 Review
Review

Affiliation:
1Department of Cardiovascular and Metabolic Sciences, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA
2Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, OH 44195, USA
3Center for Microbiome & Human Health, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA
Email: wangz2@ccf.org
ORCID: https://orcid.org/0000-0002-6455-8228
Affiliation:
4Department of Neurology, Neurological Institute, Cleveland Clinic, Cleveland, OH 44195, USA
ORCID: https://orcid.org/0000-0002-8345-6470
Affiliation:
1Department of Cardiovascular and Metabolic Sciences, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA
2Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, OH 44195, USA
3Center for Microbiome & Human Health, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA
5Department of Cardiovascular Medicine, Heart, Vascular and Thoracic Institute, Cleveland Clinic, Cleveland, OH 44195, USA
Explor Med. 2025;6:1001339 DOI: https://doi.org/10.37349/emed.2025.1001339
Received: January 16, 2025 Accepted: May 23, 2025 Published: June 23, 2025
Academic Editor: Undurti N. Das, UND Life Sciences, USA
The article belongs to the special issue Gut Microbiota Derived Metabolites and Chronic Inflammatory Diseases
Nutrients containing a trimethylamine (TMA) moiety in their structure can be metabolized by the gut microbiota through enzymatic cleavage of the C-N bond, producing TMA. In the liver, TMA is subsequently oxidized to trimethylamine N-oxide (TMAO) by flavin monooxygenases (FMOs). TMAO exerts pro-atherogenic and pro-inflammatory effects that contribute mechanistically to several chronic inflammatory diseases including cardiovascular disease, chronic kidney disease, obesity, non-alcoholic fatty liver disease, and neurodegenerative diseases. Targeting this metaorganismal pathway may offer substantial health benefits in the prevention and treatment of chronic inflammatory conditions.
Trimethylamine N-oxide (TMAO) is a small organic compound derived from gut microbial metabolism of dietary nutrients. These nutrients include phosphatidylcholine, L-glycerylphosphorylcholine, choline, betaine, Nε-trimethyllysine, δ-valerobetaine, carnitine, γ-butyrobetaine and ergothioneine [1–8]. Initially, TMAO was believed to be a simple metabolic waste product with no significant biological activity [9, 10]. However, subsequent studies have revealed TMAO’s role as a chemical chaperone [11].
TMAO has been implicated in the development and progression of chronic inflammatory diseases, including atherosclerosis, chronic kidney disease, obesity, non-alcoholic fatty liver disease (NAFLD), Alzheimer’s disease and cancer [2, 12–16]. However, the exact mechanisms by which TMAO promotes inflammation are still under investigation.
In this review, we summarize the roles of the TMAO metaorganismal pathway in chronic inflammatory diseases and discuss the potential of targeting this pathway for the prevention and treatment of chronic diseases.
The TMAO metaorganismal pathway [17], involves the cleavage of the C-N bond by gut microbial trimethylamine (TMA) lyase in dietary precursors containing TMA [18–20]. TMA is then oxidized to TMAO through the catalysis of hepatic flavin monooxygenases (FMOs) [21] (Figure 1).
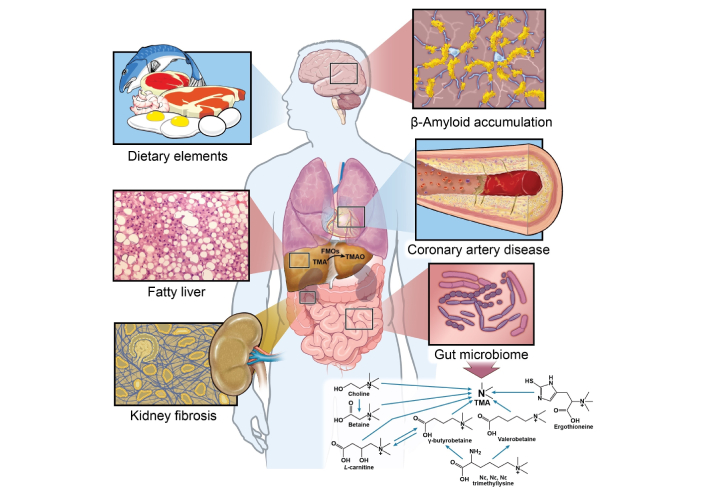
TMAO metaorganismal pathway and chronic inflammatory disease. FMOs: flavin monooxygenases; TMA: trimethylamine; TMAO: trimethylamine N-oxide. Illustration by David Schumick, BS, CMI. Reprinted with the permission of the Cleveland Clinic Enterprise Creative Services. © 2025, All Rights Reserved.
The precursors of TMAO are present in eggs, meats, and dairy products, dietary sources commonly associated with a western diet and cardiovascular disease (CVD) [22, 23]. In fact, vegans and vegetarians have less fasting plasma TMAO and chronic dietary supplementation of carnitine or red meat can induce the gut microbiota to produce more proatherogenic TMAO when compared to individuals that have a diet low in carnitine [20, 24]. Trillions of microbes reside in the human gut. Certain microbes have been identified to express TMA lyases, including the commonly reported choline TMA lyase (cutC/D), carnitine Rieske oxygenase/reductase (cntA/B), betaine reductase, TMAO reductase (TMAOase), ergothionase, and γ-butyrobetaine utilizing enzyme (Gbu) [18–20, 22, 23]. The average relative abundance of bacteria capable of producing TMA is estimated to be less than 1.2% [25]. However, it is important to note that not all bacteria in the human gut encode TMA lyase [25]. Based on the UniProt database, there are several hundred bacterial strains encoding cutC/D. In the human gut the ten most abundant bacterial strains encoding cutC/D include Collinsella tanakaei YIT 12063, Clostridium botulinum Eklund 17B, Alkaliphilus metalliredigens QYMF, Enterococcus phaeniculicola BAA-412, Desulfovibrio alaskensis G20, Enterococcus asini 700915, Clostridium hathewayi CAG:224, Clostridium tetani Massachusetts, Desulfovibrio hydrothermalis AM13, Enterococcus moraviensis BAA-383 [17]. Although cntA/B is found to be expressed in some bacterial strains [18], no single strain isolated from the human gut has been shown to catalyze the conversion of carnitine to TMA [20]. This may be because cntA/B functions as an oxygenase, which is limited by the strict anaerobic conditions of the human gut. At least two bacterium strains are required for the process. The catabolism of carnitine to TMA involves two steps. In the first step conversion of carnitine to γ-butyrobetaine occurs in gut bacteria containing the CaiTABCDE gene cluster such as Escherichia coli, Enterobacter cloacae, Klebsiella pneumoniae, and Salmonella enterica. Next, the conversion of γ-butyrobetaine to TMA occurs via a second bacterium strain containing the GbuABCDEF gene cluster such as Emergencia timonensis SN18 [20]. No single bacterium strain isolated from human gut contain the two gene clusters [20, 26]. Although yeaW/X is highly homologous to cntA/B and has been considered the same enzyme in some papers, it has a broader substrate usage, which includes choline and betaine [5, 27].
Other potential precursors for microbial TMA production include betaine, ergothioneine, and sinapine. These nutrients are abundant in some vegetables, along with their corresponding enzymes—betaine reductase and ergothionase—which have been identified in various bacterial taxa, including Clostridium XIVa, Dorea, and Escherichia coli [23, 25, 28].
TMAO biosynthesis consists of two steps. In the first step, TMA is produced from dietary precursor catalyzed by microbial TMA lyase, a rate limiting step. In the second step, TMA is oxidized to TMAO, by FMOs. Initially, only hepatic FMO3 was known to perform this function. However, there are five types of FMOs expressed in the human liver [29]. We successfully cloned FMO1, FMO2, FMO3, FMO4, and FMO5 from HepG2 or Hep3B cells and constructed them in pCG vectors. These constructs were then expressed in 293T cells. Our findings indicate that, in addition to FMO3, both FMO1 and FMO2 can also catalyze the enzymatic oxidation of TMA to TMAO. Both the specific activity and mRNA expression of FMO3 are significantly higher than FMO1 and FMO2 in human liver. Thus, FMO3 is the dominant enzyme involved in the oxidation of TMA to TMAO in humans [21].
FMO3, the predominant FMO expressed in the liver, has also been detected in other tissues such as the lung, aorta and adipose as previously reported [21, 30]. This suggests that multiple tissues might participate in the oxidation TMA to TMAO.
According to the World Health Organization (WHO), CVD is the leading cause of death worldwide and accounts for approximately one-third of all deaths annually. CVD encompasses various conditions, including coronary artery disease, stroke, arrythmias, heart failure, and peripheral artery disease.
Using untargeted metabolomics analysis, we identified a novel metabolic pathway involving the gut microbiota dependent metabolism of dietary phosphatidylcholine to TMA [2]. Gut microbes cleave the C-N bond in choline using TMA lyase to produce TMA, and TMA is rapidly oxidized by hepatic FMOs to TMAO. TMAO exhibits multiple pro-atherogenic properties resulting in the initiation, progression, and sequalae of atherosclerosis [2, 19].
Several large clinical cohort studies have shown that TMAO and its precursors are independently associated with prevalent CVD and incident major adverse cardiac events (MACE) [2–4, 30–33]. Although choline, betaine and carnitine can predict the risk of future MACE, this association appears to be primarily driven by the gut microbiota-derived metabolite TMAO [3, 4].
Supplementation of dietary precursors choline, carnitine, γ-butyrobetaine, and L-glycerophosphorylcholine in atherosclerotic susceptible mice increases TMA/TMAO and atherosclerotic plaque formation in a gut microbiota dependent manner [1, 2, 4, 5]. This effect is abolished upon suppression of the gut microbiota using broad-spectrum antibiotics [1, 2, 4, 5]. There is a strong positive correlation between circulating TMAO levels and the area of atherosclerotic plaques in these mice [2]. Additionally, direct dietary supplementation of TMAO increases atherosclerotic plaque formation in atherosclerotic susceptible mice suggesting a direct causal role of TMAO in the pathogenesis of atherosclerosis [2].
Our group and others have described multiple mechanisms linking TMAO to the pathogenesis of atherosclerosis. First, TMAO can alter cholesterol metabolism by simultaneously increasing cholesterol deposition and decreasing reverse cholesterol transport [2, 4]. TMAO increases the expression of scavenger receptors, CD36, SR-A1, and LOX-1 in macrophages, resulting in increased foam cell formation, a key mediator of atherosclerosis pathogenesis [2, 34, 35]. TMAO disrupts reverse cholesterol transport by 1) down-regulating the expression of C7A1 and C27A1, two key enzymes responsible for bile acid biosynthesis; 2) down-regulating the expression of several bile acid transporters in liver tissue; and 3) down-regulating the expression of ABCA1 and ABCG1 in macrophages, potentially due to suppression of liver X receptor (LXR) activity by the TMA/FMO3/TMAO axis [4, 36, 37]. The net effect is reduced cholesterol elimination from the body and an increase in atherosclerosis. TMAO has been shown to prime and activate the NLRP3 inflammasome in vascular endothelial cells via NF-κB signaling [38, 39].
The TMAO metaorganismal pathway may influence the severity and functional outcomes after stroke. Laboratory research has shown that stroke severity is transmissible via TMA-producing gut microbial transplantation [40]. TMAO activates platelet through mobilization of Ca2+ from intracellular calcium stores, increasing cytosolic Ca2+ and promoting thrombus formation [41]. In addition, TMAO can induce aortic endothelial cells to express tissue factor (TF), which binds and activates factor VIIa and initiates the thrombosis cascade, leading to thrombin activation. The TF-VIIa complex enhances platelet sensitivity to agonists such as adenosine diphosphate (ADP), thrombin, and collagen. Furthermore, the TF-VIIa complex upregulates endothelial cell expression of vascular cellular adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), promoting monocyte recruitment to the aorta. Together, these actions constitute the molecular mechanisms underlying TMAO’s pro-thrombotic effects [42].
Studies have shown that patients with heart failure exhibit significantly elevated levels of circulating TMAO compared to healthy individuals [32, 43, 44].
TMAO contributes to the pathogenesis of heart failure through multiple direct and indirect mechanisms. As mentioned above, TMAO promotes atherosclerosis contributing to conditions such as coronary artery disease, a common cause of heart failure. Second, TMAO impairs endothelial function and the production of the vasodilator nitric oxide (NO) resulting in constricted blood vessels and hypertension, another major cause of heart failure [45, 46]. Third, TMAO increases circulating pro-inflammatory chemokines and cytokines, which are known to lead to the progression of heart failure [1, 47, 48]. Fourth, TMAO promotes fibrosis in the heart tissue, which can stiffen the heart, making it harder for the heart to pump effectively [49, 50]. Fifth, TMAO increases oxidative stress, disrupts mitochondrial function, and promotes the processes of vascular injury and tissue damage that contribute to heart failure [51–53].
In animal models, reducing circulating TMAO has been demonstrated to reverse heart failure [54, 55]. Consequently, reducing circulating TMAO has emerged as a potential therapeutic approach for heart failure [56].
Atrial fibrillation (AF) is a cardiac arrythmia associated with significant morbidity and mortality mediated primarily through the sequalae of stroke and heart failure [57, 58]. Animal studies have demonstrated that local injection of TMAO can exacerbate AF progression in a rapid atrial pacing (RAP)-induced AF model. This exacerbation is characterized by heightened instability in atrial electrophysiology [59]. Metagenomics sequence data confirm an enrichment of TMA-producing bacteria in the gut of patients with AF [60]. TMAO has also linked to right atrium fibrosis and AF in patients undergoing cardiac surgery [61]. Mechanistically, TMAO may contribute to electrophysiological dysfunction by activating the protein kinase R-like endoplasmic reticulum kinase (PERK)/IRE1α axis, leading to increased atrial remodeling and subsequent arrhythmogenesis [62]. A systematic review and meta-analysis established a significant, dose-dependent relationship between elevated circulating TMAO levels and increased risk of AF [63].
Aortopathy includes aortic dissection, characterized by a tear of the aorta, and aortic aneurysms which are characterized by a bulge or swelling of the aorta. Aortopathies can present as life threatening emergencies, causing strokes, or rupture leading to rapid physiological decline and death [64]. A recent study, comparing plasma TMAO levels in 253 aortic dissection patients with 98 healthy subjects, revealed a significant elevation of TMAO in aortic dissection patients compared to healthy controls [65]. Furthermore, experimental studies have demonstrated that TMAO exacerbates the progression of aortic dissection by inducing endothelial dysfunction and inflammation, primarily through activation of the NF-κB signaling pathway [38, 65]. Similarly, patients with aortic aneurysm exhibit significantly elevated circulating TMAO when compared to healthy controls. LDLr-null mice fed a choline-supplemented chow diet exhibit aggravated aortic aneurysm pathogenesis in a gut microbiota-dependent manner mediated by the generation of TMAO [66].
Notably TMAO concentrations have also been linked to hypertension, a major risk factors for aortopathy [67]. Each 1 μM increase in circulating TMAO levels is associated with a 1.014% increase in the risk of hypertension [68]. Mechanistically, TMAO promotes angiotensin (Ang) II-induced vasoconstriction, thereby facilitating the development of Ang II-induced hypertension [69].
Obesity is a chronic inflammatory disease and is associated with the up-regulation of pro-inflammatory molecules such as interleukin-1 beta (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α) [70]. Obesity contributes to the development of metabolic diseases and other complications, including insulin resistance, metabolic syndrome, CVD, and cognitive impairment [71–74].
TMAO has been implicated in the pathogenesis of obesity by initiating pro-inflammatory cascades [38]. TMAO promotes macrophage infiltration and activation within adipose tissue, driving a shift in macrophage phenotype toward the pro-inflammatory M1 state, contributing to adipose tissue dysfunction, a key hallmark of obesity-related metabolic dysfunction [30, 75]. Supporting its role in fat regulation, studies in animal models have shown that FMO3-knockout mice exhibit reductions in fat mass, including visceral and subcutaneous fat [30]. Furthermore, a small-molecule inhibitor of microbial choline TMA lyase has been reported to protect mice against obesity. Interestingly, the increased relative abundance of the probiotic Akkermansia may also mediate this effect [76].
The clinical relevance of TMAO in obesity has been reported. A study involving 50 obese children aged 4 to 18 years, along with 20 control subjects, reported significantly higher serum TMAO levels in the obese group compared to controls. Furthermore, TMAO levels were positively correlated with body mass index (BMI), waist circumference, and waist-to-height ratio [77]. Similarly, a study involving 137 adults demonstrated that serum TMAO levels were positively associated with obesity-related factors, including BMI, visceral adiposity index (VAI), and fatty liver index (FLI) [78].
Patients with chronic kidney disease exhibit significantly elevated levels of TMAO, and the accumulation of circulating TMAO exacerbates glomerular fibrosis [12, 79]. The molecular mechanisms by which TMAO exacerbates renal insufficiency, include the activation of inflammatory pathways, induction of oxidative stress, SMAD signaling, and impairment of endothelial cell function [12, 38].
Targeting the TMAO metaorganismal pathway by inhibiting microbial choline TMA lyase activity shows potential benefits in slowing the progression of chronic kidney disease. In apoE–/– mice fed an adenine-rich diet, chronic kidney disease and cardiac hypertrophy were observed alongside elevated circulating TMAO levels. Treatment with the choline TMA lyase inhibitor, iodomethylcholine, significantly reduced circulating TMAO and TMAO-induced kidney disease and cardiac hypertrophy [80].
NAFLD is a common chronic liver condition characterized by excessive fat accumulation (steatosis) in the liver in the absence of alcohol consumption or other secondary causes [81–83]. NAFLD affects approximately 25% of the global adult population [82].
TMAO is an oxidative product of TMA in the liver. A systematic meta-analysis including 7 studies with 7,583 individuals revealed that patients with NAFLD have elevated levels of circulating TMAO [84].
Several studies have reported that directly supplementing TMAO into the diet of mouse and zebrafish models promotes NAFLD [14, 85]. Further mechanistic investigations demonstrated that TMAO induced NAFLD includes the activation of the PERK signaling pathway (Figure 2) and inhibition of the farnesoid X receptor signaling [14, 85].
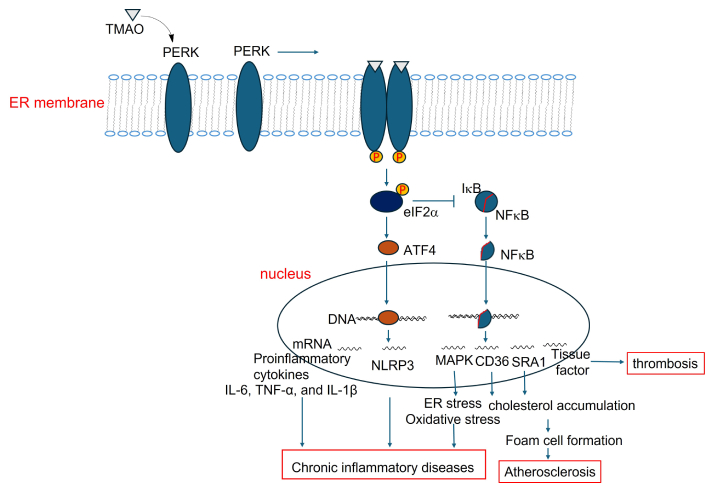
PERK signaling mediates TMAO-driven pathways contributing to chronic inflammation. TMAO: trimethylamine N-oxide; ER: endoplasmic reticulum; IL-6: interleukin-6; TNF-α: tumor necrosis factor-alpha; PERK: protein kinase R-like endoplasmic reticulum kinase
Neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease (PD), Huntington’s disease, amyotrophic lateral sclerosis (ALS), frontotemporal dementia, Lewy body dementia, and multiple sclerosis (MS) involve progressive degeneration and functional loss of neurons in the brain or spinal cord.
Alzheimer’s disease is the most common cause of dementia and is a syndrome characterized by a decline in cognitive function severe enough to interfere with daily life. The involvement of TMAO in Alzheimer's disease has been widely investigated. A study investigating TMAO levels in cerebrospinal fluid (CSF) in 40 people with Alzheimer’s disease and 335 cognitively-intact individuals (total n = 410) revealed a significant elevation in CSF TMAO levels among those with Alzheimer’s disease compared to cognitively-intact participants. Moreover, heightened CSF TMAO levels correlated with biomarkers indicative of Alzheimer’s pathology, including phosphorylated tau and the phosphorylated tau/Aβ ratio [86]. Further investigation has solidified our understanding of the molecular mechanisms linking TMAO to Alzheimer’s disease [87]. TMAO has been shown to trigger inflammation and senescence in endothelial cells, causing barrier disruption and subsequent interaction with neurons. Functioning as a chemical chaperone, TMAO modifies the conformation of Tau C-terminal, promoting microtubule formation and Tau aggregation in the presence of heparin. This results in the formation of amyloid-like structures. Additionally, TMAO upregulates clusterin expression, leading to increased levels of β-secretase and subsequent deposition of Aβ-amyloid between neurons [87].
PD is characterized by the progressive loss of dopamine-producing neurons in the substantia nigra of the brain. Dopamine is a neurotransmitter that plays a crucial role in coordinating smooth and controlled muscle movements [88, 89]. In recent decades, there has been increasing recognition of the role of the gut-brain axis in modulating dopamine levels [90]. A study of 60 patients with PD showed elevated circulating TMAO levels compared to healthy controls [91]. With a mean follow up of 4.3 years, higher baseline TMAO levels were associated with an increased risk for motor progression and a trend for cognitive deterioration [91]. A Mendelian randomization study indicated that circulating TMAO is causally linked to the progression of PD [92]. Using a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mouse model, investigators found that elevated circulating TMAO had adverse effects on motor capacity, striatal neurotransmitters, and neuroinflammation in both the striatum and hippocampus of the mice [93]. TMAO was found to promote the metabolism of striatal dopamine and aggravate neuroinflammation and regulate glial cell polarization [94].
The relationship between TMAO and other neurodegenerative disorders has not been well studied. Conclusive associations are complicated by the fact that an important substrate of TMAO, choline, is an essential nutrient that is required for normal development of the brain [95]. A study in a Chinese population found that patients with ALS, a progressive neurodegenerative disease that affects nerve cells in the brain and spinal cord, had lower levels of TMAO, choline, and γ-butyrobetaine, but higher levels of carnitine and betaine [96]. A recent study involving 2,517 participants showed that circulating TMAO is not associated with incident dementia. Instead, higher choline levels were associated with a poor prognosis [97]. Dietary supplementation of citicoline, also known as cytidine-5-diphosphocholine, another choline source, for 12 weeks improved overall memory performance, especially episodic memory, in healthy older populations with age-associated memory impairment [98]. Further research is needed to unveil the differential pathways through which TMAO and dietary choline function to regulate neurodegeneration and inflammation.
In addition to the above chronic inflammatory diseases, emerging research is now exploring the potential links between TMAO and cancer. Preliminary data suggest that elevated circulating TMAO is associated with an increased risk of colorectal, gastric, liver, pancreatic, breast, and prostate cancers [99–106].
A Genome Wide Association Study reveals a genetic link between TMAO and colorectal cancer, uncovering a total of 91 common genetic pathways associated with both [99]. A correlation between circulating TMAO and a higher risk of distal colorectal cancer was found in the prostate, lung, colorectal, ovarian cancer screening trial cohort [107]. TMAO has been shown to exert oncogenic effects in colorectal cancer by promoting cell proliferation and angiogenesis, as demonstrated through in vitro cell culture experiments and studies using tumor-bearing mouse models [16].
TMAO has been implicated in the progression of NAFLD, a cause of non-alcoholic steatohepatitis, liver fibrosis, cirrhosis, and ultimately hepatocellular carcinoma (HCC) [14, 108]. Worsening liver fibrosis is a risk factor for HCC development. Elevated TMAO levels have been linked with worsening liver fibrosis [109] by increasing the expression of miR-34a and miR-22 and activating PERK signaling [85, 110].
In addition to the above-mentioned diseases, the TMAO meta-organismal pathway is involved in the pathogenesis of other systemic inflammation including systemic lupus erythematosus, rheumatoid arthritis, psoriasis, osteoporosis, and gallbladder stones [111–116].
Circulating TMAO can enter cells via specific transporters, including organic cation transporter 2 (OCT2) and endothelial TMAO transporter 1 (ETT1) [117, 118]. As a bioactive metabolite, TMAO can activate various intracellular signaling cascades, including the MAPK and NF-κB pathways, the NLRP3 inflammasome, and PERK signaling as well—mechanisms closely linked to chronic inflammation (Figures 2, 3, and 4).
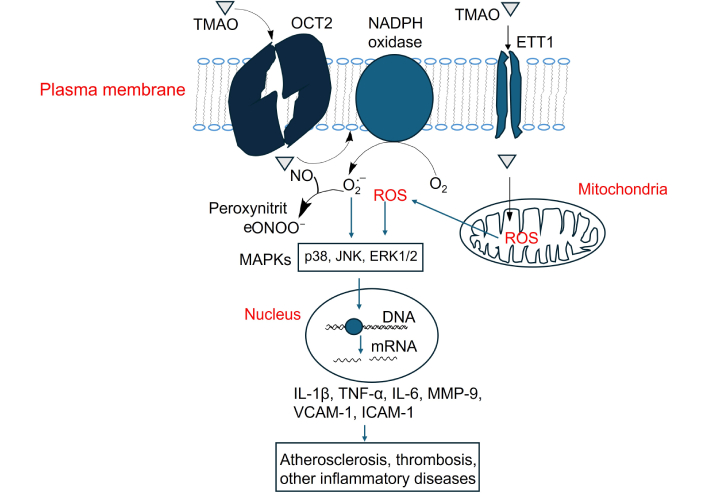
Activation of MAPK signaling by TMAO and its contribution to atherosclerosis, thrombosis, and inflammation-related diseases. TMAO: trimethylamine N-oxide; NO: nitric oxide; ROS: reactive oxygen species; IL-1β: interleukin-1 beta; TNF-α: tumor necrosis factor-alpha; MMP-9: matrix metalloproteinase-9; VCAM-1: vascular cellular adhesion molecule-1; ICAM-1: intercellular adhesion molecule-1; OCT2: organic cation transporter 2
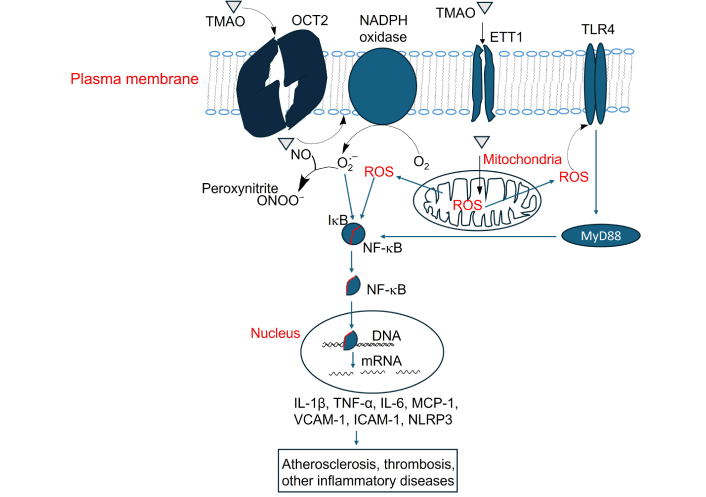
Activation of NF-κB signaling by TMAO and its contribution to atherosclerosis, thrombosis, and inflammation-related diseases. TMAO: trimethylamine N-oxide; OCT2: organic cation transporter 2; TLR4: Toll-like receptor 4; NO: nitric oxide; ROS: reactive oxygen species; IL-1β: interleukin-1 beta; TNF-α: tumor necrosis factor-alpha; MCP-1: monocyte chemotactic protein 1; VCAM-1: vascular cellular adhesion molecule-1; ICAM-1: intercellular adhesion molecule-1
TMAO increases intracellular reactive oxygen species (ROS), primarily through NADPH oxidase activation [119, 120]. In addition, TMAO can impair mitochondrial function, which also leads to increased production of ROS [75]. Elevated ROS levels serve as upstream activators of p38, ERK1/2 (extracellular signal-regulated kinase) and JNK (c-Jun N-terminal kinase) MAPK signaling [121]. p38 and JNK are associated with pro-inflammatory response and their activation leads to the transcription of proinflammatory cytokine genes like IL-1β, TNF-α, and IL-6, matrix metalloproteinase-9 (MMP-9), and adhesion molecules, VCAM-1 and ICAM-1, which promote endothelial dysfunction, atherosclerosis and thrombosis [122–128]. ERK1/2 is more associated with cell proliferation, differentiation, and survival, but can also mediate inflammation in vascular cells [129–131] (Figure 3).
Studies using human aortic endothelial cells (HAECs), human vascular smooth muscle cells (HVSMCs) and mouse models have shown that TMAO treatment increases phosphorylation of p38, JNK and ERK1/2 [38, 132]. Inhibition of MAPKs (using specific inhibitors) reduces TMAO-induced atherosclerosis [132], suggesting the causality.
TMAO-induced ROS also activates NLRP3 inflammasome and regulates NF-κB pathway [133–135], which works synergistically with MAPKs to affect inflammation (Figure 4).
In addition, NO reacts with superoxide (O2•⁻), a major ROS, to produce peroxynitrite (ONOO⁻), a potent oxidant that damages cellular components such as proteins, lipids, and DNA [136–138]. This reaction decreases NO bioavailability, promoting endothelial dysfunction and contributing to oxidative and nitrosative stress in CVD [138, 139].
TMAO activates NF-κB signaling through multiple ways. As above-mentioned, TMAO increases intracellular ROS, and ROS activates NF-κB signaling by triggering the IKK complex, leading to IκBα phosphorylation and degradation. This frees NF-κB (p65/p50) to enter the nucleus, a process involving redox-sensitive PI3K/PTEN/Akt and NIK/IKK pathways [140]. In the nucleus, NF-κB binds to the promoter regions of pro-inflammatory genes, inducing the expression of cytokines such as TNF-α, IL-6, IL-1β, and monocyte chemotactic protein 1 (MCP-1) as well as adhesion molecules including ICAM-1, VCAM-1 [141] (Figure 4).
TMAO activates macrophage cell surface receptor Toll-like receptor 4 (TLR4) [142], which activates MyD88-dependent signaling and further stimulates NF-κB [143] (Figure 4). TLR4 is not a known receptor for TMAO, as the mechanism by which TMAO activates TLR4 remains under investigation. Treatment of HAECs and HVSMCs with TMAO, as well as acute TMAO injection in mice, activates NF-κB signaling, leading to leukocyte recruitment to the aortic endothelium [38]. Inhibitors of NF-κB block the TMAO-induced enhancement of leukocyte adhesion to the aortic endothelial layer [38].
As mentioned above, TMAO induces NF-κB signaling. Activated NF-κB promotes the transcription of NLRP3 and pro-IL-1β, two key components of the NLRP3 inflammasome [144]. TMAO impairs mitochondrial function, increasing ROS production, triggering the assembly and activation of the NLRP3 inflammasome [75, 144]. During this process, NLRP3 oligomerizes and recruits ASC (apoptosis-associated speck-like protein) and recruits pro-caspase-1. Pro-caspase-1 is then auto-cleaved into active caspase-1, leading to the release of mature IL-1β [144]. IL-1β is a key pro-inflammatory cytokine that plays a central role in driving chronic inflammation [145].
TMAO-induced inflammation and endothelial dysfunction in human umbilical vein endothelial cells (HUVECs) can be reversed by treatment with the ROS inhibitor N-acetylcysteine (NAC) or by siRNA-mediated knockdown of NLRP3, suggesting that TMAO exerts its pro-inflammatory effects via the ROS/NLRP3 inflammasome [146].
PERK has been identified as a potential receptor for TMAO [147], suggesting its role in mediating TMAO signaling as endoplasmic reticulum (ER) stress, which activates the unfolded protein response (UPR). Based on the well-established PERK signaling pathway [148–151], we hypothesize that TMAO binds to PERK, triggering its dimerization and autophosphorylation, thereby activating it (Figure 2). Activated PERK phosphorylates eIF2α and enhances ATF4 translation while suppressing IκB synthesis. The resulting decrease in IκB levels allows NF-κB to translocate into the nucleus. Both ATF4 and NF-κB, nuclear transcription factors, then bind to DNA and upregulate proinflammatory mediators, including the NLRP3, cytokines (IL-6, TNF-α, and IL-1β), and scavenger receptors (CD36, SR-A1). This cascade promotes inflammation in various tissues, and contributes to CVD, fatty liver disease, and even neurodegeneration (Figure 2).
The PERK inhibitors GSK2606414 and GSK2656157 effectively reversed TMAO-induced upregulation of ICAM-1 in HAECs [152], implicating PERK signaling as a mediator of TMAO-driven atherosclerosis.
The TMAO metaorganismal pathway is a universal pathway that is causally linked to many chronic inflammatory diseases. As illustrated in Figure 1, there are three strategies to reduce circulating TMAO levels: first, optimizing diet by limiting foods high in TMA precursors; second, blocking the oxidation of TMA to TMAO; and third, altering the gut microbiota composition or developing microbial TMA lyase inhibitors.
For dietary optimization, we conducted a crossover diet study with 113 participants who followed three diet regimens—non-meat, red meat, and white meat—in random order. Each diet lasts four weeks and at the end of each diet challenge, blood was withdrawn to measure circulating TMAO. There was a two-week gap between two diet regimens. Measurements of TMAO indicated that chronic consumption of red meat can significantly increase circulating TMAO levels, while consumption of non-meat and white meat sources did not have the same effect [24]. These results suggest that modifying dietary habits can alter circulating TMAO levels.
The gut microbiota plays a critical role in TMAO metaorganismal pathway. In a hybrid mouse diversity panel, different mouse strains showed varying susceptibilities to atherosclerosis, with the aortic lesion area positively correlated with circulating TMAO levels. Mice prone to atherosclerosis exhibited higher circulating TMAO levels, while those resistant to atherosclerosis showed lower levels [153]. Interestingly, when cecal contents from atherosclerosis-prone C57BL apoE–/– mice were transplanted into germ-free recipient mice fed a choline-supplemented chow diet, the recipient mice developed significantly larger aortic lesions after 16 weeks compared to germ-free recipients that received cecal contents from atherosclerosis-resistant NZW apoE–/– mice [153]. These results suggest that distinct gut microbiota communities can transmit the susceptibility of atherosclerosis.
Certain natural products, such as resveratrol, berberine, allicin, quercetin; prebiotic, grape pomace rich in polyphenol nutraceutical; probiotics, like some Lactobacillus and Bifidobacterium strains; have been reported to modulate gut microbiota composition. These interventions reduce the relative abundance of TMA lyase-encoding bacteria, thereby decreasing TMA and TMAO production and ultimately attenuating atherosclerosis [154–160].
Choline structural analogues, such as 3,3-dimethyl-1-butanol and halomethylcholines, have been shown to inhibit microbial choline TMA lyase, reducing TMA production and consequently lowering TMAO levels. This inhibition holds potential for attenuating atherosclerosis, thrombosis, kidney fibrosis, obesity and insulin resistance as well [76, 80, 101, 161–163].
Several drugs have been reported to impact the TMAO metaorganismal pathway, which may contribute to cardiovascular health. Metformin, commonly used to treat type II diabetes, has been shown to modulate gut microbiota composition, reducing the relative abundance of bacteria that encode choline TMA lyase, and thereby attenuating atherosclerosis [164]. Meldonium, a structural analogue of carnitine used to treat arrhythmias, myocardial infarction, heart failure, and atherosclerosis, inhibits the microbial conversion of carnitine to TMA, ultimately lowering TMAO levels—an effect that offers an additional mechanism for treating atherosclerosis [165, 166]. Statins and aspirin, which lower blood lipids, reduce inflammation, and inhibit thrombosis, have also been reported to reduce circulating TMAO levels [167–169]. Thus, targeting the TMAO metaorganismal pathway may hold promise for cardiovascular health benefits.
More interestingly, TMA, produced by certain gut microbes, can be further degraded by another group of microbes through a process called methanogenesis [170]. Studies have shown that mice fed a methane-producing bacterium such as Methanobrevibacter smithii and Methanoimicrococcus blatticola exhibited decreased levels of circulating TMAO, which subsequently led to a reduction in atherosclerosis [171]. This suggests a potential therapeutic avenue where manipulating gut microbiota to promote methanogenesis could mitigate the cardiovascular risks associated with elevated TMAO levels.
TMAO is excreted in the urine, and renal insufficiency can lead to increased circulating levels of TMAO. Increasing the glomerular filtration rate may reduce circulating TMAO. This strategy is still under investigation. Although loop diuretics are expected to enhance glomerular filtration rate, they actually inhibit the renal excretion of TMAO [172].
TMAO, acting as a chemical chaperone, can stabilize protein conformation and help restore misfolded proteins and alleviate ER stress [173]. This mechanism could provide therapeutic benefits in treating various protein misfolding diseases, including lens opacity formation and neurodegenerative disorders [173, 174]. Furthermore, TMAO has been shown to modulate immune function by promoting immunostimulatory macrophages and CD8+ T cells [175]. In an orthotopic mouse model of pancreatic ductal adenocarcinoma (PDAC), TMAO was evaluated for its potential antitumor effects, which were characterized by delayed tumor growth [175]. The health benefits of TMAO require further investigation.
Due to the distinct production and target sites of TMAO, it is now recognized as a hormone. It plays a key role in triggering numerous inflammatory cascades, contributing to the pathogenesis of various chronic diseases. Consequently, inhibiting TMAO production has been proposed as a potential therapeutic strategy for improving health outcomes. Numerous natural products, probiotics, and prescription drugs have demonstrated promise in inhibiting TMAO biosynthesis. Additionally, several synthesized structural analogs have shown potential as TMAO biosynthesis inhibitors. The development of potent pharmacological inhibitors of TMAO biosynthesis could be utilized to treat a variety of human diseases.
The TMAO metaorganismal pathway has been implicated in a range of chronic inflammatory diseases, and targeting this pathway holds promise as a therapeutic strategy. Several molecular mechanisms have been identified, revealing the involvement of multiple inflammatory signaling pathways. However, our understanding remains incomplete and further in-depth investigations are needed to clarify how TMAO activates pro-inflammatory signaling.
AF: atrial fibrillation
CSF: cerebrospinal fluid
CVD: cardiovascular disease
ER: endoplasmic reticulum
FMOs: flavin monooxygenases
HAECs: human aortic endothelial cells
ICAM-1: intercellular adhesion molecule-1
IL-1β: interleukin-1 beta
MMP-9: matrix metalloproteinase-9
NAFLD: non-alcoholic fatty liver disease
NO: nitric oxide
OCT2: organic cation transporter 2
PD: Parkinson’s disease
PERK: protein kinase R-like endoplasmic reticulum kinase
ROS: reactive oxygen species
TF: tissue factor
TLR4: Toll-like receptor 4
TMA: trimethylamine
TMAO: trimethylamine N-oxide
TNF-α: tumor necrosis factor-alpha
VCAM-1: vascular cellular adhesion molecule-1
We thank David Schumick for the artwork (Figure 1) and Xiayan Ye for the references’ editing.
SM and RK: Conceptualization, Writing—original draft. ZW: Conceptualization, Writing—original draft, Formal analysis, Visualization, Writing—review & editing.
Zeneng Wang reports being named as coinventors on pending and issued patents held by the Cleveland Clinic relating to inflammation and cardiovascular diagnostics and therapeutics and having received royalty payments for inventions or discoveries related to diagnostics or therapeutics from Procter & Gamble and Cleveland Heart Lab, a fully owned subsidiary of Quest Diagnostics. In addition, Zeneng Wang, who is the Editorial Board Member and Guest Editor of Exploration of Medicine had no involvement in the decision-making or the review process of this manuscript.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by National Heart, Lung, and Blood Institute grants [P01 HL147823] (ZW); the National Institutes of Health grant [P01 HL158502] (RK). Additional support was also provided to RK by a generous gift from Richard Morrison (used for this work). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Olha Denefil ... Khrystyna Loza
Weinan Du ... Juxiu Li
Alejandra Vargas ... David A. Johnson
Shin Takasawa ... Maiko Takeda
