 Perspective
Perspective

Affiliation:
1Office of the Center Director, Center for Drug Evaluation and Research, U.S. Food and Drug Administration, Silver Spring, MD 20993, USA
†The authors contributed equally to this work.
Email: Cassandra.Taylor@fda.hhs.gov
ORCID: https://orcid.org/0000-0001-7476-2049
Affiliation:
2Office of Executive Programs, Center for Drug Evaluation and Research, U.S. Food and Drug Administration, Silver Spring, MD 20993, USA
†The authors contributed equally to this work.
ORCID: https://orcid.org/0000-0003-1022-7893
Explor Med. 2023;4:813–821 DOI: https://doi.org/10.37349/emed.2023.00179
Received: June 23, 2023 Accepted: August 17, 2023 Published: October 31, 2023
Academic Editor: Richard M. Sherva, Boston University School of Medicine, USA
The article belongs to the special issue Beyond Weed: Clinical Applications of Cannabis and Cannabinoids
Since the early 1970s, the U.S. Food and Drug Administration (FDA) has received over 800 investigational new drug applications (INDs) for, and pre-INDs pertaining to, research of cannabis or cannabis-derived products. The current data show that applications for research of these products submitted by both academic researchers and commercial developers focus on four major clinical areas: addiction and pain medicine (53%), neurology (19%), immunology and inflammation (14%), and psychiatry (9%). The product types studied have expanded greatly in recent years and include a wide variety of topical, inhalable, injectable, and oral products. In this article, the authors present a breakdown of cannabis and cannabis-derived applications received by the FDA over the past 50 years. The authors also provide a summary of their experience and challenges in reviewing applications for research of cannabis and cannabis-derived products, as well as recommendations for those interested in studying cannabis and cannabis-derived products in human clinical trials. This perspective article includes a discussion on important IND criteria, the pre-IND consultation program, drug master files (DMFs), and various guidance documents and resources. Lastly, the authors provide their perspective for the future of cannabis drug development.
The U.S. Food and Drug Administration (FDA) continues to observe increasing interest in botanical drug development, which includes the evaluation of cannabis and cannabis-derived products (CCDPs) in human clinical trials [1, 2]. Researchers who want to study cannabis as part of human research and/or drug development do so under an investigational new drug application (IND) [3]. The IND provides a mechanism for those developing a new drug to conduct studies in humans (clinical studies) and ship their investigational drug to clinical trial sites. Prior to submission of an IND, a researcher may seek input from FDA on the studies to be conducted under the IND through a pre-IND submission [4]. When FDA receives a submission for research of human drug products, it is assigned for review to a specific clinical review division within the Center for Drug Evaluation and Research (CDER) based upon the therapeutic use to be evaluated. Generally, the FDA’s Botanical Review Team (BRT) provides consultation on CCDP submissions received, regardless of clinical indication.
When INDs for CCDPs are submitted, CDER reviews the proposed research, including the product to be evaluated in the research, like it does INDs for any other product regulated as a drug (i.e., subject to the same authorities and requirements as drug products containing any other substance) [5]. Botanical products, such as cannabis, are complex mixtures that often have more than one active constituents that may contribute to their potential therapeutic effects. As part of CDER’s review team, BRT considers how chemically complex these products are when assessing if an IND satisfies the FDA’s requirements. To conduct clinical trials of CCDPs under an IND, all FDA requirements must be met, regardless of the source of cannabis or any other botanical product under study in the trial(s) [6].
To date, the FDA has not approved a marketing drug application for cannabis for the treatment of any disease or condition. However, clinical investigations of botanicals have led to the isolation of many naturally occurring compounds or derivatives of botanical compounds that have become well-known pharmaceuticals, as described in Wu et al. [7]. There are four examples of this related to cannabis drug development. Marinol (1985), Cesamet (1985), and Syndros (2016) are FDA-approved human drug products that are synthetic forms of a cannabinoid [delta-9 tetrahydrocannabinol (delta-9 THC)], which is a botanical compound found in the Cannabis sativa L. plant. The FDA has also approved Epidiolex (2018), which is a naturally occurring cannabinoid [cannabidiol (CBD)] that has been isolated from the plant and highly purified.
Applications to study CCDPs, including related synthetics, in humans began arriving at the FDA in the early 1970s. These early applications included protocols to evaluate mainly smokable forms of cannabis or single-molecule synthetic active pharmaceutical ingredients formulated into capsules or tablets. Over the last 50 years, more than 800 INDs evaluating CCDPs, including synthetic active pharmaceutical ingredients, have been submitted to the FDA. Notably, the past 10 years have brought a proliferation of new product types proposed for use in human clinical trials as well as an increase in applications. In this article, we provide a landscape of cannabis drug development in the United States by presenting the history and sharing our experiences in reviewing CCDP applications. We also include a perspective on what the future of cannabis drug development may look like.
As mentioned previously, more than 800 INDs have been submitted to the FDA since the early 1970s, with a dramatic increase in applications received in the past 10 years. During the first 40 years of this period, the FDA received over 400 applications; nearly identical to the number of applications received in the past 10 years. The beginning of this clinical research correlates with enactment of Title II of the Comprehensive Drug Abuse Prevention and Control Act of 1970, commonly called the Controlled Substances Act (CSA), enacted on October 27, 1970 [8]. The CSA established “a federal policy to regulate the manufacturing, distributing, importing/exporting, and use of regulated substances” [9] and placed all substances that were in some manner regulated under existing federal law into one of five schedules [10]. Certain cultivars and parts of the Cannabis sativa L. plant have been controlled under the CSA since 1970 under the drug class “Marihuana” [11].
The FDA’s BRT has supported this growth in research by leading the development and publication of multiple guidance documents [12, 13] and manuscripts [5, 7]. In 2016, the FDA issued Botanical Drug Development, revised final guidance for industry [12]. This guidance is intended to aid applicants in the development of botanical drug products and provides the FDA’s current thinking regarding growing conditions and manufacturing quality controls for botanical raw materials (BRMs), such as aerial parts, roots, rhizomes, flowers, leaves, and/or the whole plant. In 2023, the FDA issued Cannabis and Cannabis-Derived Compounds: Quality Considerations for Clinical Research, final guidance for industry [13]. This guidance reflects the FDA’s current thinking on clinical research of human drugs containing CCDPs, with a focus on manufacturing high-quality drug products and provides recommendations for those interested in developing these types of products for use in drugs for clinical research. The FDA continues to support robust scientific research needed to develop new drugs from cannabis and is committed to supporting the development of these new drugs through the IND and drug approval processes.
CDER categorizes INDs as either commercial or research (non-commercial). INDs must contain information in three broad areas: animal pharmacology and toxicology studies, manufacturing information, and clinical protocols and investigator information [6]. Once an IND is submitted, the sponsor must wait 30 calendar days before initiating any clinical trials, unless FDA explicitly authorizes earlier initiation of the clinical trials [6]. During this time, the FDA reviews the IND for safety, ensuring that research subjects will not be subjected to unreasonable risk [3]. Once the FDA has reviewed the IND and determined that it is safe to proceed, the IND is considered “active”. Currently, the FDA has over 150 active commercial and research INDs that evaluate CCDPs, including related synthetics.
In the past 50 years, nearly all clinical review divisions in CDER’s Office of New Drugs [14] have received at least one cannabis product application (IND or pre-IND). The clinical trials conducted (INDs) or proposed for consideration (pre-INDs) under these applications evaluate the use of CCDPs for a wide variety of clinical indications and are categorized as approximately three-fourths research and one-fourth commercial. Research INDs are typically sponsored by individual investigators (primarily physicians) or groups of investigators from academic and research institutions. Commercial INDs are often sponsored by pharmaceutical manufacturers with clear intentions to eventually seek approval to market the drug product. The research under the majority of CCDP INDs focuses on four major therapeutic areas: addiction and pain medicine, neurology, inflammation, and psychiatry (Figure 1). At least 53 percent of these INDs include clinical trials with addiction and pain medicine indications, 19 percent have neurological clinical indications, 14 percent have immunology and inflammation indications, and 9 percent have psychiatry indications.
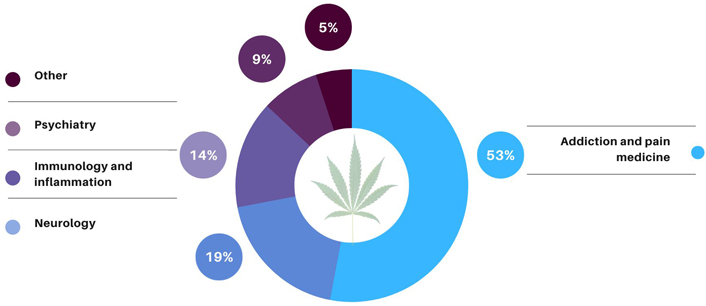
Cannabis clinical research areas. Most of the CCDP applications are focused on four major research areas: addiction and pain medicine, neurology, immunology and inflammation, and psychiatry. [Background image: Greola84/iStock/Getty Images Plus via Getty Images.]
Investigators have begun studying a wider variety of routes of administration (ROAs) over the last 50 years under clinical trials (Figure 2). In the 1970s and 1980s, much of the cannabis used under clinical trials was inhaled via cigarettes obtained from the National Institute on Drug Abuse [15], with a few oral product types being studied. The drug approvals of Marinol (May 1985) and Cesamet (Dec. 1985), which are oral formulations of synthetic delta-9 THC, occurred relatively soon after the enactment of the CSA in 1970.
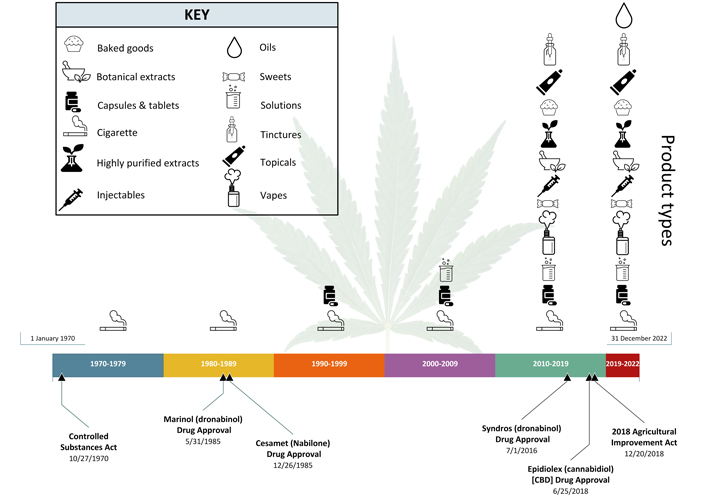
Changes to product types over the past 50 years. Timeline spans from January 1, 1970, to December 31, 2022, and is divided into 10-year timeframes (as applicable). It includes two major pieces of federal legislation, the CSA of 1970 and the 2018 Agricultural Improvement Act as well as significant FDA drug product approvals. These include one cannabis-derived drug product (Epidiolex) and the three synthetic cannabis-related drug products (Marinol, Syndros and Cesamet). These events are presented in conjunction with the general product types commonly studied under clinical trials for each timeframe. The trend of studying varied product types dramatically increased from 2010 to 2019 and continues today. [Background image: Greola84/iStock/Getty Images Plus via Getty Images.]
Over time other oral product types began to be proposed for clinical trials in the 1990s and 2000s, but inhalation via cigarette remained the primary ROA studied. However, in the 2010s the variety of product types dramatically expanded in clinical trials to include a wider array of oral product types and other ROAs. Broadly, the product types proposed for use included baked goods, botanical extracts, capsules and tablets, oils, purified (or semi-purified) extracts, sweets (e.g., candies), tinctures, and vaporized products (e.g., vapes). A significant amount of these product types are botanicals and are heterogeneous in nature. The increase in the types of products and ROAs being studied in clinical trials correlates with the drug approvals of Syndros (2016) and Epidiolex (2018), which are both oral formulations. This expansion also tracks with the increased availability of products in the state-level marketplace [16], along with the passage of the Agricultural Improvement Act (i.e., the Farm Bill) on December 20, 2018 [17]. The diversification of ROAs and product types in clinical trials has continued since 2020 to present and includes, but is not limited to, products for inhalation, oral ingestion, topical application, and injection (Figure 2).
The emergence of interest in studying these various ROAs under clinical trials is likely due to changes in the cannabis consumer in the United States. In recent years, consumers have transitioned from exclusively smoking dried cannabis flower to consuming other non-flower forms, such as edibles [18]. This is especially prevalent among younger consumers [19]. Overall, edibles have grown significantly as a product type [18]. This growth could be attributed to the ease of use and access of edible products, as well as a general reduced stigma around cannabis use in current society. In addition, the dramatic expansion of the marketplace allows a consumer to shop around for a preferred product type based upon their individual situation (e.g., at home versus in social settings). The consumer preference has provided an interest and opportunity for researchers to utilize multiple ROAs and for the product(s) to be tailored to clinical trial participants. However, there remains a complexity and uniqueness to developing cannabis product types for use in clinical research. These research challenges include, but are not limited to, absent and/or inadequate quality and manufacturing information, unknown safety profiles and unknown benefits/risks for emerging compounds [e.g., hexahydrocannabinol (HHC), THC acetate (THC-O)], and the complex matrix effects on testing of final product formulations.
Over the course of 50 years, the FDA has accumulated valuable experience by assessing more than 800 INDs that evaluate cannabis products. The FDA recognizes the uniqueness of botanicals, including CCDPs, and the technical challenges associated with botanical drug development and research. Due to the heterogeneous nature of botanicals, quality controls and therapeutic consistency of botanical drug substances and botanical drug products are known challenges. In general, these can be supported by the “totality of the evidence” approach as outlined in the Botanical Drug Development guidance for industry [12]. Research and drug development using CCDPs have the same challenges as other botanicals with the addition of considerations of control status under the CSA. The CSA and its implementing regulations are within the Drug Enforcement Administration’s (DEA) jurisdiction. Those proposing drug development activities involving controlled substances should consult DEA about the applicable requirements. It may be useful for researchers to calculate the delta-9 THC content in their proposed CCDP early in the development process to gain insights into the product’s potential abuse liability and control status (Section Ⅲ.C in [13]). Those using cannabis BRMs in drug development activities must comply with 7 CFR 990 [20] (or any superseding regulations) and should reference the United States Department of Agriculture’s guidelines for sampling [21] and testing [22] methods for evaluating the levels of delta-9 THC.
During the review of IND submissions, we have commonly encountered research programs that are unable to meet the IND requirements [6] related to the BRM source, the chemistry, manufacturing, and controls (CMC) for the proposed botanical drug substance and/or botanical drug product, as well as the nonclinical (i.e., animal pharmacology and toxicology studies) and clinical requirements. As part of an IND for any investigational human drug (including those that contain CCDPs), sponsors are expected to provide information to show that they can consistently manufacture a quality product. In each phase of clinical investigation, sponsors must submit sufficient information to demonstrate the identity, quality, purity, and potency or strength of the investigational drug [6]. The amount of information appropriate to meet this expectation increases with successive stages of drug development. We recommend reviewing the Botanical Drug Development guidance for industry [12] and the Cannabis and Cannabis-Derived Compounds: Quality Considerations for Clinical Research guidance for industry [13] for examples of IND requirements that need to be satisfied to begin clinical research, including BRM controls, CMC information [23–30], and devices [31, 32]. In addition, these guidance documents provide considerations for previous human experience (e.g., pharmacology and toxicology studies). The FDA’s website contains information, including guidance documents, to assist sponsors in preparing INDs both generally [3] and for cannabis specifically [33]. We recommend utilizing the Agency’s resources to avoid delays in drug development and research plans.
Another tool for researchers is CDER’s pre-IND Consultation Program [4]. This program fosters early communications between researchers and the Office of New Drugs’ review divisions to provide specific feedback on the data necessary to include in an IND submission. The FDA encourages researchers to request a pre-IND meeting [34] to discuss questions related to the development of specific CCDPs. The FDA websites also contain a wealth of resources, specifically, FDA and Cannabis: Research and Drug Approval Process [33] provides background knowledge and steps to assist researchers proposing to study CCDPs. The FDA Regulation of Cannabis and Cannabis-Derived Products, Including CBD page [35] highlights FDA news releases and statements, consumer information, FDA remarks and testimony, science and research, other regulatory questions, and frequently asked questions and answers on CCDPs. In addition to these resources, there are a handful of other CDER programs available to help speed up drug development if certain criteria are met [36].
Lastly, another option to assist with meeting the manufacturing IND requirements is utilizing the well-established drug master file (DMF) pathway. DMFs are submissions to the FDA used to help speed drug development, provide confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of human drug products [37]. DMFs utilize a standardized electronic submission platform and can be updated at any time. During the FDA’s May 31, 2019, public hearing [38] on CCDPs, the Agency heard concerns from stakeholders about their ability to protect proprietary information while participating in drug development. Since then, the FDA has continued to encourage stakeholders to submit DMFs for cannabis products to the Agency for review. To use the information provided in a DMF, researchers must obtain a letter of authorization (LOA) from a DMF holder to support their proposed clinical trial under IND or to support a new drug application (NDA) [39, 40]. The FDA will then conduct a technical review of the DMF contents for adequacy. The reviewed contents in a DMF are neither approved nor disapproved. DMF content is only shared between the FDA and the DMF Holder, meaning the contents of a DMF are not shared with a sponsor/researcher who provides a LOA. Many researchers can reference the same DMF at the same time, allowing for ease of information sharing from the DMF holder to support multiple clinical trials under FDA review simultaneously. In addition, there is no cost to submit a DMF if it is in support of an IND or NDA. An effective way to satisfy IND requirements may be by obtaining and submitting a LOA to reference an existing IND, NDA, or supplier’s DMF that contains the required information for the product under development.
We predict that the FDA will continue to see an increase in INDs evaluating CCDPs. As more medical and adult-use (i.e., non-medical) programs at the state-level evolve, there will likely be increased interest in clinical research to study CCDPs that are commonly used by consumers in state-level programs. With this, it is also expected that FDA will see new, innovative products for evaluation under INDs submitted to the Agency. This may include the use of newly identified or less common cannabinoids, as well as other components of the cannabis plant, such as terpenes. Researchers may also begin to expand on their use of edible CCDP product types, as well as utilize novel delivery systems for CCDPs under IND. There may also be more stakeholders submitting DMFs to the FDA in the future. Stakeholders may utilize this well-established pathway to help support drug development.
The FDA will continue to encourage robust clinical research of CCDPs. The Agency supports sound, scientifically based research into the therapeutic uses of CCDPs and will continue to work with companies interested in bringing safe, effective, and quality drug products to market. The FDA believes the drug development and approval process represents the best way to ensure that safe and effective new medicines are available to patients in need of appropriate medical therapy. This includes drugs that contain cannabis and cannabis-derived compounds.
BRMs: botanical raw materials
BRT: Botanical Review Team
CCDPs: cannabis and cannabis-derived products
CDER: Center for Drug Evaluation and Research
CSA: Controlled Substances Act
DMF: drug master file
IND: investigational new drug application
LOA: letter of authorization
NDA: new drug application
ROAs: routes of administration
THC: tetrahydrocannabinol
U.S. FDA: United States Food and Drug Administration
Disclaimer: This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
CLT and SAP: Conceptualization, Data curation, Formal analysis, Investigation, Visualization, Writing—original draft, Writing—review & editing. Both authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Gregory Giordano ... Angela D. Bryan
Lucile Rapin ... Michael Dworkind
Elizabeth N. R. Schjelderup ... Alasdair M. Barr
Hannah Thurgur ... David J. Nutt
Amanda Stueber, Carrie Cuttler
Gerhard Nahler
Trevor R. Norman
