 Case Report
Case Report

Affiliation:
1Center for Biomedical and Pharmaceutical Research, Institute of Biology, Vietnam Academy of Science & Technology, Hanoi 10000, Vietnam
2Faculty of Science, Monash University, Melbourne 3800, Australia
ORCID: https://orcid.org/0009-0009-8969-2800
Affiliation:
3Hanoi Medical University Hospital, Hanoi 100000, Vietnam
Affiliation:
1Center for Biomedical and Pharmaceutical Research, Institute of Biology, Vietnam Academy of Science & Technology, Hanoi 10000, Vietnam
ORCID: https://orcid.org/0009-0002-4648-4226
Affiliation:
1Center for Biomedical and Pharmaceutical Research, Institute of Biology, Vietnam Academy of Science & Technology, Hanoi 10000, Vietnam
ORCID: https://orcid.org/0009-0002-9272-5967
Affiliation:
1Center for Biomedical and Pharmaceutical Research, Institute of Biology, Vietnam Academy of Science & Technology, Hanoi 10000, Vietnam
ORCID: https://orcid.org/0009-0008-0383-1092
Affiliation:
1Center for Biomedical and Pharmaceutical Research, Institute of Biology, Vietnam Academy of Science & Technology, Hanoi 10000, Vietnam
ORCID: https://orcid.org/0009-0005-5109-6115
Affiliation:
1Center for Biomedical and Pharmaceutical Research, Institute of Biology, Vietnam Academy of Science & Technology, Hanoi 10000, Vietnam
Email: nguyenhaiha@igr.ac.vn; nguyenhaiha78@gmail.com
ORCID: https://orcid.org/0000-0002-5431-5935
Explor Neurosci. 2025;4:1006102 DOI: https://doi.org/10.37349/en.2025.1006102
Received: May 09, 2025 Accepted: July 02, 2025 Published: July 27, 2025
Academic Editor: Yamei Tang, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, China
Neurofibromatosis type 1 (NF1) is a hereditary, autosomal dominant condition marked by the development of tumors along the nervous system due to uncontrolled cell proliferation. The current case reports a 31-year-old male patient diagnosed with NF1 with the involvement of bilateral pheochromocytomas and colonic inflammatory polyps/leiomyoma. A genetic profile was explored through whole-exome sequencing to identify pathogenic variants, and segregation analysis was subsequently performed in the patient’s family. Sequencing analysis revealed a novel heterozygous frameshift variant, NF1 c.7301dupA (p.S2435Efs*11), which was identified as the pathogenic variant in the patient. Additionally, two identified variants, PMS2 c.2T>C (p.M1T) and MUTYH c.850-2A>G, may be associated with colonic tumor conditions in the patient. These findings provide insights into the molecular etiology underlying this rare presentation of multiple tumors in a Vietnamese male and may contribute to improved treatment planning and patient management.
Neurofibromatosis type 1 (NF1) (OMIM 613113), historically known as von Recklinghausen disease, is a common autosomal dominant neurogenetic disorder and affects approximately 1 in 3,000 to 1 in 4,000 individuals worldwide [1]. NF1 manifests as a multisystem disorder with an extraordinarily variable clinical spectrum, making diagnosis and management highly complex. According to the National Institutes of Health (NIH) diagnostic criteria for NF1, a definitive diagnosis requires the presence of at least two major clinical features: (1) six or more café-au-lait macules (CALMs); (2) axillary or inguinal freckling; (3) two or more neurofibromas, or one plexiform neurofibroma; (4) optic glioma; (5) two or more Lisch nodules; (6) distinctive osseous lesions; or (7) a first-degree relative with a confirmed diagnosis of NF1 [2].
NF1 is caused by pathogenic variants in the NF1 gene, located on chromosome 17q11.2. This gene encodes neurofibromin, a tumor suppressor protein essential for regulating cellular proliferation and differentiation. Neurofibromin functions as a Ras-GTPase-activating protein (Ras-GAP), catalyzing the hydrolysis of active Ras-GTP to its inactive GDP-bound form, thereby suppressing the Ras-MAPK signaling pathway and preventing excessive cell proliferation [1]. Pathogenic variants in the NF1 gene disrupt neurofibromin’s function by altering its structure or reducing its expression. These disruptions lead to hyperactivation of the Ras-MAPK pathway, which drives the uncontrolled proliferation of nerve cells, Schwann cells, and perineurial cells, predisposing patients to tumorigenesis along nerve pathways [1, 3].
NF1 presents with age-dependent manifestations, often progressing from early childhood into adulthood. Initial signs include CALMs and axillary or inguinal freckling, typically evident around the age of 7, followed by the development of cutaneous neurofibromas and Lisch nodules during adolescence [4]. Optic pathway gliomas occur in 15–20% of patients, often before age 7, and may lead to visual impairment. Skeletal abnormalities, such as tibial pseudarthrosis, sphenoid wing dysplasia, and scoliosis, affect a subset of patients and can significantly impact the quality of life [4, 5]. Malignant complications, including malignant peripheral nerve sheath tumors (8–13%), pheochromocytomas (PHEOs), and gastrointestinal stromal tumors, tend to emerge later and contribute to morbidity and mortality [1]. Although numerous NF1 manifestations have been reported, molecular etiology evaluation of rare cases remains limited.
In this report, we present the first documented case of NF1 complicated by both malignant PHEO and colorectal tumors in a Vietnamese patient. Rare tumors such as PHEO, reported in approximately 0.1–5.7% of NF1 cases, along with gastrointestinal stromal tumors and colorectal cancer, represent uncommon manifestations of the disease [6, 7]. Consequently, routine early screening for these tumors is generally considered ineffective for the broad NF1 cohort [8]. The identification of genotype–phenotype correlations in our present cases can support personalized treatment for the patient. Furthermore, the genetic segregation findings may benefit the surveillance and risk assessment for the patient’s offspring.
A Vietnamese male, 31 years old, was admitted to the tertiary care unit of Hanoi Medical University Hospital with a primary complaint of persistent, severe pain in the left lumbar region for one month. The patient exhibited no symptoms of sympathetic hyperactivity, including palpitations, diaphoresis, headaches, or abnormalities in urinary or bowel function. The medical history of the patient shows that he has premature joint ossification, deformity of the left lower limb, and the onset of neurofibromas at approximately 15 years of age. He underwent the left adrenalectomy five years ago for PHEO of the adrenal gland scaled score (PASS) of 0 for PHEO. The patient’s family history has no report of consanguineous marriage or substance abuse.
Upon physical examination, the patient was alert and oriented, with normal vital signs, including blood pressure and heart rate. Musculoskeletal abnormalities were also recognized, including deformity of the left talocrural joint, shortening of the left lower limb relative to the right, and spinal deformities characterized by scoliosis and kyphosis. A dermatological examination revealed numerous neurofibromas of heterogeneous sizes, diffusely distributed across the body, with the largest measuring 33 cm in diameter. Additionally, multiple hyperpigmented macules of varying dimensions were observed on the skin (Figure 1A). Family members, including his parents and his spouse, had no history of neurofibroma or PHEO. However, his daughter (7-year-old) and son (5-year-old) were observed to have CALMs and neurofibromatosis. In addition, the patient’s child appeared to have a large Mongolian spot extending in the lumbosacral and gluteal regions (Figure 1B and C).
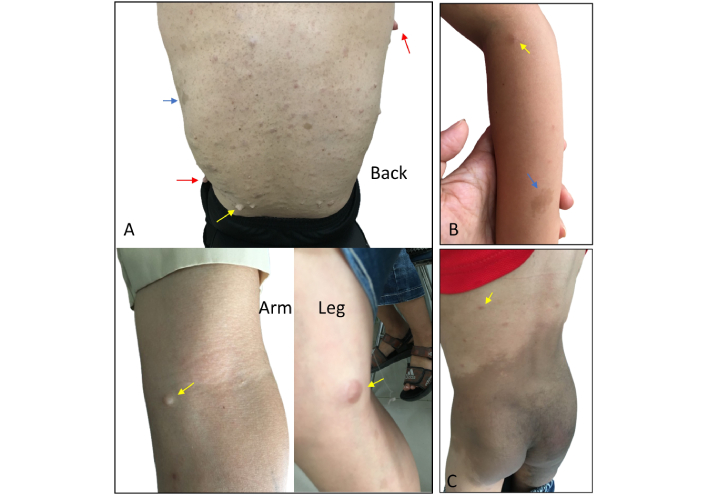
Dermatological manifestations observed in the patient and his children. (A) Cutaneous nodular neurofibromas (red arrows), subcutaneous plexiform neurofibromas (yellow arrows), and café-au-lait macules (blue arrows) on the torso of the 31-year-old male patient. (B) Café-au-lait macules (blue arrows) were observed on the left arm of the patient’s 7-year-old daughter with subcutaneous plexiform neurofibromas (yellow arrow). (C) An enormous Mongolian spot was observed on the lumbosacral region extending onto the gluteal area of the patient’s 5-year-old son with subcutaneous plexiform neurofibromas (yellow arrow)
Biochemical analyses, including blood and urine (Table 1), revealed several abnormalities. Plasma catecholamine levels were significantly elevated, with dopamine nearly twice the upper limit of the reference range (178.00 pg/mL; reference 0.00–100.00 pg/mL), adrenaline exceeding the reference range by more than twice folds (1,436.33 pg/mL; reference 0.00–600.00 pg/mL), and noradrenaline more than fivefold the upper limit (565.67 pg/mL; reference 0.00–100.00 pg/mL). Adrenocorticotropic hormone (ACTH) levels were also elevated at 96.80 pg/mL, exceeding the upper reference limit (63.30 pg/mL) by over 50%. Urinary catecholamine analysis revealed elevated dopamine levels (210.20 µg/24 h; reference range 0.00–20.00 µg/24 h), exceeding the upper limit by more than tenfold. An abdominal ultrasound was then performed, identifying a right adrenal mass consistent with PHEO. A CT-guided biopsy of the right adrenal gland further revealed a hypercellular tumor characterized by darkly stained enlarged nuclei and frequent mitotic figures (5 per 10 high-power fields), including atypical mitoses. Histopathological examination of the resected adrenal gland supported the suspicion of malignant PHEO with a PASS of 6, indicating high malignant potential. Additionally, endoscopic examinations of the gastrointestinal tract revealed gastritis, and colonoscopy showed colorectal tumors with multiple colonic polyps, including one at the hepatic flexure, and another located 18 cm from the anal verge. Biopsy analysis of these lesions confirmed colonic inflammatory polyps and leiomyoma.
Preoperative laboratory testing prior to right adrenalectomy (age at surgery: 31-year-old)
| Laboratory test | Values | Reference range | Unit |
|---|---|---|---|
| Blood laboratory tests | |||
| Catecholamine (ELISA) | |||
| Dopamine | 178.00 | 0.00–100.00 | pg/mL |
| Adrenaline | 1,436.33 | 0.00–600.00 | pg/mL |
| Noradrenaline | 565.67 | 0.00–100.00 | pg/mL |
| Endocrinology—hormones | |||
| ACTH | 96.80 | 7.20–63.30 | pg/mL |
| Cortisol | 368.00 | 138.00–635.00 | nmol/L |
| Urine laboratory tests | |||
| 24-h urine catecholamine (ELISA) | |||
| Dopamine | 210.20 | 0.00–20.00 | µg/24 h |
| Adrenaline | 185.24 | 0.00–600.00 | µg/24 h |
| Noradrenaline | 325.46 | 0.00–90.00 | µg/24 h |
Laparoscopic right adrenalectomy was performed to remove the entire affected adrenal gland. After the adrenalectomy, the ACTH levels significantly reduced to 2.55 pg/mL, which was over 60% lower than the lower limit of the reference range (7.20 pg/mL) (Table 2). The patient had an uneventful recovery and was discharged in stable condition two days after the procedure.
Postoperative laboratory testing following right adrenalectomy
| Laboratory test | Values | Reference range | Unit |
|---|---|---|---|
| Blood laboratory tests | |||
| Endocrinology—hormones | |||
| T3 (triiodothyronine) | 0.63 | 0.98–2.34 | nmol/L |
| FT4 (free thyroxine) | 13.86 | 9.01–19.05 | pmol/L |
| TSH (thyroid stimulating hormone) | 0.63 | 0.35–4.94 | µIU/mL |
| ACTH (adrenocorticotropic hormone) | 2.55 | 7.20–63.30 | pg/mL |
| Cortisol | 37.40 | 138.00–635.00Morning: 133.00–537.00Afternoon: 68.20–327.00 | nmol/L |
Peripheral blood samples from the patient and his family members were collected following informed consent for genetic analysis, prompted by a clinical suspicion of NF1. The whole-exome sequencing (WES) was performed on the patient’s sample, and the resulting data were processed and filtered using a cancer gene panel. Three heterozygous candidate variants were identified: (1) c.7301dupA (p.S2435Efs*11) in the NF1 gene (NM_001042492); (2) c.2T>C (p.M1T) in the PMS2 gene (NM_000535); and (3) c.850-2A>G in the MUTYH gene (NM_001048174) (Table 3). The NF1 c.7301dupA (p.S2435Efs*11) is a frameshift variant leading to a premature stop codon after 11 amino acid residues. This variant is novel because it has not been reported in any public database. The PMS2 c.2T>C variant results in the loss of the translation start codon of the PMS2 gene. According to the ClinVar database, it was classified as a pathogenic or likely pathogenic variant associated with hereditary nonpolyposis colorectal cancer type 4. The MUTYH c.850-2A>G is a splice-site variant, reported in the ClinVar as “conflicting classifications of pathogenicity” to the familial adenomatous polyposis 2 condition.
The candidate variants of the patient detected by WES
| Gene | Nucleotide change (amino acid change) | Consequence | ClinVar classification | Zygosity | Related diseases |
|---|---|---|---|---|---|
| NF1 | c.7301dupA (p.S2435Efs*11) | Frameshift | - | HET | Neurofibromatosis type 1, autosomal dominant |
| PMS2 | c.2T>C (p.M1T) | Missense | Pathogenic/likely pathogenic | HET | Lynch syndrome, autosomal dominant |
| MUTYH | c.850-2A>G | Splicing | Conflicting classifications of pathogenicity | HET | Familial adenomatous polyposis 2, autosomal recessive |
The reference sequences: MUTYH: NM_001048174; NF1: NM_001042492; PMS2: NM_000535 according to NCBI. -: not available; WES: whole-exome sequencing; HET: heterozygous
The appearance of three variants and their segregation in the patient’s family were examined by Sanger sequencing (Figure 2). As a result, the NF1 c.7301dupA was absent in both parents of the patient, suggesting it was a de novo variant. Additionally, this variant was transmitted to both his daughter and son. Both PMS2 c.2T>C and MUTYH c.850-2A>G variants were inherited from the patient’s father. The PMS2 c.2T>C variant was transmitted to both children, while the MUTYH c.850-2A>G variant was found only in the daughter. A notable discrepancy in the zygosity of the PMS2 c.2T>C variant was observed between WES and Sanger sequencing, with WES reporting a heterozygous state and Sanger sequencing indicating homozygosity. Figure 3 shows the timeline outlining the key clinical events in the case report.
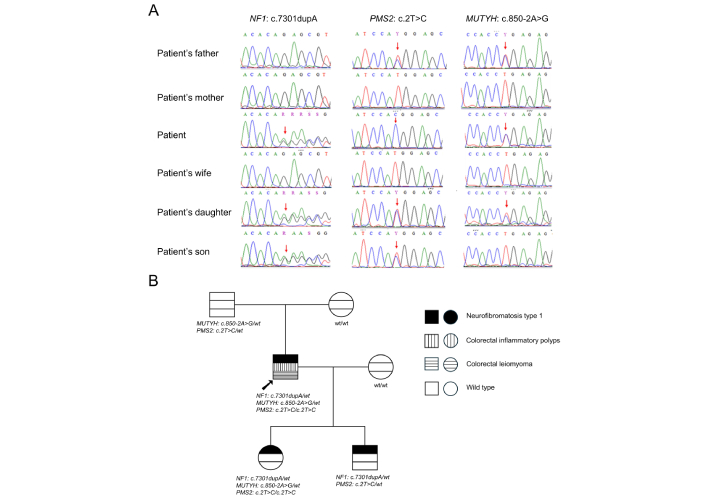
Sanger validation of variants and their segregation within the proband’s pedigree. (A) Sanger sequencing chromatograms of the patient’s family members. The red arrows showed the positions of mutations. (B) The family pedigree follows the segregation of the variants. The black arrow indicates the proband. wt: wild type
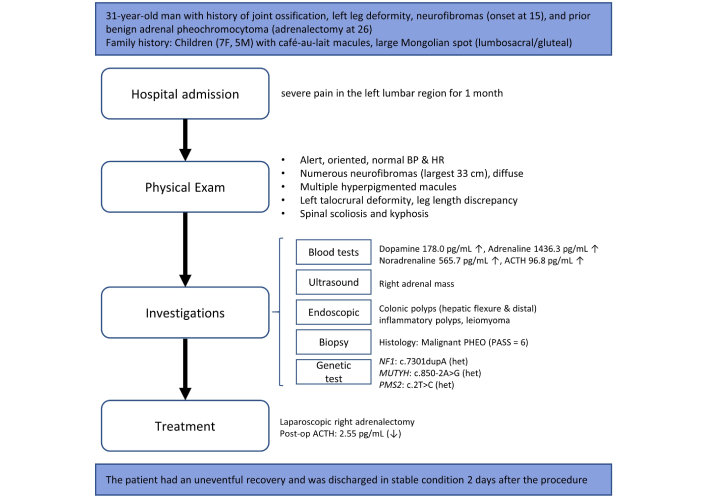
Case report’s timeline. ↑: elevated; ↓: decreased; BP: blood pressure; HR: heart rate; ACTH: adrenocorticotropic hormone; PHEO: pheochromocytoma; PASS: PHEO of the adrenal gland scaled score; het: heterozygous
Neurofibromatosis is a relatively common autosomal dominant disorder and manifests in a wide range of clinical aspects. Cases of NF1 associated with PHEOs have been occasionally reported, with a prevalence ranging from approximately 0.1–5.7% [9]. PHEOs are uncommon catecholamine-producing neoplasms that originate from chromaffin cells of the adrenal medulla, with an estimated incidence in the general population is approximately 1 per 200,000 individuals [10]. The presence of PHEOs in patients with NF1 around adulthood (average age at 39 years old), with only 15% present bilateral adrenal tumors [9]. Although colonic leiomyomas have not been reported in patients with NF1, a limited number of colonic polypoid lesion types have been documented [11, 12].
The frameshift variant in exon 51 of the NF1 gene introduces a premature termination codon that is predicted to trigger nonsense-mediated decay and result in loss of neurofibromin function. Notably, this variant has not been reported in ClinVar, LOVD, or any other public database, underscoring its novelty. According to the ACMG/AMP 2015 guidelines [13], the variant meets the criteria for pathogenic (PVS1, PS2, PM2, and PP4). The truncation of the neurofibromin protein’s C-terminal domain could disrupt its interaction with syndecans due to the absence of the syndecan-binding region. This diminishes Ras signaling regulation and contributes to NF1 clinical features, such as tumors and neurological deficits [14]. Additionally, interactions with focal adhesion kinase and collapsin response mediator protein 2 could also be disrupted, contributing to the more severe pathological conditions associated with NF1.
NF1 colonic involvement is considered a rare clinical manifestation with limited reports [15]. In this case, the patient incidentally discovered colonic inflammatory polyps and a leiomyoma during a colonoscopy, without any symptoms such as bleeding, volvulus, or intussusception. Through Sanger validation, our patient was confirmed to have two variants associated with syndromes marked by the development of colorectal adenomas: homozygous PMS2 c.2T>C and heterozygous MUTYH c.850-2A>G. By segregating PMS2 c.2T>C in Sanger validation, the patient is the only family member found to be homozygous for the variant. Two possibilities have been proposed: (1) the patient may carry an additional copy number variant involving PMS2, such as a whole-gene or partial-gene deletion; or (2) preferential duplication of the mutant allele may have occurred during cell division. Multiplex ligation-dependent probe amplification is planned for future testing to further investigate these hypotheses.
PMS2 encodes a crucial component of the DNA mismatch repair (MMR) system, and pathogenic variants in this gene have been associated with Lynch syndrome as well as clinical features overlapping with NF1 [16]. Pathogenic variants in PMS2 disrupt the MMR system, leading to uncontrolled cellular proliferation and an increased risk of colorectal cancer [17]. Possession of homozygous pathogenic PMS2 c.2T>C or loss of heterozygous probably causes the phenotypes of colorectal tumors presented in the patient. Besides, MUTYH c.850-2A>G has been reported in several types of cancer, including colorectal cancer [18], but also in unaffected individuals (in the present case, the patient’s father), causing the conflict in classification. The pathogenic significance of heterozygous MUTYH variants remains uncertain compared to biallelic mutations. However, the co-occurrence of homozygous PMS2 c.2T>C and heterozygous MUTYH c.850-2A>G may collectively contribute to the development of these colonic clinical features in the patient.
Since the NF1 c.7301dupA variant is transmitted to the patient’s children, they have begun to exhibit typical clinical features of NF1, including subcutaneous plexiform neurofibromas and CALMs, observed in the daughter’s arm and the son’s back. According to the diagnostic criteria established by the NIH, both children fulfill the clinical diagnosis of NF1. Based on the patient’s current condition, the ongoing monitoring and further clinical evaluations of his children are strongly recommended to assess potential tumor development or other NF1-related complications. In the present case, the patient underwent two surgical procedures to remove PHEOs and colorectal polyps, and leiomyomas without complications, and brought the level of ACTH into a manageable range. Patients with NF1-PHEO tend to present cardiovascular remodeling with heart palpitations and hypertension, and persist even after tumor removal, underscoring the importance of long-term cardiovascular monitoring [9]. The continuous growth of cutaneous and subcutaneous tumors in NF1 can cause a significant psychosocial burden, as individuals may feel self-conscious about their appearance, regardless of age or gender. In such cases, there are currently no effective pharmacological treatments, and surgical removal remains the only available option.
In conclusion, this rare case of NF1 with concurrent PHEOs and colonic involvement illustrates the association between genotype and phenotype in a complex manifestation of NF1. Genetic screening supports the underlying genetic etiology of colonic involvement and provides a basis for establishing a surveillance strategy for the patient and his offspring.
ACTH: adrenocorticotropic hormone
CALMs: café-au-lait macules
MMR: mismatch repair
NF1: neurofibromatosis type 1
NIH: National Institutes of Health
PASS: pheochromocytoma of the adrenal gland scaled score
PHEOs: pheochromocytomas
WES: whole-exome sequencing
We sincerely thank the patient and his family for their participation in this study.
HTTL: Visualization, Writing—original draft. HTT: Investigation. LKN: Investigation. LTD: Investigation. QNKL: Investigation. THNH: Visualization, Writing—review & editing. HHN: Conceptualization, Writing—review & editing, Supervision. The submitted version has been read and approved by all authors.
The authors declare that they have no conflicts of interest.
The study was approved by the Institutional Review Board of the Institute of Genome Research, Vietnam Academy of Science and Technology (No. 4-2024/NCHG-HĐĐĐ). This study complies with the Declaration of Helsinki (2013 version).
Informed consent to participate in the study was obtained from the participants and the parents of the participants.
Informed consent to publication in the study was obtained from the participants and their parents.
The datasets supporting the study can be provided by the corresponding author upon reasonable request.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 3143
Download: 50
Times Cited: 0
