 Review
Review

Affiliation:
1School of Human and Social Sciences, “Kore” University of Enna, 94100 Enna, Italy
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0003-4149-9488
Affiliation:
2Department of Clinical and Experimental Medicine, Internal Medicine, Garibaldi-Nesima Hospital, University of Catania, 95122 Catania, Italy
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0003-4483-3365
Affiliation:
2Department of Clinical and Experimental Medicine, Internal Medicine, Garibaldi-Nesima Hospital, University of Catania, 95122 Catania, Italy
ORCID: https://orcid.org/0000-0001-6593-8823
Affiliation:
2Department of Clinical and Experimental Medicine, Internal Medicine, Garibaldi-Nesima Hospital, University of Catania, 95122 Catania, Italy
ORCID: https://orcid.org/0000-0002-5436-8279
Affiliation:
3Department of Anaesthesiology and Intensive Care Medicine, IRCCS Gemelli, 00168 Rome, Italy
Affiliation:
2Department of Clinical and Experimental Medicine, Internal Medicine, Garibaldi-Nesima Hospital, University of Catania, 95122 Catania, Italy
Email: francesco.purrello@unict.it
ORCID: https://orcid.org/0000-0003-3313-8543
Affiliation:
2Department of Clinical and Experimental Medicine, Internal Medicine, Garibaldi-Nesima Hospital, University of Catania, 95122 Catania, Italy
ORCID: https://orcid.org/0000-0002-1781-0902
Explor Med. 2020;1:364–376 DOI: https://doi.org/10.37349/emed.2020.00025
Received: September 08, 2020 Accepted: November 06, 2020 Published: December 31, 2020
Academic Editor: Alessandro Mantovani, University and Azienda Ospedaliera Universitaria Integrata, Italy
The article belongs to the special issue Exploring Type 2 Diabetes Mellitus
Glycemic homeostasis is an essential mechanism for the proper working of an organism. However, balance in blood lipid and protein levels also plays an important role. The discovery of the hormone insulin and the description of its function for glycemic control made fundamental scientific progress in this field. However, since then our view of the problem has been deeply influenced only in terms of glucose and insulin (in an insulin-centric and glucose-centric way). Based on recent scientific discoveries, a fine and sophisticated network of hormonal and metabolic interactions, involving almost every apparatus and tissue of the human body, has been theorized. Efficient metabolic homeostasis is founded on these intricate interactions. Although it is still not fully defined, this complex network can undergo alterations that lead to metabolic disorders such as diabetes mellitus (DM). The endocrine pancreas plays a crucial role in the metabolic balance of an organism, but insulin is just one of the elements involved and each single pancreatic islet hormone is worthy of our concern. Moreover, pancreatic hormones need to be considered in a general view, concerning both their systemic function as direct mediators and as hormones, which, in turn, are regulated by other hormones or other substances. This more complex scenario should be taken into account for a better understanding of the pathophysiology and the therapeutic algorithms of DM. As a consequence, improvements in modern medicine could help to contemplate this new perspective. This review is focused on some aspects of gut-pancreas interaction, aiming to integrate this synergy into a wider context involving other organs and tissues.
In 1921 Banting et al. [1] successfully prevented the death of a dog suffering from induced diabetes by administering a pancreatic extract and thus addressing the alterations caused by the absence of the pancreas. Their findings changed the history of diabetes mellitus (DM): they were conscious that the pancreatic extract should contain a factor, later known as insulin, capable of preventing the death of the animal, but they could not imagine that for the following century no one would have dared to think differently than them. In 1921 the knowledge of diabetes pathogenesis was centered on the reduced amount and action of insulin and everything else was considered not as a contributor but a secondary factor. This concept has driven scientists’ efforts, pharmaceutical companies’ investments, and patients’ hopes for years. The acknowledgment of the correct interpretation of Banting and Best’s results arrived in 1923 [2] when the authors received a Nobel prize for their findings, then proposed in human models as well. In those years, for the first time, a child affected by type 1 diabetes mellitus was treated and saved thanks to the pancreatic extract containing insulin.
In 1923 the diabetes pathophysiology was explained taking into account only two elements, glucose and insulin; no other scientific findings were considered to support or to discredit this pathophysiological model. At that time, this model was considered the only valid point of view, although the gut was already identified as an organ able to produce hormones affecting glucose homeostasis, such as secretin [3], and glucagon was discovered as another Langerhans islet hormone. This latter hormone, even if capable of counter-regulating insulin’s excessive action, was considered only as an antidote for insulin action.
Recalling these critical steps in our knowledge makes us aware of the pitfalls lying in wait, as we go beyond the “Pillars of Hercules”, we need to be unbridled.
Our knowledge about this field was reassessed with the introduction in clinical practice of incretin and glucagon-like peptide 1 (GLP-1), which questioned the insulin-centric view destabilizing Banting and Best’s milestones. Nowadays, it would be restrictive to consider glucagon just as a counter-regulator of insulin action, which acts only to correct a hypoglycemic crisis. Pancreatic islets include different cellular populations cooperating to satisfy the metabolic needs of the organism, but it also represents an endocrine organ reflecting different embryological origins. The islets of Langerhans derive from a common precursor able to differentiate into several cell types. This process involves specific transcription factors driving the transition from a stem cell to a mature one. To date, some molecular pathways associated with differentiation, de-differentiation and trans-differentiation processes have been described for pancreatic islet cells. These knowledges concerning endocrine pancreas, could be useful not only for diabetes treatment but also in the field of regenerative medicine.
Cell-type commitment in the pancreas is controlled by a set of master regulatory transcription factors regulating the transition from one progenitor cell state to the next, resulting in mature endocrine cell type specification in islets. Specifically, α cells differentiation involves coordinated and tightly controlled specific transcription factor activation/repression that drive the transition from a stem cell to a mature α cell during embryonic development and the postnatal period until weaning. Since α and β cells had developed from common progenitor cells, evidence from animal and human studies suggest that mature α cells could regress backwards to a precursor-like stage and/or transdifferentiate into β cells; this process, in some circumstances, could increase β cells mass (Figure 1) [4–6]. Nevertheless, to date, the factors regulating trans-differentiation process and the specific molecular mechanisms involved are still under investigation. Intestinal hormones, particularly glucose-dependent insulinotropic polypeptide or gastric inhibitory peptide (GIP) and GLP-1, greatly influence islet cells to such an extent that, in some circumstances, they affect endocrine release of some pancreatic hormones. This network of hormone production inside the islet could be interpreted as a way to coordinate the secretory activity of Langerhans hormone-producing cells or as a maturing or trans-differentiative stimulus that makes cell populations more prone for the secretory support under particular conditions such as obesity, pregnancy, and metabolic or inflammatory stress. It is intriguing to notice that both pancreatic islets and endocrine intestinal cells share a similar hormone production apparatus able to release time after time gut hormones into the pancreas. These anatomic and functional features, overlooked for years, open new pathophysiological perspectives and question previous certainties.
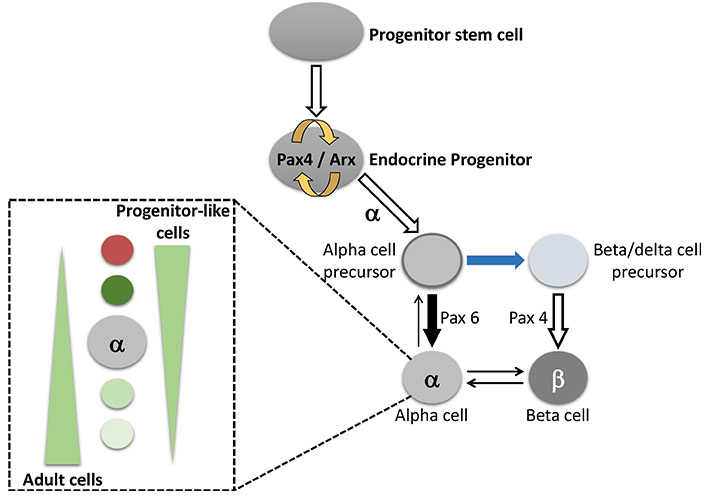
Schematic representation of the α cell differentiation. This process involves specific transcription factors that drive the transition from a stem cell to a mature α cell. The α cell has stem-like proprieties and could transdifferentiate into β cells. Furthermore, alpha cells inside the islet can have different differentiation grades (as shown in the left sub-panel). This phenomenon, widely reported for α and β cells, indicates that inside the same cell population there can be a spectrum of cell differentiation. More in details, α cell can be in a stem-like state able to differentiate into other cell types or into a finally committed cell. Pax4: paired box 4; Pax6: paired box 6; Arx: aristaless related homeobox
In 2011, Lee et al. [7, 8], supporters of the importance of glucagon in DM pathogenesis, produced data on murine models revolutionizing the whole diabetes knowledge of the last century. They prevented diabetes onset in rats treated with streptozotocin, a toxic agent for β cells, by blocking the glucagon receptor. Using rats with a total deletion of the glucagon receptor gene (GcgR–/–), thereby inhibiting glucagon action, they observed that the clinical phenotype of knockout rats was comparable to wild type (WT) rats. As expected, the transgenic rats showed a greater number and size of α cells as well as higher GLP-1 circulating levels. Both WT and GcgR–/– rats had comparable levels of plasmatic insulin. The most surprising result was obtained through the selective destruction of the β cell population using streptozotocin. Following the treatment with this compound, WT rats developed worsening hyperglycemia, polyurea, polydipsia, and death after only a few days. Conversely, rats lacking the glucagon receptor, did not develop any diabetic phenotypes during the treatment and the follow-up of 12 weeks, although serum insulin levels were undetectable. Furthermore, to verify any residue of β cells, the two rat groups (streptozotocin treated and untreated GcgR–/– rats) were subjected to oral glucose tolerance test (OGTT) (2 g/kg). This further analysis, including glycemia and insulinemia detection, showed that the trend of glycemia both in treated rats and in the control group was comparable, despite insulin levels not being measurable. These data show that, at least in rats, if glucagon action is blocked there is no diabetes development under an insulin-deprived condition. A year later, these results were strengthened by the same team with further data on a similar in vivo model [8]. Using GcgR–/– rats treated with streptozotocin, the authors induced the re-expression of the glucagon receptor through an adenovirus transient infection (lasting 8 weeks). They found that the presence of the glucagon receptor restored glucagon action and induced diabetes development. At the end of the 8 weeks of infection, the loss of the glucagon receptor was associated with normoglycemia. In other words, these results showed that the effect of insulin absence, even when complete, remained harmless if glucagon action was blocked. According to our actual knowledge, a therapy for “type 1 DM like” patients would be rather unimaginable without insulin supplementation, but in light of these experimental results it is clear that similar mechanisms could be believable in humans as well. Therefore, some questions arise spontaneously. For example, can patients with pancreatitis undergo massive destruction of the endocrine component of the organ? Why should these patients lacking part of the organ and insulin and glucagon production develop diabetes? Why can we treat these subjects just with exogenous insulin? Is the phenomenon described by Lee et al. [7, 8] specific to the rat model or are we overlooking some details? The following paragraphs try to answer these open questions.
In the 1970s literature provided many articles focusing on the endocrine function of the gut. Several research groups worked on this topic, including some Italian groups, and these initial data have become very interesting in light of our actual knowledge concerning incretin [9]. In that period, it was found that dog and human gut extracts contained substances able to cross-react with the anti-glucagon antibody [10]. The isolation technique available at that time was able to identify glucagon in the gastric fundus, duodenum, and colon of the dog, while according to the antibody cross-reaction, the expression of other peptides could be only hypothesized. For these technical limitations, further validations were needed. Later, these supposed substances were identified as glucagon-like-peptides. Nowadays it is widely known that the gut is capable of producing glucagon that originates from proglucagon [11]. Proglucagon was characterized in 1983 [12] when it was observed that its amino acid sequence comprises not only glucagon and GLP-1, but also oxyntomodulin, GLP-1, glicentin, and some other elements that are still not well defined and whose functions are not yet fully understood. However, it is known that specific enzymatic cleavages are determinant for the biogenesis of the specific end-product. Pro-hormone convertases 2 (PC2) is highly expressed in pancreatic α cells and it causes proglucagon processing into glucagon, glicentin-related pancreatic polypeptide, whereas pro-hormone convertases 1/3 (PC1/3) is more abundant in intestinal L-cells and neurons where it leads to glicentin, oxyntomodulin, GLP-1 and 2 formation [13]. Glucagon is produced not only by α cells to contrast hypoglycemia and insulin action, but also by the mammalian gut. This information could represent a new and entirely unopened field of research and it could be useful to solve some of the questions introduced in the previous paragraphs. Indeed, patients with pancreatitis develop diabetes despite the massive destruction of pancreatic islets because they still produce glucagon in the gut. Moreover, the unknown components originating from proglucagon cleavages could also play a role in the pathogenesis of new-onset diabetes for these patients. Moreover, GcgR–/– rats showed higher GLP-1 circulating levels, probably because of compensatory reasons. Is this phenomenon mediated by an augmented compensatory expression of the entire precursor or by an increased anti-hyperglycemic response to counteract insulin absence? To date, these aspects remain to be fully elucidated.
The scientific history of proglucagon is a paradigm for how endocrinology findings can progress.
The most relevant aspect is the complex and unexploited network of interactions between pancreatic and gut hormones. The model proposed by Unger and Lee [7, 8] consisting in an increase of GLP-1 circulating levels by glucagon receptor blocking, was confirmed later by other experimental data [14, 15]. Interesting information was provided by an in vivo mouse model in which both the action of glucagon and that of GLP-1 was inhibited. This model showed maintenance of carbohydrate tolerance, high sensibility to GIP action, increased cholecystokinin (CCK) action and induction of the expression of incretin-like compounds, probably derived from proglucagon secreted by intestinal endocrine cells [16]. Another murine model showing selective inhibition of both GLP-1 and GIP action, supported the evidence of gut-pancreas interactions [17]. In this experimental model, the lack of the two most important gastrointestinal hormones affecting glucose homeostasis, led to a compensatory increase of the other proglucagon-derived hormones. These data present some important and interesting food for thought about the management and the therapeutic approach to patients with DM. These findings were also confirmed in humans. An Australian team investigated the behavior of the measurable gastrointestinal hormones in subjects with a previous history of pancreatitis [18]. They found that oxyntomodulin more than others was able to regulate other intestinal hormone balance [glicentin and vasoactive intestinal peptide (VIP)] and metabolic homeostasis. Despite the fact that the authors focused on gastroenterological aspects and on the alterations of the intestinal microbiota, they discovered how intestinal hormonal balance was modified as a consequence of pancreatitis and how the study of intestinal hormones needs to undergo a systematic review to better understand their overall functions. The authors highlighted that these hormones are also produced in the brain opening new challenging areas of investigation. Gut-brain-pancreas interactions can no longer be overlooked in a modern conception of diabetes and in modern medicine.
To better understand gut-pancreas interactions it is important first to analyze cell-cell connections inside the pancreatic islet (Figure 2). In 1975, fifty-four years after insulin function discovery, Unger and Orci [19] published an article where the critical role of glucagon in the pathogenesis of diabetes was finally recognized. In the early 1970s, the scientific production of Unger highlighted the importance of glucagon, the role of insulin, the interactions between α and β cells, somatostatin control of both insulin and glucagon, and the similarities between glucagon and some gastrointestinal hormones [20–24]. In the last few years, these findings, along with the discovery of the incretin effect in humans and the use of GLP-1 for the treatment of hyperglycemia in diabetic patients, became crucial for a better understanding of diabetes pathophysiology. β cells account for 65–80% of islet cell subtypes and they produce insulin in response to food intake; an increase in blood glucose, proteins, and lipid levels induce a greater release of insulin, while fasting and hypoglycemia reduce hormone secretion. These events exert the same influence on glucagon secretion from pancreatic α cells. More in detail, α cells are sensitive to the increase in post-prandial glucose levels and meal-induced insulin spike. These conditions physiologically inhibit glucagon release, whereas hypoglycemia and lack of insulin induce its secretion. Up to the 1970s, α cells of diabetes patients were shown to not be responsive to such metabolic variations. For example, the concepts that post-meal hyperglycemia and hyperinsulinemia, in non-diabetic subjects, regulate glucagon secretion, and that the consequent hyperglucagonemia induces and sustains hyperglycemia in diabetes were well consolidated. However, a few years later, when the administration of GLP-1 in diabetic patients was found to reduce glucagon levels during hyperglycemia and to increase its secretion in response to hypoglycemia [25], all these aspects regarding pancreatic physiology and pathophysiology attracted the interest of the scientific community. α cells are very similar to β cells: both share secretion apparatus, ion channels, transcription factors, and morphology. These features are related to their common embryological origin. However, in terms of differentiation, α cells are less specialized and more genetically and epigenetically unstable [26]. This feature is particularly interesting since under specific conditions α cells can stop working as specialized cells producing glucagon and can convert into stem-like supporting cells or into more differentiated cells, such as β cells. Indeed, in murine models where the pancreatic β cell component was completely destroyed by streptozotocin treatment, it has been demonstrated that α cells control and coordinate pancreatic β cells repopulation [27]. These data, extremely important for regenerative medicine, drew scientists’ attention to these previously underestimated cells and to their crosstalk with other distinct cell subtypes within pancreatic islets. Increasing evidence has shown that some conditions responsible for metabolic injuries to β cells, such as lipotoxicity [28], cause an impairment of α cells as well. α cells chronically exposed to palmitate develop cell-specific insulin resistance and this could explain the unresponsiveness of this cell population to increased blood insulin and glucose levels [29]. Another finding of great interest is the ability of α cells to produce, on their own, GLP-1 when exposed to particular metabolic conditions [30]. GLP1 secretion by α cells occurs thanks to the expression of the proglucagon gene. In murine and human models, it has been seen that α cells produce glucagon from proglucagon, through PC2 activation, whereas they obtain GLP-1 through the action of PC1/3 [31]. GLP-1 production within pancreatic islets could be functional to regulate the secretion of different hormones (not only insulin), to sustain cellular identity, for stem-cell differentiation, or to prevent toxic/metabolic damage. GLP-1, at least in murine models, exerts a defensive role by protecting pancreatic islets from injury and death. Other distinct cell populations within pancreatic islets have been identified along with α and β cells. According to the common nomenclature, we can distinguish δ cells producing somatostatin, PP cells (known as F cells) producing pancreatic polypeptide and ε cell secreting ghrelin. Pancreatic islets contain other less characterized cell populations that could represent cells at an early phase of differentiation or cells not well characterized using our current estimating methodologies and knowledge [32]. For example, ghrelin positive cells are detected only in some pancreatic islets and they could represent cells at a specific stage of differentiation during the development process of the islet, contributing to maintain pancreatic islet architecture. Moreover, ghrelin positive cells are also found in the placenta, stomach, pituitary gland, testicular and thyroid: in pancreatic islets ghrelin is found co-localized with insulin and glucagon. A better understanding of the role of these “minor” islet endocrine cells and their alteration in diabetes, could provide new insights into islet pathophysiology and scientific advances in developing novel therapeutic strategies for diabetes.
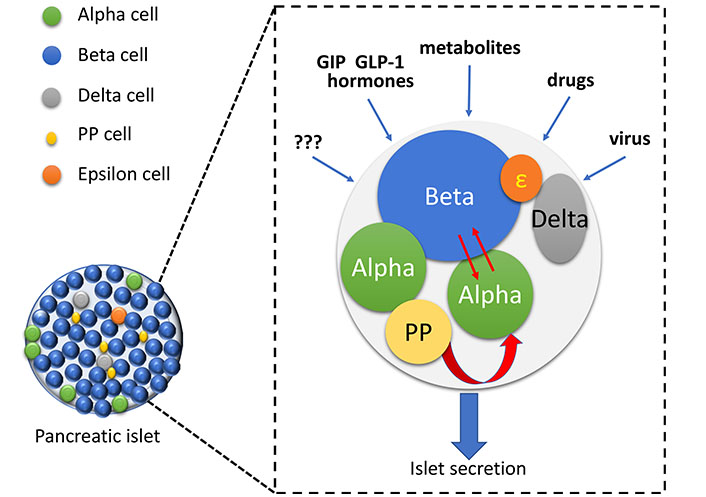
Schematic representation of Langerhans islet and a possible example of cell-cell connection inside the organ. Pancreatic islet has at least five different mature cell types. Each of these cell populations, in turn, represents a family including cells with several differentiation grade. Recently, it has been demonstrated that islets contain other less characterized cell populations that could represent cells at an early phase of differentiation or trans-differentiation. Several external factors can regulate and affect cell interactions inside pancreatic islet. These cell to cell interactions could modulate islet secretory homeostasis, cell proliferation and trans-differentiation
The ability of GLP-1 and receptor agonists/analogs to modulate glucagon secretion is well known. However, to date, the proof of a direct action of GLP-1 on α cells is lacking. Although the regulating action of GLP-1 on glucagon secretion is universally recognized, some doubts remain on the presence of the GLP-1 receptor on the α cell. The scientific production on the topic is not homogeneous with some reports finding the GLP-1 receptor expressed in a subpopulation of α cells and some others excluding this presence. To better clarify this issue, GLP-1 action was studied on PP islet cells, expressing the GLP-1 receptor. PP cells are located close to blood capillaries of human islets and they influence both insulin and glucagon secretion. Moreover, PP also regulated the intestinal motility and gastric filling [33, 34]. Chia et al. [35] recently demonstrated that PP cells express receptors not only for GLP-1 but also for GIP. The direct action of these two hormones on PP secretion could represent a link between gut and pancreas and could also have a direct effect on pancreatic islet physiology. While GLP-1 receptor expression is uncertain for α cells, GIP receptor expression has been widely demonstrated on α, β, and PP cells. Therefore, GIP is a gut-pancreas mediator of crucial importance. Donath et al. [36] employing murine and human experimental models, found that GIP promotes intestinal secretion of GLP-1 and acts as a mediator of gut-pancreas interaction. Moreover, GIP induced α cells to locally produce interleukin 6 (IL-6), which in turn stimulated insulin and GLP-1 production. These data could suggest that GIP makes the islet ready to GLP-1 action before that GLP-1 reaches the islets of Langerhans. Furthermore, interleukin production and inflammation could play a role in intra-islet GLP-1 production, hormone secretion and stem cell differentiation. The loss of GIP influence on pancreatic islets, as previously demonstrated in pre-diabetes subjects, could precede the loss of the incretin effect. Therefore, GIP could represent an early biomarker of pancreatic damage to be analyzed in populations at risk of diabetes [35–38].
These findings indicate that gut-islet cross-talk is essential to control islet secretion and to maintain cellular morphology. In particular, gastrointestinal hormones, acting on pancreatic islet cells, especially in cells expressing GIP and GLP-1 receptors, could contribute to maintaining and/or preventing cellular identity. In this way, we could imagine a physiological condition, where the balance is maintained (equilibrium state) (Figure 3A), or a condition (dis-equilibrium state) were pancreatic islet cells become insensitive to intestinal hormones (Figure 3B). In the latter state, cells lose their functional identity determining alterations of hormonal secretion and cellular morphology. Accordingly, the islet loses the normal architecture of the endocrine organ.
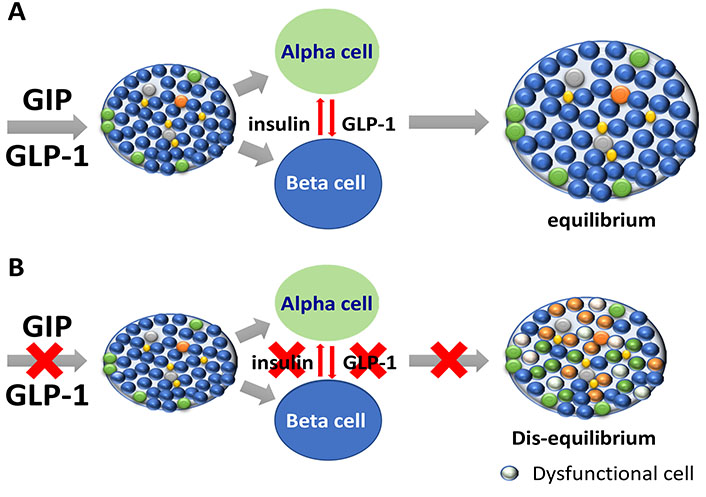
(A) Gastrointestinal hormones and pancreatic islets: in physiology. Gastrointestinal hormones, in particular GIP and GLP-1, regulate hormonal secretion and architecture in pancreatic islets. GIP, secreted by the gut, interacts with islet cell population and acts as mediator of gut-pancreas interaction. Some evidences indicate that GIP makes the islet ready to GLP-1 action and to release hormones and GLP-1. In this way, gastrointestinal hormones could contribute to preserve the islet cytoarchitecture. (B) Gastrointestinal hormones and pancreatic islets, hypothetical aspects of pathophysiology. Impaired GIP and GLP-1 actions on islet could lead to reduce the positive effects of gastrointestinal hormones in islets. If GIP and GLP-1 abnormally interact with islet, it is possible to imagine a “dis-equilibrium” in terms of hormonal secretion and cell to cell interactions. In these conditions, islet cytoarchitecture could be affected
GLP-1 is produced by intestinal L-cells, which are predominantly located in the ileum and colon [39]; moreover, L-cells are in direct contact with dipeptidyl peptidase-4 (DPP-4) producing cells. As soon as GLP-1 is secreted, it is almost completely degraded by DPP4. Since an ingested substance takes some hours to reach the colon, it is unlikely that the early GLP-1 secretion stimulated by nutrient ingestion could be sustained only by L-cells. Moreover, there is no evidence of a certain, direct colon-islet connection. It has been demonstrated that the fragments derived from GLP-1 and GIP inactivation act as mediators themselves, interacting with the central nervous system, through vagal afferents and hepatic receptors located near the portal vein. The connection existing between enteroendocrine cells, liver, and central nervous system and the resulting influence on endocrine pancreatic secretion and glycemic homeostasis supposes the existence of a connection axis known as the brain-neural-pancreas axis [40–42]. These new findings could reassess DPP-4 inhibitor therapy. These drugs could influence the network between gut, brain, and pancreatic islets, with an action that goes beyond the simple increase of circulating levels of GIP and GLP-1. Therefore, this class of drugs could reveal new, positive implications in the future.
To date, several interactions among different hormones and several systems in the body have been described. GLP-1 specific actions into blood vessels, skeletal and cardiac muscles, bone tissue, liver, kidney and adipose tissue have been widely studied and there are plenty of literature data [43].
Moreover, several benefits in terms of safety and death reduction of GLP-1 receptor agonists or analogous have been reported by cardiovascular outcomes trials (CVOTs). In addition of CVOTs, some molecular aspects of GLP-1 actions and interactions have been explored by new research lines.
Among pleiotropic effects of gastrointestinal hormones and especially GLP-1, it is important to report the emerging role of GLP-1 as anorexigenic and neuroprotective hormone [43–45]. Currently, other novel peptide hormones have been discovered. These novel intestinal peptides pave the way to novel molecular pathways and crass-talk pathways in the body; for instance, studies conducted in enteroendocrine cells led to the identification of novel mediators, such as the insulin-like peptide 5 (INSL-5) mainly expressed in brain and colon L-cells. It seems that this new identified hormone could act as an energetic sensor mediating brain-gut cross-talk. Its secretion is probably enhanced by short chain fatty acids (SCFAs) deriving from enzymatic digestion of gut fatty acids. For this reason, in this perspective, gut microbiota could exert a pivotal role because of its ability to hydrolyse fatty acids in different SCFAs. It has been reported that intraperitoneal, but not intracerebroventricular, administration of INSL-5 dose-dependently could increase food intake in mice, indicating that this peptide may exert its orexigenic effect by acting on peripheral targets, rather than via the central nervous system (CNS) [46]. Therefore, INSL-5 could represent an important link between the gut, brain, microbiota and host in the context of metabolism [47].
Furthermore, it has been recently reported that gut microbiota and the gastrointestinal hormones are involved in many neurological and neurodegenerative conditions including Parkinson’s disease and Alzheimer’s disease. However further additional studies should be conducted in order to elucidate the mechanisms underlying the microbiota-gut-brain-pancreas axis interaction and also the potential microbial-based intervention and therapeutic strategies for neurodegenerative disorders [48].
The above-mentioned findings support the hypothesis that intestinal hormones play a pivotal role both in the function and the integrity of pancreatic islet architecture. It is clear that pancreas-gut interactions are not mutually exclusive. Indeed, also pancreatic hormones could influence the physiology of the gut. In particular glucagon, principally produced by pancreatic alpha cells but also by enteroendocrine cells, could represent the hormonal factor connecting the gut and endocrine pancreas. GLP-1 and GIP are the most studied gastrointestinal hormones and their action on hormonal secretion, function, and survival of pancreatic islets is extensively known. The importance of this connection is underlined by experiments conducted in diabetic murine models where it was possible to isolate the effect of a single hormone or the action of multiple hormones. To date, over 20 gastrointestinal hormones have been identified and the number of their active fragments is enormous considering that several fragments may arise from the cleavage of the primary gastrointestinal hormone sequence. Furthermore, the presence of different isoforms of the receptor of each gastrointestinal hormone has recently been hypothesized [49]. In light of these considerations, in addition to glucose homeostasis, novel studies should be performed to analyze other effects of proglucagon-derived peptides. In the next few years, it will also be useful to better investigate the role of other hormones, such as CCK or gastrin, that we know regulate mainly the mechanical functions of the gut. It has recently been seen that CCK secreted by I cells in the duodenum has a crucial function in maintaining basal insulin and glucagon secretion [50–52]. Gastrin, expressed in the G cells of the gastric mucosa, is involved in differentiation, trans-differentiation, and neogenesis of ductal precursors towards pancreatic β cell [53, 54]. However, in the last few years, the role of novel hormones, called “decretins”, which behave in an opposite way to incretins, is emerging. Decretins, as limostatin and neuromedin U, are induced by fasting and suppressed insulin secretion. Neuromedin U, so named for its potent contractile activity on the uterus, has also been isolated in the intestine and the brain, therefore it could be involved in the gut-brain axis. Receptors of neuromedin U have also been identified in pancreatic islets, supporting its potential role in the regulation of pancreatic secretory machinery [50, 55–57]. All these findings pave the way for future research regarding the role of gastrointestinal hormones on the interaction with pancreatic islets and vice versa.
Recent scientific progress has profoundly changed the scenario of diabetes. Nowadays, nobody would base their own knowledge only on the binomial insulin-glycemia. The discovery of gastrointestinal hormones has allowed us to revise some scientific aspects that were already known but neglected for a long time, and to reconsider the milestones of our research within diabetes. For example, the brain should be taken into account in the context of a pancreas-gut interaction. The stimuli from food intake could play their proper role only when the brain-gut-pancreas system is working properly. Therefore, this newly identified axis seems to be the best overview to refer to a better understanding of diabetes pathophysiology.
In order to fully understand gastrointestinal hormone role and their interactions with other organs, it is essential to comprehend their mechanisms of action. Concerning this latter aspect, during the last years, novel findings regarding the physiology and pathophysiology of these hormones have been described that will lead to the development of novel therapeutic strategies at least for diabetes patient management. Among these novel findings, it is important to report the almost “ubiquitary” expression of GLP-1, GIP and glucagon receptors and their pleiotropic actions. However, there are different isoforms of these receptors accounting for tissue-specific responses [49]. The isoform expression could be influenced by metabolic state or energetic condition of tissues. These new paradigms could change our actual vision of incretin systems and gastrointestinal hormones. Moreover, according to the specific expressed receptor isoform in different tissues, the specific ligands, involved in the stimulus, could favour different metabolic responses paving the way for new points of view of obesity and diabetes [49]. This absolutely new evidence challenges the current knowledge and raise doubts concerning the therapeutic strategies based on GLP-1 receptor agonists/analogues commonly used in clinical practise. Nevertheless, the understanding of these aspects lays the foundation for the targeted use of a specific hormone rather than another, in order to have a specific action. Double and triple analogues/agonists/antagonists acting on GIP, GLP-1 and Glucagon receptors are in an advanced study phase and near to commercialisation. The possibility to modulate these hormone actions certainly will allow more targeted diabetes therapeutic strategies. Certainly, these multi-agonists are absolutely promising for the treatment of DM and obesity [58].
In light of these considerations, our journey should not be considered over. We must continue to explore, going beyond the boundaries of our knowledge, and avoiding that previous scientific achievements influence our future studies.
CCK: cholecystokinin
DM: diabetes mellitus
GIP: gastric inhibitory polypeptide
GLP-1: glucagon like peptide 1
INSL-5: insulin-like peptide 5
WT: wild type
We wish to thank the Scientific Bureau of the University of Catania for language support.
RM, AS, AF, SDM, AM, FP and SP wrote sections of the manuscript. All Authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This study was funded by PRIN 2017 and 2016/2018 Department Research Plan University of Catania, Department of Clinical and Experimental Medicine (project #A). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2020.
Copyright: © The Author(s) 2020. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Alessandro Mantovani ... Giovanni Targher
Agostino Milluzzo ... Laura Sciacca
Erika Pedone ... Amelia Caretto
Saula Vigili de Kreutzenberg
Mauro Maurantonio ... Luca Roncucci
