interactions with other CNS cells [57], as shown in Figure 2. This also has implications for mitochondrial function, given the capacity of pineal melatonin to optimize mitochondrial function, including by upregulating sirtuin-3, which contributes to the suppression of oxidant production at three points on the mitochondrial electron transport chain [58]. Melatonin can also act on mitochondrial melatonin receptors (MT1r and MT2r) to modulate mitochondrial function [59, 60]. Aging accelerators, such as type 2 diabetes mellitus (T2DM) and obesity [61], also decrease pineal melatonin [62], thereby modulating the capacity of pineal melatonin to dampen and reset at night.
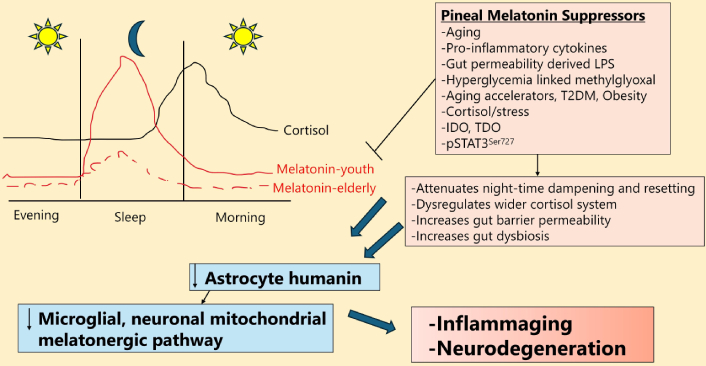
Suppressors of pineal melatonin attenuate astrocyte humanin to accelerate aging. Shows the dramatic suppression of pineal melatonin over aging and how this is regulated by many other factors to decrease astrocyte humanin, thereby suppressing the inflammation resolution driven by the mitochondrial melatonergic pathway, culminating in inflammaging and neurodegeneration. Thicker arrows indicate an accumulation of different processes acting to modulate whatever the arrow is pointing to. LPS: lipopolysaccharide; T2DM: type 2 diabetes mellitus; IDO: indoleamine 2,3-dioxygenase; TDO: tryptophan 2,3-dioxygenase; pSTAT3Ser727: phosphorylated signal transducer and activator of transcription 3 at Serine727.
The consequences of suppressed pineal melatonin are complicated by the impact this may have on the rise in cortisol in the second half of sleep that culminates in the cortisol awakening response (CAR), which peaks in the first 30 min after awakening, as shown in the graph in Figure 2. As melatonin suppresses the GR-α nuclear translocation [63], the suppression of pineal (and local) melatonin may disinhibit GR-α nuclear translocation and the influence of cortisol on night-time dampening and resetting [64]. Heightened GR-α activation, as is common under stress conditions, upregulates GR-β, which is proposed to function as a decoy receptor to limit cortisol effects. However, as the GR-β also regulates transcription [65], GR-β induction under conditions of heightened GR-α activation will also have distinct effects on patterned gene expression. The GR-α is not exclusively sited in a cytoplasmic complex but may also be expressed on the plasma membrane [66], with the canonical, cytoplasmic GR-α and plasma membrane GR-α interacting with canonical, nuclear pSTAT3Tyr705 [67], thereby potentially modulating the induction or suppression of the melatonergic pathway. The GR-α can also be expressed on the mitochondrial membrane [68] and within the mitochondrial matrix [69], indicating potentially diverse effects on mitochondrial function [70]. Whether suppressed pineal melatonin over aging and aging accelerating conditions modulates the GR subtype and/or localization site requires investigation, as any change would indicate dramatic alterations in circadian and stress cortisol effects over aging. This is exemplified by the mitochondrial membrane GR-α modulating mitochondrial transcription and metabolism [68]. Whether pineal and/or local melatonin regulates the GR-α mitochondrial translocation will be important to determine, including by melatonin epigenetically upregulating Bcl-2 associated athanogene (BAG)-1 [71], which can repress GR-α transcription and increase GR-α mitochondrial translocation [72]. BAG-1 is decreased over aging, concurrent with an increase in BAG-3 [73], being another aging-associated change in GR-α regulation.
Compared to melatonin, cortisol has distinct effects on the function of astrocytes [74, 75] and other CNS cells [76], with stress-associated cortisol decreasing astrocyte lactate availability for neurons, thereby compromising neuronal mitochondrial function and metabolism [77]. An aging-associated decrease in the night-time melatonin/cortisol ratio has significant impacts on individual cells and their interactions, including mitochondrial function as well as wider CNS processes, such as the glymphatic system [75]. This adds to the complexity of changes over the course of aging and underpins a night-time driven initiation of many aging-linked systemic and CNS diseases [70], including neurodegenerative conditions [21], as shown in Figure 2. Intrinsic mitochondrial rhythms add to this complexity and are discussed next.
There is a growing appreciation of the role of the circadian rhythm in regulating mitochondrial function and the two-way interactions of nuclear circadian genes with mitochondrial function and products [78, 79]. Many mitochondria-encoded proteins follow a circadian rhythm, as does mitochondrial reactive oxygen species (ROS), OXPHOS, and ATP production [78, 79], suggesting that variations in humanin production by the mitochondrial genome may also be circadian regulated. This requires future investigation, including the roles of circadian melatonin and cortisol, both of which express receptors on the mitochondrial membrane that modulate mitochondrial gene transcription. This may be further complicated by the capacity of seemingly all cells to induce and suppress melatonin production (including via STAT3 interactions with NF-κB) and upregulate local cortisol production via 11β-hydroxysteroid dehydrogenase (HSD) 1, as well as the factors that regulate the expression of mitochondrial melatonin and GRs. Whether these classical circadian regulators, pineal melatonin and adrenal cortisol, impact directly on the regulation of MDPs, such as humanin, will be important to clarify. This may occur indirectly, e.g., melatonin (and gut microbiome derived butyrate) can increase mitochondrial sirtuin-3, which is regulated over the circadian rhythm and thereby the acetylation status of its mitochondrial target proteins, including the pyruvate dehydrogenase complex (PDC) and therefore OXPHOS and ATP production [80]. Sirtuin-3, like other nicotinamide adenine dinucleotide (NAD+) dependent sirtuins, has effects that are determined by variations in the circadian regulation of NAD+ [81], which can be induced by fasting and exercise and is generally increased at night in humans [81].
In other cell types, melatonin increases sirtuin-3, thereby attenuating the acetylation level in mitochondrial proteins, leading to enhanced PDC activity and increased conversion of pyruvate to acetyl-CoA as well as raised mitochondrial metabolism [82]. These impacts of night-time melatonin and elevated night-time human sirtuin-3 levels at night are coupled with more optimized mitochondrial metabolism and function. Melatonin and sirtuin-3 may therefore underpin how mitochondrial function optimizes night-time dampening and resetting for the coming day. This becomes problematic over the course of aging, where both melatonin and sirtuin-3 are decreased [82, 83]. As nuclear DNA repair [requiring NAD+ consuming poly(ADP-ribose) polymerase 1 (PARP1)] as well as protein synthesis are enhanced in the wake phase, whilst redox and mitochondrial remodeling are enhanced during sleep (via raised melatonin and sirtuin-3 levels), wakefulness has been classed as nucleo-restorative whilst sleep may be regarded as mitochondria restorative [84]. How night-time optimization/resetting of mitochondrial function interacts with MDPs, such as humanin, in the course of changes in night-time dampening and resetting over aging will be important to clarify. Clinically, this may be especially pertinent in the pathoetiology of aging-linked medical conditions such as Alzheimer’s disease and amyotrophic lateral sclerosis, where there is a growing appreciation of the relevance of astrocytes and mitochondrial function [85, 86]. This is also pertinent to recent work indicating that the pathoetiologies of the major aging-linked and typically fatal medical conditions, such as cancer, cardiovascular diseases, and neurodegenerative conditions, are importantly determined by alterations in night-time processes [87–89].
The interactions of the circadian rhythm and mitochondrial function are further complicated by mitochondria having a nucleus independent ultradian rhythm (12 h). These autonomous 12-h rhythms in mitochondrial DNA (mtDNA) transcription are synchronized by metabolic and/or endoplasmic reticulum stress cues, and therefore independent of classical, circadian light/dark cues that entrain the nuclear circadian clock [78]. Mitochondria have a short half-life (8–24 days) [90], determined by variations in fission and fusion [91]. Melatonin inhibits the mitochondrial fission that is driven by dynamin-related protein 1 (DRP1) recruitment from the cytosol to the mitochondrial outer membrane [92]. DRP1-driven fission is indicative of impaired mitochondrial function and consequently raised ROS production, ultimately culminating in mitochondrial network fragmentation and mitophagy. Fission is therefore typically a wakefulness process [93]. Melatonin prevents these fission-linked processes and increases fusion [94], with mitochondrial fusion and fatty-acid β-oxidation during sleep optimizing mitochondrial function and efficient ATP production, both in the CNS [95] and systemically [96]. Melatonin’s circadian driven effects, therefore, provide distinct day and night mitochondrial phenotypes upon which ultradian processes act. The suppression of circadian processes over aging influences mitochondrial ultradian rhythms, as shown in preclinical models, further altering mitochondrial function [97]. See Figure 3.
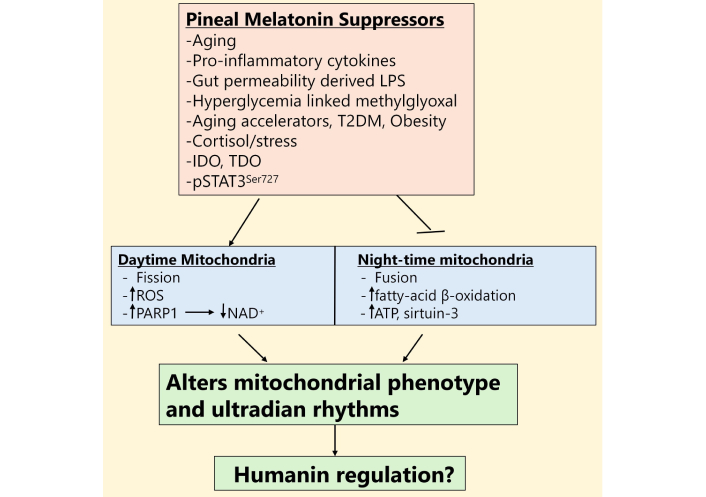
Distinct daytime and night-time mitochondrial ‘phenotypes’ are differentially regulated by pineal melatonin and its suppression. LPS: lipopolysaccharide; T2DM: type 2 diabetes mellitus; IDO: indoleamine 2,3-dioxygenase; TDO: tryptophan 2,3-dioxygenase; pSTAT3Ser727: phosphorylated signal transducer and activator of transcription 3 at Serine727; ROS: reactive oxygen species; PARP1: poly(ADP-ribose) polymerase 1; NAD+: nicotinamide adenine dinucleotide; ATP: adenosine triphosphate.
The interactions of astrocytes, microglia, and neurons are also powerfully regulated by systemic processes, including factors regulating gut dysbiosis and gut permeability, which, as noted, are also modulated by pineal melatonin at night [98]. Pineal melatonin exits the pineal gland via the pineal recess into the third ventricle, which lies immediately above the hypothalamus and is lined by tanycytes [99]. As well as regulating the circadian rhythm via the modulation of SCN neurons and astrocytes, including via astrocyte STAT3 [100], melatonin also significantly upregulates oxytocin [101]. Oxytocin is classically associated with bonding, with effects partially mediated by the positive allosteric modulation of the μ-opioid receptor [102]. Oxytocin projects to all areas of the brain and regulates cortisol effects on affect (amygdala), brain reward/motivation (nucleus accumbens/ventral tegmental area), and cognition (hippocampus) [64] as well as projecting to cortex astrocytes, microglia, and neurons [103, 104], with effects that include the optimization of mitochondrial function [105].
Pineal melatonin induced oxytocin also activates the vagal nerve [106], which releases acetylcholine (ACh) that activates ACh receptors, especially the alpha-7 nicotinic ACh receptor (α7nAChR). As α7nAChRs are increased by melatonin [107] and can be expressed on the mitochondrial outer membrane [108], the α7nAChR may be an intimate aspect of night-time dampening and resetting, including from α7nAChR increasing intracellular Ca2+ [109] that can drive GR-α translocation from its cytoplasmic complex to the plasma membrane [110]. Activation of the α7nAChR increases SPMs (possibly via humanin induction) that change NF-κB dimer composition to upregulate the mitochondrial melatonergic pathway and melatonin release, thereby allowing vagal nerve activation to dampen inflammatory activity only when a capacity to upregulate local melatonin is evident, as shown preclinically and clinically in the gut [111–113]. The role of gut permeability/dysbiosis and gut associated inflammation may therefore be regulated directly by pineal melatonin as well as indirectly via the melatonin/oxytocin/vagal/α7nAChR pathway. This has consequences for the gut modulation of astrocyte, microglia, and neuronal mitochondrial interactions.
The short-chain fatty acid, butyrate, produced under more optimal gut conditions is a powerful pan-histone deacetylase inhibitor (HDACi), which optimizes mitochondrial function, increases mitochondrial sirtuin-3 [114], upregulates the mitochondrial melatonergic pathway (as shown in intestinal epithelial cells [115]) and can be used as a preferential (vs. acetate) metabolic substrate for astrocytes [116]. Melatonin, including via the oxytocin/vagal/α7nAChR pathway, can modulate core CNS processes via impacts on the gut microbiome, as well as decreasing gut permeability to prevent lipopolysaccharide (LPS) from entering the circulation [117]. Pineal melatonin and the oxytocin/vagal/α7nAChR pathway are proposed to mediate their effects on gut enteric glial cells, which are powerful determinants of coordinated changes in cells around the gut, including the mucosal immune system, intestinal epithelial cells, and enteroendocrine cells as well as the enteric nervous system [118]. Enteric glial cells have a number of astrocytic features, and whether enteric glial cells are a significant source of gut humanin will be interesting to determine. Overall, such data highlights how circadian and systemic processes may interact to modulate core processes in key CNS cells. See Figure 4.
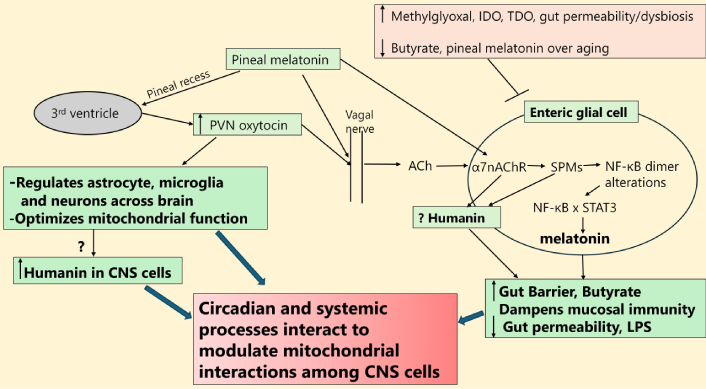
Interactions of circadian and systemic processes in the regulation of CNS mitochondria. The bolder/thicker arrows were meant to represent the accumulated influence of factors. PVN: paraventricular nucleus; ACh: acetylcholine; IDO: indoleamine 2,3-dioxygenase; TDO: tryptophan 2,3-dioxygenase; α7nAChR: alpha-7 nicotinic ACh receptor; SPMs: specialized pro-resolving mediators; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; STAT3: signal transducer and activator of transcription 3; LPS: lipopolysaccharide.
As highlighted throughout, the interactions of astrocytic, microglial, and neuronal mitochondria may be intimately linked to wider circadian and systemic processes that are powerfully changed over the course of aging due to the dramatic decrease in pineal melatonin. Gut and cortisol system dysregulation, coupled to an attenuated capacity of suppressed pineal melatonin to stimulate oxytocin and vagal nerve activation lead to alterations in daytime and night-time mitochondrial phenotypes and their function. As core processes of cellular and intercellular regulation, the change in mitochondrial phenotypes alters interactions of astrocytes, microglia, and neurons in the CNS, with significant implications for aging-associated neurodegenerative conditions, as shown in Figure 5.
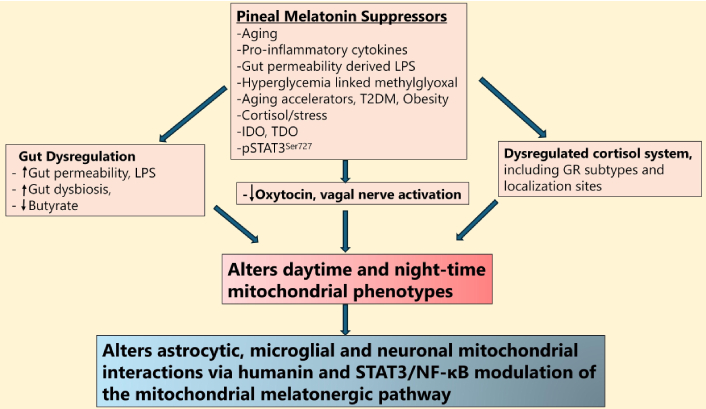
Pineal melatonin regulates mitochondrial phenotypes to determine glial-neuronal interactions. Shows how pineal melatonin suppressors, especially aging, impact the gut, oxytocin, vagal, and cortisol system to modulate diurnal mitochondrial phenotypes to alter glial-neuronal interactions. LPS: lipopolysaccharide; T2DM: type 2 diabetes mellitus; IDO: indoleamine 2,3-dioxygenase; TDO: tryptophan 2,3-dioxygenase; pSTAT3Ser727: phosphorylated signal transducer and activator of transcription 3 at Serine727; GR: glucocorticoid receptor; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells.
The ubiquitous use of ‘should’, ‘potentially’, ‘suggestive’, etc. highlight the major limitation of this article, which arises from the lack of direct data, especially pertaining to astrocyte mitochondrial function and regulation, including in the induction of humanin and the melatonergic pathway. However, this does provide a number of future research implications.
1. Does pineal and/or astrocyte melatonin modulate astrocyte mitochondrial humanin induction? Is humanin, like many mitochondrial proteins and processes, regulated over the circadian rhythm? Is humanin production linked to the shift from daytime fission to night-time mitochondrial fusion and, therefore, linked to variations in pineal melatonin over aging?
2. Do mitochondrial GPCRs, including MT1r, MT2r, GR-α, TrkB, and cannabinoid receptors modulate humanin production? Does the decrease in the night-time melatonin/cortisol ratio over aging suppress humanin production by astrocyte mitochondria? Does the suppression of humanin over aging contribute to increased inflammaging and associated neurodegenerative disorders?
3. Does astrocyte-derived humanin act on the STAT3 and NF-κB interactions to modulate local melatonin production, including in microglia, neurons, and possibly oligodendrocytes? Would this indicate that humanin acts as an interface between pineal melatonin (induction of astrocyte humanin) and local melatonin production and inflammation resolution in microenvironment cells, including microglia and neurons? Do the raised levels of night-time pineal melatonin and sirtuin-3 increase humanin production and/or release from astrocyte mitochondria?
4. Do factors that suppress the tryptophan-melatonin pathway, such as pro-inflammatory cytokine induced indoleamine 2,3-dioxygenase (IDO), cortisol/GR-α induced tryptophan 2,3-dioxygenase (TDO), and T2DM/hyperglycemia induced methylglyoxal (via protein-protein interactions with tryptophan [119]), attenuate the capacity of astrocyte-derived humanin to induce the mitochondrial melatonergic pathway in microglia (thereby maintaining a pro-inflammatory M1-like phenotype) and neurons (thereby attenuating neuronal function, long-term depression (LTD) and long-term potentiation (LTP) [120])? Preclinical data indicate that the putative induction of the hippocampal mitochondrial melatonergic pathway by pineal melatonin induced astrocyte humanin optimizes the induction of LTP, cognition, and memory [121]. This is in sharp contrast to the detrimental effects of cortisol at the GR-α [122], indicating that how night-time pineal melatonin and cortisol are regulated may be intimately linked to both humanin induction (pineal melatonin) and suppressed capacity to induce the local melatonergic pathway (cortisol at the GR-α). How this is determined by variations in canonical and non-canonical pSTAT3 interactions with NF-κB dimer composition will be interesting to clarify. How CNS processes are dampened and reset at night may be intimately linked to alterations in basic processes of learning and memory that are determined by the capacity of astrocyte-derived humanin to induce the local mitochondrial melatonergic pathway in neighboring cells of the local microenvironment.
5. Does pSTAT3Ser727 in the mitochondrial matrix, vs. MAMs, differentially regulate NF-κB and p65 mitochondrial translocation? How would this differential siting of pSTAT3Ser727 modulate astrocyte mitochondrial humanin production?
6. Does pSTAT3Ser727 modulate 14-3-3ζ availability for the stabilization of AANAT and the initiation of the mitochondrial melatonergic pathway via impacts in the mitochondrial matrix, vs. MAMs?
7. Does released mitochondrial melatonin have ‘autocrine’ effects on mitochondrial melatonin receptors?
8. As melatonin can increase Ca2+ influx into cells, with differential impacts on mitochondrial and endoplasmic reticulum Ca2+ [123], whilst raised Ca2+ drives the plasma membrane translocation of the GR-α in preclinical models [110], does the loss of melatonin over aging modulate GR-α site of localization? As the α7nAChR is induced by melatonin [107] and increases intracellular Ca2+ [109], does the α7nAChR increase GR-α expression at the plasma membrane, especially at night, thereby changing the consequence of GR-α by cortisol at night and during the morning CAR?
9. Does the vagal nerve modulate mitochondrial humanin levels and release? If so, is this mediated via α7nAChR/SPMs/NF-κB/STAT3 and local melatonergic pathway induction in the course of inflammation resolution?
10. Are enteric glial cell mitochondria a significant source of humanin release into the gut microenvironment in the course of inflammation resolution, including in the course of vagal nerve activation? Does enteric glial cell released humanin increase the melatonergic pathway in other cells of the gut microenvironment? Is this relevant to the pathoetiology of Parkinson’s disease, where it is proposed that enteroendocrine cells are the major sources of α-synuclein? Enteric glial cells have direct contact and provide trophic support to enteroendocrine cells [118]. Would enteric glial cell humanin increase the mitochondrial melatonergic pathway in enteroendocrine cells, thereby increasing melatonin production, which suppresses α-synuclein production and oligomerization into more toxic forms?
Conceptualizations of how astrocyte, microglial, and neuronal mitochondria interact with each other require integration with wider circadian and systemic processes, as highlighted throughout. The dramatic but gradual decrease in pineal melatonin over aging changes the nature of how systemic mitochondria are dampened and reset at night in preparation for the coming day. This has implications for all aging-associated conditions across diverse organs and tissues, including neurodegenerative conditions mediated by the influence of circadian and systemic processes on mitochondrial function in astrocytes, especially but also in microglia, neurons, pericytes, oligodendrocytes, and endothelial cells. Alterations in dampening and resetting of mitochondria have implications for individual cells, their microenvironment interactions, and systemic systems. The induction of humanin and its interactions with the regulation of the mitochondrial melatonergic pathway forms a novel basis for understanding inflammation resolution systemically, including in the CNS interactions of glia and neurons. This provides a series of novel future research directions, the investigation of which should provide new treatment implications for conditions that are currently poorly managed.
AANAT: aralkylamine N-acetyltransferase
ACh: acetylcholine
ATP: adenosine triphosphate
BAG: Bcl-2 associated athanogene
CAR: cortisol awakening response
DRP1: dynamin-related protein 1
GH/IGF-1: growth hormone/insulin-like growth factor 1
GRs: glucocorticoid receptors
LETMD1: leucine zipper EF-hand containing transmembrane protein 1-domain-containing protein 1
LTP: long-term potentiation
MAMs: mitochondria-associated membranes
MDPs: mitochondria-derived peptides
MT1r: melatonin receptor 1
NAD+: nicotinamide adenine dinucleotide
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
NLRP3: NLR family pyrin domain containing 3
OXPHOS: oxidative phosphorylation
PDC: pyruvate dehydrogenase complex
pSTAT3Ser727: phosphorylated signal transducer and activator of transcription 3 at Serine727
pSTAT3Tyr705: phosphorylated signal transducer and activator of transcription 3 at Tyrosine705
ROS: reactive oxygen species
SCN: suprachiasmatic nucleus
SPMs: specialized pro-resolving mediators
STAT: signal transducer and activator of transcription
T2DM: type 2 diabetes mellitus
TFAM: mitochondrial transcription factor A
Trk: tropomyosin receptor kinase
α7nAChR: alpha-7 nicotinic acetylcholine receptor
GA: Conceptualization, Writing—original draft, Writing—review & editing. The author read and approved the submitted version.
The author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.