 Review
Review

Affiliation:
Microbiology and Immunology, Drexel University College of Medicine, Philadelphia, PA 19129, USA
Email: cma49@drexel.edu
ORCID: https://orcid.org/0000-0002-9060-0581
Explor Med. 2025;6:1001326 DOI: https://doi.org/10.37349/emed.2025.1001326
Received: January 30, 2025 Accepted: April 30, 2025 Published: May 27, 2025
Academic Editor: Alessandro Granito, University of Bologna, Italy
The inflammasome is a critical inflammatory signaling cascade. Diverse events, including infections or tissue damage, activate it. It is essential in regulating the early stages of wound healing but is overall downregulated in the later stages. If uncontrolled inflammasome expression occurs, healing tends to stall and chronic wounds ensue due to the release of proinflammatory cytokines, such as interleukin (IL)-1β and IL-18. Furthermore, if left unchecked, chronically activated inflammasomes can promote a healing milieu when none is warranted and this can cause fibroproliferative skin disorders such as keloids and hypertrophic scars. This review examines the role of the inflammasome and what is known about its regulation in each phase of wound healing. This review also discusses the contribution of inflammasome activation during stalled wound healing, such as that found in chronic wounds and diabetic ulcers. It discusses the hyperactivation of the inflammasome during fibrotic skin pathology. A better understanding of the contribution of inflammasome signaling in various stages of wound healing could lead to identifying more effective therapeutics for treating different aspects of abnormal wound healing.
Wound healing is a fundamental event needed for the reconstruction of damaged tissues. It is a finely orchestrated, complex, and dynamic process [1, 2]. Most wounds are minor and usually close within 12–14 days; however, resolving more extensive wounds may take several months to a year to complete the healing process [2, 3]. While the skin is most often studied because it is easily monitored, wound healing can occur in any organ. In normal skin, epidermal and dermal layers exist in homeostasis [4], and this protects the body from external insults, pathogens, and the skin’s microbiome [5, 6]. Any trauma that breaches this protective barrier will initiate wound healing [7].
Inflammasomes are essential inflammatory complexes that have been extensively studied in the last decade, primarily in the context of disease. However, immediately after wounding, they become activated and play a significant role in the healing process by secreting cytokines that kick-start the healing process. At the same time, activating these inflammatory complexes is involved in the clearance of pathogens that may contaminate the wound bed. The activity of the inflammasomes is essential in the early phases of healing and is downregulated in the later stages. This review discusses the role of the inflammasome at each stage of healing. It will be presented from the perspective of skin healing because it has well-defined stages that have been extensively studied. At the end of the review, we will discuss targeting nucleotide-binding domain, leucine-rich repeat, and pyrin domain-containing protein (NLRP)3 as a therapeutic modality for treating chronic wounds, using diabetic ulcers and burn wounds as examples. We will also discuss the contribution of inflammasome activation in developing fibroproliferative scars and targeting its activation to alleviate this aberrant healing process.
There are four well-described phases attributed to wound healing: hemostasis (lasting 0–several hours post-injury), the inflammatory phase (1–3 days), the proliferative phase (4–12 days), and the remodeling phase (21 days–1 year) [2, 3, 8–10]. Each phase is tightly regulated, but there is some overlap between them [2, 3, 8–10]. Dysregulation in these steps can result in impaired healing or excessive scarring.
Due to the injury and damage to the cells, inflammasomes become activated and contribute to wound healing by secreting proinflammatory cytokines that attract cells [11–15], which aids in the repair process. These secreted cytokines also recruit inflammatory cells, such as macrophages [11, 12] and T cells [13, 14], to clear away the damaged cells and pathogens. The release of tissue factor is crucial in the clotting cascade and mediating tissue inflammation [16]. Fibroblasts are also recruited after wounding [15, 17], and secrete products to aid the healing. They are much more than a source of extracellular matrix scaffolding. They are essential sentinel cells that, when damaged, instigate the wound repair process and participate by producing and responding to cytokines and chemokines. They have functional inflammasomes that become activated during critical wound-healing phases, and this helps to promote healing.
Depending on the initiating mechanism, activation of the inflammasome can run a well-defined course with the resolution of inflammation and healing of the injury or be continuous, resulting in fibrosis [18] or a chronic wound [19]. The acute response to an initiating event (tissue injury) with a resolution of the injury versus a chronic reaction leading to unresolved disease and fibrosis is not entirely known. We speculate that acute injury may resolve on the clearance of the initiating signal (pathogen, virus, chemical, etc.). In contrast, chronic wounds result from a pathogen or irritant that is not cleared, causing continuous inflammasome activation and interleukin (IL)-1β and IL-18 processing and release.
Inflammasomes are activated in the first two phases of wound healing, which start the healing cascade. Still, in the later phases of healing, they are downregulated to promote wound resolution. Several inflammasomes have been implicated in the initial stages of efficient wound closure. These include NLRP1, NLRP3, and NLR family caspase activation and recruitment domain-containing (NLRC)4. Hu and colleagues [20] found that the mice’s interstrain differences in wound healing correlated with various NLRP1 paralogs, which altered the production of macrophage inflammatory protein-1a by keratinocytes. Other studies have shown the importance of NLRP3 in wound healing due to the activation of caspase-1 and the release of IL-1β. The released cytokines attract fibroblasts and inflammatory cells to the site to aid wound healing. If there is a reduced inflammatory response, wound healing is delayed [21, 22]. To confirm this observation, delayed wound healing happens in NLRP3-KO mice, and this was recapitulated when NLRP3 chemical inhibitors were used. In addition, the absence of the AIM2 (absent in melanoma 2) inflammasome also slows the rate of wound closure [23]. However, in contrast, the sustained activation of NLRP3 can hinder wound healing [24]. This is because the regenerated tissues continue to be damaged due to the release of cytokines and the recruitment of inflammatory cells [25].
When exposed to the environment during skin injury, the exposed collagen starts the intrinsic and extrinsic clotting cascades [26]. The bleeding causes aggregation of platelets and the formation of a fibrin clot that usually occurs within minutes of the injury. The serine protease signaling cascade regulates blood clotting [27, 28]. It is a tightly regulated cascade, ensuring coagulation occurs only when needed. The damaged tissues leak adenosine diphosphate, causing platelet aggregation and adhesion to damaged collagens. Platelets initiate wound healing [29] by secreting thrombin and fibrin, which interact with and enhance the clotting cascade [28]. The formation of a fibrin clot further strengthens platelet aggregation and stabilizes the clot. Platelets secrete platelet-derived growth factor and TGF-β [29], which causes the recruitment of inflammatory cells and fibroblasts to the injured site [7, 30]. Platelets also enhance the activation of the inflammasome in macrophages and monocytes [31], kickstarting the inflammatory response. Platelets do not have DNA and do not transcribe NLRP3 on demand, but they express NLRP3 [32]. Studies investigating platelets in NLRP3-KO mice noted that bleeding times were approximately 4 times longer, even though the number of platelets was unchanged compared to that found in their wild-type counterparts [33]. There was also reduced platelet accumulation at the wound site [33, 34]. Further confirming NLRP3 expression in platelets, a study investigated patients with sickle cell disease [35]. It was found that platelets in sickle cell disease were activated, and there was increased expression of caspase-1, which was further elevated during sickle cell crisis [35]. These studies were further recapitulated in Towne sickle cell disease mice, demonstrating that clotting was elevated in homozygous mice compared to wild-type and heterozygous [35].
Nevertheless, platelets influence nearby cells, as shown in a study by Rolfes et al. [31], demonstrating that platelet products activate toll-like receptor-4, the receptor for advanced glycation end products and CD36, causing the activation of NLRP3 in nearby cells (Figure 1). Co-culture methods show that platelets enhance inflammasome activation in innate cells. In the absence of platelets, NLRP3 activation and production of IL-1α, IL-1β, and IL-18 from macrophages, monocytes, and neutrophils were reduced [31]. Inhibition of these components lowered IL-1β, TNF-α, and caspase-1 activity in human monocyte-derived macrophages [31]. The inflammasomes play a prominent role in the pro-inflammatory aspects of platelets [32, 36].
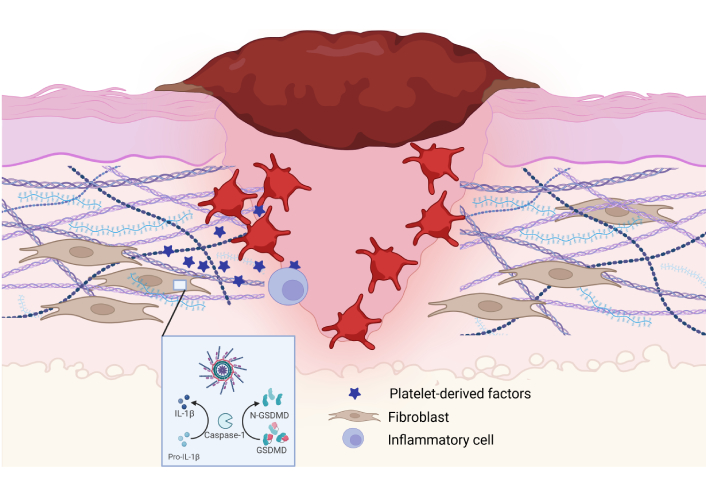
Inflammasome activation and signaling in the early stages of wound healing. Platelets secrete molecules that activate the NLRP3 inflammasome in the early stages of wound healing. This helps to promote factors (platelet-derived factors, IL-1β, IL-18, etc.) that enhance cell recruitment and replication to start the wound healing process. IL: interleukin; NLRP: nucleotide-binding domain, leucine-rich repeat, and pyrin domain-containing protein. Created in BioRender. Artlett, C. (2025) https://BioRender.com/p12v164
Inflammasome products secreted from other cells also mediate platelet activation [37], resulting in cyclical events and adding to the signaling milieu in the damaged tissues. Ischemic reperfusion can also play a prominent role in tissue damage at the local wound site [38], further activating various inflammasomes [39]. If IL-1 is blocked with antibodies or the inflammasome is inhibited, reperfusion injury is ablated [40, 41]. Another study found that the inhibition of caspase-1 significantly reduced fibrin deposition in the clot [42]. The release of thrombin activates NLRP3 in various tissue cells [43, 44], further correlating inflammasomes with the hemostasis phase of wound healing (Figure 2).
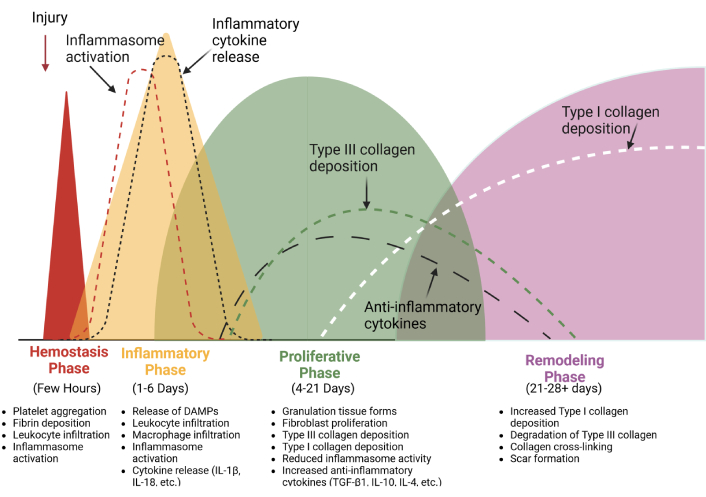
Inflammasome activation during the normal process of wound healing. The activity of the inflammasome is prominent in the hemostasis and the inflammatory phases, where it is required to secrete cytokines that promote cell recruitment and kick-start the healing process. It is downregulated in later phases of the wound-healing process. IL: interleukin. Created in BioRender. Artlett, C. (2025) https://BioRender.com/a90o856
Once the fibrin clot has formed, the inflammatory phase begins, and depending on the size of the injury, this phase may last up to 4 days [7, 30]. The extravasation of neutrophils from blood vessels to the injury site is an automatic response. This process also occurs with sterile injury. Due to the initial tissue damage, there is a loss of blood and secondary tissue injury, triggering the release of endogenous danger signals called damage-associated molecular patterns [45–47]. These alarmins further facilitate the activation of inflammasomes, causing the release of cytokines that further attract inflammatory cells to the injured site. This phase primarily involves recruiting inflammatory cells into the wound in response to the release of chemokines such as intracellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin. Cytokines such as IL-1β and IL-18, processed by an activated inflammasome, indirectly attract cells. IL-1β induces monocyte chemoattractant protein-1 [48], while monocyte chemoattractant protein-1 induces IL-1β expression in monocytes [49].
In the early stages of wound healing, inflammatory cells capable of phagocytosis infiltrate the site. Phagocytosis is one of the main mechanisms of the innate immune response and is critical in wound healing. Although most cells can phagocytose, not all are “professional” phagocytes. Neutrophils, macrophages, and monocytes are considered to be professional phagocytes that infiltrate damaged tissue and phagocytose cellular debris, such as proteins and fragments from damaged cells. They also phagocytose any microorganisms found within the injured tissue [50]. This process provides the first line of defense against infection. One indirect result of phagocytosis is that fibrin is broken down, further attracting phagocytes to the injured site [51]. The recruitment of macrophages into the tissues provides an additional line of defense in preventing or limiting infection. Macrophages phagocytose any bacteria that neutrophils cannot remove. Furthermore, the macrophages secrete various chemotactic attractants and growth factors, initiating healing. Growth factors and chemoattractants are critical regulators of the wound-healing process. They cause the proliferation and migration of fibroblasts to the injured tissue, starting the second phase of wound healing [7, 51–54]. Initial studies suggest an enhanced inflammatory response during the early stages is required for efficient wound healing [55]. Without NLRP3, there is delayed angiogenesis, re-epithelialization, and the development of granulation tissue [56]. These observations were also recapitulated in caspase-1 deficient mice [56], suggesting that the downstream products of an activated inflammasome are essential in the early stages of wound healing. The lack of NLRP3 reduces the inflammation in wounded tissue with less IL-6 and tissue repair factors such as matrix metalloproteinase-9, epidermal growth factor, and vascular endothelial growth factor in NLRP3-KO skin [57].
The inflammatory phase begins with the transient expression of IL-1β. IL-1β is an essential cytokine involved in wound healing. IL-1β secretion peaks on day one and declines between days 3 and 7 post-injury [55, 58]. IL-1β is necessary for the initiation of wound healing as it enhances the secretion of TGF-β1 [59], IL-6 [60], and vascular endothelial growth factor [60], and these cytokines are also needed for collagen deposition, cell replication, and wound repair. The expression of these proinflammatory cytokines starts to decline after 5 days [55]. However, the extended duration of IL-1β released into a wound during prolonged healing can promote fibrosis. A study investigating deep incisional wound healing underscored this observation. Mice lacking the IL-1 receptor had improved wound healing with less fibrosis, and they found that this was due to reduced TGF-β, IL-6, and the vascular endothelial growth factor in the wounds [61]. Furthermore, the authors identified more collagenolytic activity at the wound site [61]. These observations suggest that extended wound healing time is more likely to be pro-fibrotic, which will be discussed below.
While most studies investigate NLRP3, Qin et al. [62] investigated the role of NLRC3 during cutaneous wound healing. They found that the ablation of NLRC3 promoted an enhanced inflammatory response in the wounds. This observation was specific to neutrophils, while macrophages remained unchanged. This suggests that NLRC3 suppresses inflammation in the early stages of wound healing [62].
Granulation tissue formation is necessary to enhance wound healing, and this feature characterizes the proliferative phase. It generally starts about four days after the initial injury. If the wound is relatively minor, this process can last up to 21 days [30]. The proliferative phase is characterized by red granulation tissue at the base of the wound. During this stage, M2 macrophages that function in repair should dominate the wound environment [63]. The chemo-attractants and various growth factors secreted in the wound bed recruit fibroblasts to the injury [64–66]. To further enhance this phase of wound healing, fibroblasts secrete fibroblast growth factor-2, platelet-derived growth factor, and TGF-β1. This sustains fibroblast proliferation and migration via autocrine signaling [67]. The primary role of the fibroblast is to pull together the wound margins and rebuild the damaged extracellular matrix. The fibrin clot is gradually replaced with granulated tissue. Type I collagen, which is stronger and more stable, will eventually replace this temporary matrix patch [7, 50–52, 68].
Fibroblasts are critical cells in the wound-healing process. They deposit collagen and other extracellular matrix proteins that different cell types use to migrate along. This starts the process of re-epithelialization and angiogenesis. Intriguingly, fibroblasts from the fascia migrate into the wound, dragging the fascia extracellular matrix substrates into the wound site [69]. Correa-Gallegos et al. [69] proposed that the fascia comprised a “…specialized prefabricated kit of sentry fibroblasts, embedded within a movable sealant…”. This kit becomes activated with the injury and can seal the wound. These specialized fibroblasts are tagged with engrail-1 [70], which depends on p120 (catenin delta-1) expression and is enhanced after wounding. Silencing p120 reduced the trafficking of the facia fibroblasts and extracellular matrix into the wound site, which augmented wound healing [70]. Two additional proteins were found to be involved in the mobilization of the fascia into the wound, including N-cadherin [71] and connexin-43 [72].
While fibroblasts are migrating into the wound, keratinocytes proliferate and migrate along the extracellular matrix, helping to seal the wound’s surface from the environment [30]. Keratinocytes migrate between the scab and the new granulation tissue to seal the wound from the external environment [73]. This helps to reestablish the skin’s barrier function. Keratinocytes also produce extracellular matrix proteins, including laminin and type IV collagen IV, further enhancing this protective barrier [7, 30]. Angiogenesis begins when endothelial cells migrate along fibronectin into the site [7, 30, 74]. Angiogenesis provides the necessary oxygenation in the injured tissue that fibroblasts and keratinocytes require [50, 75], as the injury is hypoxic [76].
At this stage of wound healing, pro-inflammatory cytokine expression (IL-1β, IL-6, and TNF-α) is typically downregulated, progressively declining from about day 5 onwards [55]. After completing this phase, all pro-inflammatory cytokines have returned to basal levels [55]. However, at the same time, anti-inflammatory cytokine expression increases, including TGF-β1, IL-10, and IL-4 [77], and collagen synthesis is increased (Figure 2).
Myofibroblasts contract the wound margins during this phase, pulling the edges together and secreting collagen and other extracellular matrix proteins to fill the wound. They use cytoskeletal stress fibers and focal adhesions to bind to fibronectin and collagen, pulling the matrix together and contracting the wound size [50, 51, 75, 77]. The secretion of types I and III collagens reinforces the patch and strengthens the extracellular matrix [78, 79]. Early in this process, myofibroblasts secrete more type III collagen, especially in the early stages of tissue granulation [80], but as the healing progresses, myofibroblasts switch to type I collagen [81]. Type III collagen provides a flexible and temporary scaffold for new tissue formation, allowing for rapid cell migration and angiogenesis. This is because type III collagen is loosely organized. Later, in the healing process, type I collagen replaces type III collagen during wound maturation, and the extracellular matrix gains tensile strength. Studies have found that type III collagen regulates wound repair and matrix remodeling by attenuating myofibroblast activation [80].
While the activation of most inflammasomes progressively declines, intriguingly, there is still a role for the inflammasome in this stage of healing. Zhao et al. [57] reported that myofibroblast-macrophage interactions were crucial for wound healing in these final stages and that prostaglandin E might be a downstream effector of NLRP3 activation. Prostaglandin E is essential in many stages of wound healing, such as cell recruitment in the early stages [82], and is needed for angiogenesis and cell proliferation in the later stages of healing [83].
In the proliferative phase, NLRC3 inflammasome slowed cell proliferation and hindered migration. In their studies, Qin et al. [62] found that wound healing was accelerated without NLRC3 because cell proliferation and migration were enhanced. They also show that overexpressing NLRC3 in HEK293T cells proliferation and migration reduced. This suggests that NLRC3 activity controls the rate of re-epithelization of the wound. In other studies by Naik et al. [84], the authors showed the role of AIM2 inflammasome activation in accelerated skin healing, but only after repeated wounding, suggesting that the cells in wounded skin have a memory of prior wounding. This enhanced re-epithelization was dependent on IL-1β only [84].
Fibroblasts are a heterogeneous population of cells supporting the integrity of tissues. They are also the primary cells that orchestrate the remodeling phase of wound healing, where the collagen fibers become organized and cross-linked and have increased tensile strength in the fiber. To conclude, the strength of the new collagen within the wound is only about 80% as strong as the collagen laid down under homeostasis [50, 51]. A recent study examining the fibroblast population within wounds found that those expressing the CD26 cell surface protein secreted most collagen into the wound [85]. Once the remodeling phase is complete, myofibroblasts undergo apoptosis [86, 87], while some return to quiescent fibroblasts [86]. The inflammasome does not appear to play any significant role in this stage of wound healing (Figure 2).
The most crucial cytokine in this phase of wound healing is TGF-β1, as it enhances collagen deposition into damaged tissue and downregulates inflammation during the remodeling phase. During this phase of healing, the extracellular matrix is also cross-linked and reorganized into stronger stabilized fibrils within the injured tissues [88]. However, excessive or extended expression of TGF-β1 during this phase promotes scar tissue development [89]. Importantly, IL-10 is produced by CD4 T cells [90], but is also involved in the downregulation of inflammation in this phase. By influencing inflammation, extracellular matrix remodeling, and fibroblast activity, IL-10 has been linked to reduced scar formation [91]. TGF-β1 and IL-10 are temporally expressed, and TGF-β1 transiently activates myofibroblasts, whereupon they become de-differentiated in the presence of IL-10 [92].
Wound healing treatments have been discussed for over 5,000 years, and various therapies have been described [93–95]. Basic principles to enhance wound healing, such as washing the wound, applying plasters, and dressing the wounds, have long been known [94, 95]. In regenerative medicine, the success or failure of a healing wound is contingent upon extracellular matrix synthesis, cell proliferation, and the migration of fibroblasts and keratinocytes into the wound bed; however, these processes are delayed in chronic wounds such as venous stasis ulcers or when chronic diseases such as diabetes are present. A wound is considered to be chronic when it fails to heal in a timely manner [96].
Excessive inflammation due to an overactive inflammasome in the wound bed impairs wound healing, and effective therapies for chronic wound healing must disrupt this pro-inflammatory environment. Targeting inflammasomes to promote wound healing has recently gained popularity (Table 1). However, this targeting must be done strategically and not early when it is needed for healing. For example, in a burn model, the systemic administration of the NLRP3 inhibitor, glyburide, during the early stages of wound healing reduced inflammation and delayed healing [97]. However, when glyburide was given after the inflammatory phase, wound healing was not impeded [97]. This suggests that inflammation and inflammasome activation are beneficial and necessary for wound healing only in the early phases, as discussed above.
The effects of targeting NLRP3 during wound healing
| Chemical | Effect on NLRP3 | Outcome | Reference |
|---|---|---|---|
| Genistein | Increased | Delayed healing | [136] |
| Zol | Increased | Delayed healing | [137] |
| Metformin | Decreased | Faster wound healing | [138] |
| Paeoniflorin | Decreased | Promoted healing | [139, 140] |
| Calcitriol | Decreased | Promoted healing | [141] |
| Sanguisorba officinalis | Decreased | Promoted healing | [110] |
| MF-094 | Decreased | Accelerated healing | [108] |
| Glyburide/Glibenclamide | Decreased | Improved or delayed wound healing depending on time of administration | [97] |
| MFG-E8 | Decreased | Accelerated healing | [142] |
| Fenofibrate | Decreased | Accelerated healing | [143] |
| N-Acetylcysteine | Decreased | Improved healing | [144] |
| MCC950 | Inhibited | Improved healing or no effect | [109, 145] |
| Melatonin | Inhibited | Increased keratinocyte proliferation | [146] |
| 3,4-Methylenedioxy-β-nitrostyrene | Inhibited | Improved healing | [129] |
| Bletilla striata polysaccharide | Inhibited | Accelerated healing | [147] |
| Lactobacillus plantarum | Inhibited | Improved healing | [111, 112] |
| Bay 11-7082 | Inhibited | Improved healing | [144] |
Adapted from [118], CC BY. NLRP: nucleotide-binding domain, leucine-rich repeat, and pyrin domain-containing protein
Treating diabetic wounds has traditionally been done by reducing hyperglycemia, antimicrobials, debridement, grafts, and off-loading techniques [98]. Diabetic wounds do not heal quickly as they have been stalled at the inflammatory phase [99–102]. The extended release of proinflammatory cytokines is detrimental to the wound healing process, and in severe cases, patients are at risk for foot amputation [103]. The prolonged dysregulation of NLRP3 fosters the chronic inflammation that delays the healing [24] which is particularly evident in diabetic ulcers, where these wounds fail to heal. The hyperglycemia environment promotes NLRP3, caspase-1, and IL-1β expression in inflammatory cells [104], contributing to the inflammation. Furthermore, a comparison of NLRP3 expression showed 3-fold more in diabetic wounds than in non-diabetic wounds [99], and the presence of neutrophil extracellular traps promotes NLRP3-mediated inflammation [105]. These factors contribute to the proinflammatory environment in the wound that slows healing.
In diabetic wounds lasting longer than 3 months, there was a high expression of NLRP3, caspase-1, IL-1β, and IL-18 [24]. Recent studies have found that directly targeting and/or downregulating the NLRP3 inflammasome activity promotes healing. Wound dressings that lower IL-1 expression and simultaneously increase vascular endothelial growth factor have enhanced skin healing [106, 107]. A study employing MF-094, an inhibitor of the deubiquitinase enzyme, ubiquitin-specific protease-30 [108], was beneficial in promoting healing. NLRP3 activation depends on its deubiquitylation for activation, and when MF-094 inhibited this process, NLRP3 activity was reduced [108]. Downstream proteins such as active caspase-1 and IL-1β were also decreased, and wound healing was enhanced in a rat model of diabetes [108]. Another compound, MCC950, has also shown promise in promoting the healing of diabetic wounds. MCC950 targets NLRP3 activation and inhibits the release of IL-1β and IL-18 into the wound [109]. Sanguisorba officinalis is a medicinal root used in Chinese medicine for wound healing. Although its exact mechanism of action is unknown, a study using extracts from Sanguisorba officinalis demonstrated increased healing of diabetic wounds via reduced NLRP3, caspase-1, and IL-1β expression [110]. Intriguingly, Lactobacillus plantarum was found to promote the healing of diabetic foot wounds by inhibiting NLRP3 activation and pyroptosis [111, 112]. Some plant extracts are efficacious at reducing NLRP3 expression, including garcinol [113], aloe vera [114], curcumin [115], bitter melon [116], and carnosic acid extracted from rosemary [117], to name a few. Other natural and synthetic compounds inhibit the NLRP3 inflammasome and promote animal wound healing (Table 1). Ding et al. [118] reviewed other inhibitors of NLRP3 and their efficacy in diabetic wounds. Whether these substances successfully make it through clinical trials is yet to be determined.
Burn wounds also represent a significant clinical problem [119], and activation of the NLRP3 and NLRP6 have been reported [120, 121]. In successfully healing burn wounds, NRLP3 activation is beneficial in the early stages of healing [97, 122], which returns to baseline levels in the later phases [123]. However, the persistent activation of the NLRP3 inflammasome is associated with a poor prognosis in patients with septic burns [123, 124]. In burn wounds that do not heal, there is an increased incidence of pro-inflammatory M1 macrophages strongly expressing NLRP3 [125].
Animal models have been used to determine the success of various NLRP3 inhibitors for the treatment of chronic burn wounds. Bay 11-7082 irreversibly inhibits IBa phosphorylation [126], and lowers NLRP3 expression [127]. In a burn model, Bay 11-7082 reduced acute lung injury and lowered inflammatory cytokine levels [128]. As discussed above, the timely administration of glyburide to the burn was critical in its successful healing [97]. 3,4-Methylenedioxy-β-nitrostyrene inhibits NLRP3 assembly while sparing AIM2 and NLRC4 activity [129]. This was shown to reduce NLRP3 activation in burn wounds and promote the healing time of the wounds [130].
Keloid and hypertrophic scars are fibroproliferative scars that develop during skin healing [131]. There is a genetic component and familial component to the development of keloids and hypertrophic scars [132]. The direct role of the inflammasome has not been extensively studied in these abnormal healing processes. However, in systemic sclerosis, skin fibrosis is dependent on the aberrant expression of the inflammasomes and elevated caspase-1 activity [18]. Recently, it was found that the aberrant expression of NLRP3 activation contributes to the development of keloids and hypertrophic scars. This is thought to be due to the Warburg effect and the abnormal expression of glycolytic enzymes [133]. Targeting the abnormal glucose metabolism with the molecule shikonin, a pyruvate kinase M2 inhibitor, lowered NLRP3 activity and improved the healing of the skin without the overt development of the fibroproliferative scar [133]. Further studies have shown that the elevated NLRP3 signaling is mediated by Notch1 expression [134]. Notch1 expression is controlled by autophagy [135]. Thus, Lee et al. [134] found that using rapamycin to induce autophagy in keloid fibroblasts and this suppressed Notch1 and NLRP3 proteins, effectively resolving fibrosis and lowering myofibroblast expression. These studies suggest that elevated NLRP3 contributes to these types of scars and that targeting the inflammasome might contribute to better management in genetically susceptible individuals.
The inflammasome is critically involved in the proper sequence of wound healing phases. It is highly elevated in the hemostasis and inflammatory phases stages, modestly expressed in the proliferative phase, and largely absent in the remodeling phase. The chronic expression of the NLRP3 inflammasome plays a significant role in wounds that have stalled in their healing. This causes the prolonged expression of inflammatory cytokines. Blocking the inflammasome in the later stages of wound healing may benefit chronic wounds and help enhance their healing. This will lower inflammatory cytokines secreted by an activated inflammasome and drive the expression of anti-inflammatory cytokines such as TGF-β and IL-10, which is needed for collagen deposition in the wound.
IL: interleukin
NLRC: NLR family caspase activation and recruitment domain-containing
NLRP: nucleotide-binding domain, leucine-rich repeat, and pyrin domain-containing protein
CMA: Conceptualization, Investigation, Writing—original draft, Writing—review & editing.
The author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

