 Review
Review

Affiliation:
Department of Food Science and Nutrition, Nara Women’s University, Nara 630-8506, Japan
Affiliation:
Department of Food Science and Nutrition, Nara Women’s University, Nara 630-8506, Japan
ORCID: https://orcid.org/0000-0001-9024-7434
Affiliation:
Department of Food Science and Nutrition, Nara Women’s University, Nara 630-8506, Japan
ORCID: https://orcid.org/0000-0002-1803-2620
Affiliation:
Department of Food Science and Nutrition, Nara Women’s University, Nara 630-8506, Japan
Email: smatsuda@cc.nara-wu.ac.jp
ORCID: https://orcid.org/0000-0003-4274-5345
Explor Med. 2023;4:822–838 DOI: https://doi.org/10.37349/emed.2023.00180
Received: May 25, 2023 Accepted: August 10, 2023 Published: October 31, 2023
Academic Editor: Calogero Caruso, University of Palermo, Italy
The article belongs to the special issue Determinants of Exceptional Longevity
Progeroid syndromes are characterized by clinical signs of premature ageing, which may contain several diseases such as Werner syndrome, Bloom syndrome, Rothmund-Thomson syndrome, Hutchinson-Gilford progeria syndrome, and Cockayne syndrome. These disorders may also exhibit some pathological involvements reminiscent of primary mitochondrial diseases. Emerging evidence has linked mitochondria even to physiological ageing. In addition, alterations in the maintenance pathway of mitochondria have been also deliberated as relevant in age-related diseases. In particular, mitophagy and its regulatory pathway might be key process for the homeostasis of mitochondria. Therefore, chronic DNA damage and/or the activation of poly[adenosine diphosphate (ADP)-ribose] polymerase 1 (PARP1) could be a threat to the mitochondrial alterations. The PARP1 is an enzyme responding to the DNA damage, which might be also involved in the mitophagy. Interestingly, the PARP1 has been reported to play an important role in the longevity of lifespan, which has attracted growing attention with the social development. This review may provide a rationalized overview of the involvement of mitochondrial oxidative stresses in genetically defined accelerated ageing, progeroid syndromes, physiological ageing, and/or age-related diseases for the innovative therapeutic approaches.
The increasing proportion of elderly people in society characterizes an accumulative financial burden because of age-associated diseases and the significant associated cost of health and well-being maintenance [1]. There are broad-ranging suggestions on finding cost-effective interventions for age-associated diseases. Several genetically defined accelerated ageing disorders have been profoundly considered in the comprehension of ageing. Interestingly, all these disorders are associated with some defects in the maintenance of genome. In other words, mutation in DNA repair genes may be involved in the pathogenesis of these genetically ageing disorders [2]. In addition, it is becoming gradually evident that direct or indirect DNA damage by highly reactive oxygen species (ROS) may play central roles in ageing [3]. In fact, the clinical symptoms of progeroid syndromes might be associated with the molecular features of ultraviolet radiation (UVR)-induced oxidative stresses [4]. Moreover, some of these disorders may show neurological involvements reminiscent of which are realized in several mitochondrial illnesses [5]. Mitochondria are the energy factories of the cells. Evolving evidence has connected this organelle to ageing and understanding mitochondrial dysfunction in accelerated ageing diseases [6]. Therefore, an accumulation of damage to mitochondria may trigger the process of accelerated ageing. Furthermore, nuclear DNA damage may lead to enlarged energy consumption, which may also direct to alterations in mitochondrial energy production [7, 8]. Therefore, mitochondrial modifications could secondarily occur in the process of extra DNA repair after the excess DNA damage.
Mitochondria are an essential part of cells, which may have key roles in a group of metabolic processes including energy production and/or free radical generation. In addition, mitochondria have been considered as significant components in the process of ageing and cell death [9]. Exhausts of cellular energy reserves might lead to mitochondrial dysfunction and/or altered cellular metabolisms [10]. However, mitochondria are flexible organelles capable of varying their function under certain conditions caused by diverse elements of stressors, exercise, and/or diet [11]. In light damage, mitochondria could be repaired by the fusion with intact mitochondria. Otherwise, mitochondria might be segregated and transported to lysosomes for breakdown in the case of severe damage. This mitochondrial recycling is called mitophagy. Mitochondrial dynamics are thought to have important physiological roles in maintaining homeostasis [12]. Therefore, mitophagy has been recognized to be a key process for the homeostasis of mitochondria.
Several disorders characterized by chronic DNA damage and/or poly[adenosine diphosphate (ADP)-ribose] (PAR) polymerase 1 (PARP1) activation may be at risk for mitochondrial alterations. Consequently, the PARP1 may be involved in the pathogenesis of these disorders with the alteration of mitophagy, which might respond to DNA damage [13]. As the PARP1 is a typical representative of the PARP enzyme family, it would be mentioned hereafter for the explanation of PARP function. The PARP1 could irreversibly cleave nicotinamide adenine dinucleotide (NAD+) generating nicotinamide and monomeric ADP-ribose. The ADP-ribose unit could be poly-elongated in a process of poly(ADP-ribosyl)ation (PARylation), which might be involved in genome repair [14]. Interestingly, increased activation of PARP1 might be involved in the increased adenosine triphosphate (ATP) consumption in the situation of senescence [8]. The PARP1 has been reported to play an important role in longevity [15]. Upon activation of PARP1, some metabolites may inhibit pathways which are usually protective for cells. Then, PARP inhibitors have been suggested for alleviation of inflammation in chronic diseases. Therefore, the inhibition of PARP1 could reverse the phenotypes associated with accelerated ageing [15]. This implies that modulation of the PARP1 pathway might be therapeutic tactics for treating accelerated ageing diseases and/or disorders. In particular, NAD+ levels may have a deep impact on mitochondrial function and/or senescence. It could be speculated that NAD+ levels, PARP1 activation, mitochondrial dysfunction, neurodegenerative diseases, neuron senescence, and/or ageing may propagate a vicious cycle instead of guiding causal and resulting relationship.
Progeroid syndromes are a cluster of diseases characterized by indications of premature ageing, which may mimic in part physiological ageing [16]. These syndromes comprise several diseases such as Bloom syndrome, Werner syndrome, Hutchinson-Gilford progeria syndrome, Rothmund-Thomson syndrome, and Cockayne syndrome [16], which may exhibit some pathological involvements reminiscent of primary mitochondrial diseases (Figure 1). Mitochondrial illnesses are also a group of genetic diseases categorized by some defects in mitochondrial function [17]. In general, mitochondria are power plants of cells generating cellular energy ATP. Emerging evidence has linked this organelle to physiological ageing and found mitochondrial dysfunction in accelerated ageing disorders. An accumulation of damage to the mitochondria may trigger the process of ageing. Nuclear DNA damage may lead to increased energy consumption, and therefore, the mitochondrial alterations may be secondary to faults in nuclear DNA repair and/or DNA damage [8, 18]. Alterations in mitochondrial ATP production may be caused by the activation of PARP1, an enzyme that responds to DNA damage [19]. Activated PARP1 utilizes key metabolites which may attenuate pathways for the protection of cells. Notably, pharmacological inhibition of PARP1 could reverse the phenotypes related to accelerated ageing [20]. PARP1 may be characterized as a multitasking protein that achieves some signaling functions in the situation of cellular stress response as well as senescence and aging [20]. This suggests that modulation of PARP1 function and/or the related signaling may become a key target of therapeutic tactics for treating accelerated ageing.
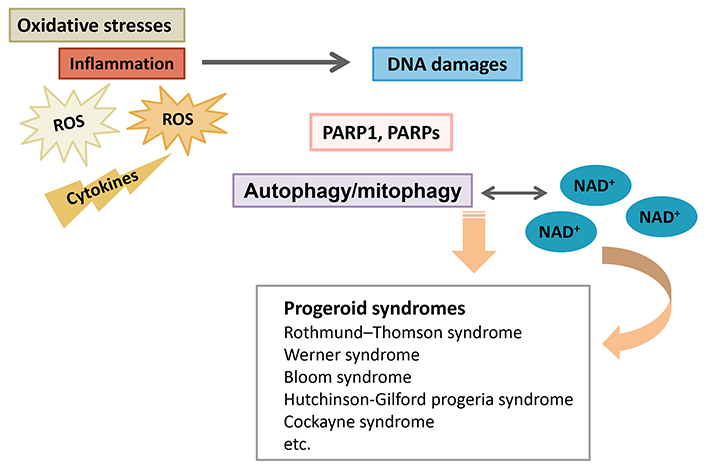
Schematic overview for the pathogenesis of progeroid syndromes such as Rothmund-Thomson syndrome, Werner syndrome, Bloom syndrome, Hutchinson-Gilford progeria syndrome, and Cockayne syndrome. The PARP1, PARPs, and/or NAD+ pools might contribute to the pathogenesis of several progeroid syndromes via the alteration of autophagy and/or mitophagy. Inflammation with several cytokines and/or ROS may be also involved in the modification of autophagy/mitophagy
Rothmund-Thomson syndrome is a rare autosomal recessive disorder characterized by premature ageing including skin atrophy, hyperpigmentation, loss of hair, cataracts, and/or skeletal anomalies, which may be associated with increased susceptibility to various cancers. A subset of mutations in the RecQ like helicase 4 gene (RECQL4) has been linked to two types of additional disorders [21]. Gene product of the RECQL4 belongs to a family of DNA helicases that plays an important role in maintaining genomic stability [22]. RECQL4 could interact with PARP1, a nuclear enzyme that also promotes genomic integrity, through its involvement in DNA repair and/or their signaling pathways. The carboxy (C)-terminal region of RECQL4 might be a substrate for PARP1, suggesting an interaction between RECQL4 and PARP1 in response to oxidative stresses [23] (Figures 1 and 2).
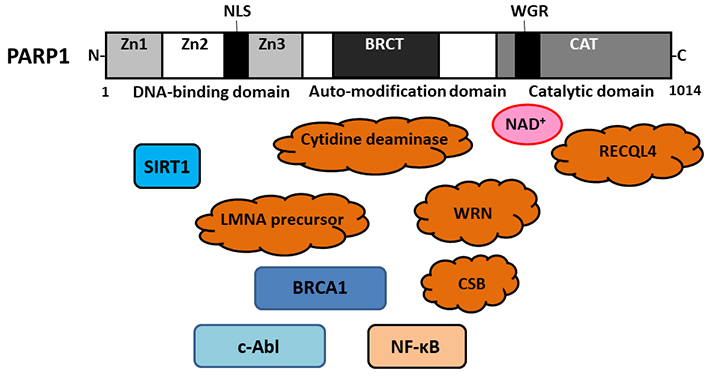
Schematic representation of the modular organization of human PARP1 domains and subdomains. The N-terminal DNA-binding domain contains 3 Zn fingers, which are responsible for DNA binding and some protein-protein interactions. A NLS can be found in this DNA-binding domain. The automodification domain is a central regulating segment with a BRCT, which may serve protein-protein interactions. The C-terminal region accommodates the catalytic centre of PARP1. The WGR domain in the catalytic domain may be important for NAD+ binding. Several key molecules responsible for progeroid syndromes including RECQL4 (Rothmund-Thomson Syndrome), WRN (Werner syndrome), cytidine deaminase (Bloom syndrome), LMNA (Hutchinson-Gilford progeria syndrome), CSB (Cockayne syndrome), might be associated with the PARP1. N: amino; Zn: zinc; NLS: nuclear-localization signal; BRCT: BReast CAncer gene 1 C-terminal; WGR: tryptophan glycine and arginine rich; WRN: Werner syndrome protein; LMNA: lamin A/C; CSB: Cockayne syndrome protein B; CAT: catalase; SIRT1: sirtuin 1; BRCA: BReast CAncer gene; c-Abl: cellular Abelson tyrosine kinase; NF-κB: nuclear factor-kappa B
Werner syndrome is also a rare autosomal premature ageing disease connected with a predisposition to cancers and/or genomic instability, which might be initiated by some mutations in the gene encoding the WRN, a member of the RecQ family helicases with a potential role in sustaining genomic stability [24]. Loss of the function of a truncated WRN protein in Werner syndrome may result in high levels of early apoptosis exhibited by intensifications of cleaved caspases and/or PARP1. WRN might play an important role in the protection against DNA damage [25]. It has been shown that PARP1 could bind to the N-terminus of WRN and to the C-terminus encompassing the RecQ-conserved domain [26]. In addition, ADP-ribosylation of PARP1 could restore the activity of WRN exonuclease in the presence of NAD+ [26] (Figures 1 and 2).
Bloom syndrome is also a rare genetic disease characterized by high levels of chromosomal instability and/or a great increase in cancers risk. Cytidine deaminase expression might be downregulated in cells of Bloom syndrome, possibly leading to decreasing the PARP1 activity [27]. Decrease in the basal activity of PARP1 may contribute to the incomplete sister chromatid disjunction [27, 28]. Therefore, it is possible that PARP1 is involved in sister chromatid disjunction during mitosis [27, 28]. The genetic instability associated with the phenotype of Bloom syndrome may result from a defect of cytidine deaminase [29]. Cytidine deaminase is an enzyme of the pyrimidine salvage pathway, which could decrease the basal activity of PARP1 [30]. The decrease in the PARP1 activity might reduce the efficacy of downstream checkpoints, leading to the increase of unreplicated DNAs during mitosis (Figures 1 and 2).
Hutchinson-Gilford progeria syndrome is a rare autosomal dominant disorder, which is also a fatal developmental disorder characterized by accelerated ageing including bone resorption and/or atherosclerosis [31]. Hutchinson-Gilford progeria syndrome may be caused by sporadic mutations in the LMNA gene, which might produce a mutant LMNA precursor of progerin [32]. Interestingly, the NAD+ salvage pathway has been altered in cells carrying the LMNA mutation, leading to altered PARP1-mediated PARylation [33]. In addition, LMNA could promote the PARP1 mono(ADP-ribosyl)ation in response to DNA damage [34] (Figures 1 and 2).
Cockayne syndrome is a rare autosomal recessive disease described as a segmental premature-aging syndrome. One of the foremost clinical hallmarks of Cockayne syndrome is neurological abnormalities. Mutations in CSB gene are present in the majority of Cockayne syndrome patients. The CSB might be an ATP-dependent chromatin remodeler without helicase activity, which could discharge oxidative stresses by regulating DNA repair pathway. Significantly, the PARP1 could enhance the kinetics of chromatin association with the CSB. In addition, CSB could associate with PARP1 [35]. After the oxidative stresses, CSB and PARP1 could colocalize in the nucleoplasm. poly(ADP)ribosylated (PARylated) PARP1 might be prerequisite for retaining the CSB protein at sites of oxidative DNA damage to facilitate DNA repair [36, 37]. In general, unrepaired oxidative DNA damages have been recognized to increase cancer incidence and premature ageing phenotypes. The regulation of PARP1 by the CSB protein might be a key function in cells with oxidative stresses [38] (Figures 1 and 2).
As PARP1 utilizes NAD+ as substrate, its extreme activation has an inclusive effect on the NAD+ metabolism in addition to the related intracellular signaling. In general, almost all members (17 members) in the PARPs family could modulate NAD+ metabolism, energy pathways, and/or oxidative metabolism. Reversible PARylation is a pleiotropic controller of several cellular functions [39]. However, uncontrolled PARPs activation may cause cell death. Therefore, noncovalent PARylation could be deliberated as a posttranslational modification of various factors to mitochondrial cell death. Utilizing the oxidized form of NAD+ as a substrate, PARPs could catalyze the covalent binding of ADP-ribose units onto aspartate, glutamate, lysine, serine, and tyrosine residues of various target proteins [40]. ADP-ribosylation might be a well-known posttranslational modification of various proteins with a noticeable regulation of many biological procedures including cellular responses to DNA damage. PARP1 has been defined as one of the fundamental DNA damage-responsive elements required for the preservation of genomic integrity. In response to several DNA damage affected by some genotoxic agents, PARP1 could promote a speedy production of PAR chains required for chromatin relaxation to organize the PAR-binding proteins, DNA repair proteins, and/or several transcription factors [41]. In fact, PARP1 is considerably involved in triggering DNA repair mechanisms [42].
Mammalian PARP1 consists of three domains such as DNA-binding domain, auto-modification domain, and catalytic domain in the C-terminus [43] (Figure 2). The auto-modification domain in the middle has a BRCA related region, which could promote the recruitment of DNA-repair enzymes. Termed with tryptophan, glycine, and arginine amino acid residues, the WGR domain in the catalytic domain might be important for NAD+ binding [44]. Moreover, c-Abl protein could interact with PARP1, which might be crucial for the expression of inflammatory genes [45]. Furthermore, the PARP1 could influence the expression of inflammatory cytokines including interleukin (IL)-1β and/or tumor necrosis factor (TNF)-α, thus stimulating inflammation [46]. Therefore, for example, excessive activation of PARP1 in microglia may contribute to several neuronal damage, which may be connected to the chronic neuro-inflammation [47]. Besides, acetylation of PARP1 plays an imperative role in several transcriptional regulations [48], which also supports inflammation via the regulation of NF-κB signaling [49]. Together, post-translational modification might be significant in the regulation of PARP1 activity. It has been shown that PARP1 could protect neurons from neuronal cell death under mild oxidative stresses as a result of successful DNA repair [50]. When severe DNA damage occurs, PARP1 may be excessively activated, leading to exhaustion of NAD+ and ATP, and eventually to cell death [51]. SIRT1, which is a NAD+-dependent protein deacetylase, and plays indispensable roles in metabolic control, may be repressed after PARP1 activation. Actually, PARP1 may cooperate with SIRT1 in conditions such as mitochondrial metabolism, DNA damage repair, and/or inflammation [52]. During the course of physiological ageing, SIRT1 activity might be decreased, leading to increased acetylation of PARP1 as well as raised activity of NF-κB [53]. By the same token, it has been demonstrated that PARP1 decreases with cumulative level of progerin in cultured progeria cells [54]. Remarkably, inhibition of PARP1 can improve mitochondrial function through activating the SIRT1 [55]. In addition, PARP1 could contribute to the preservation of blood-brain barrier (BBB) [56] and/or several circadian rhythms in brain [57], which might be linked to the pathogenesis of several neurodegenerative diseases [58]. Accordingly, PARP1 could participate in a diversity of intracellular processes to influence cellular function and/or inflammatory conditions.
The interaction between PARP1 and adenosine monophosphate-activated protein kinase alpha (AMPKα) has been defined for DNA damage dependent activation of PARP1, while PARP1 might be a target of AMPKα [59]. In addition, an anti-inflammatory crosstalk connecting AMPK to PARP1 activity has been revealed [59]. Consequently, full AMPK activity might be required for PARP1 activation during nutrient deprivation [60]. In PARP1-deficient cells, a decreased production of ROS has been shown even at early time point after starvation [60]. A nuclear quantity of α isoform of AMPK has been defined to be able to interact with PARP1 [15]. Deficiency of effective PARylation might keep the stability of the PARP1/AMPKα complex in PARP1-inactivated cells. Accordingly, the AMPKα could not be transported from the nucleus to the cytosol during the starvation. In consequence, an inefficient activation of the cytosolic AMPKα may incompletely maintain the activity of mammalian/mechanistic target of rapamycin complex 1 (mTORC1) compromising the activation of unc-51 like autophagy activating kinase 1 (ULK1) with the initiation of phagophores [60]. Therefore, the mutual interaction between PARP1 and AMPKα may direct to the optimization of autophagy initiation as a complicated molecular switch via the modification of AMPKα by PARPs [61]. During nutrient deprivation, ROS distributed from mitochondria could induce DNA damage, then PARP1 might make out this DNA damage to encourage DNA repair. In case PARP1 is extremely activated, the cellular condition might consume NAD+ and/or ATP as substrates for synthesizing PAR in cells [62]. As a result, the PARylation of AMPKα by the AMPKα/PARP1 complex might be a key factor in beginning autophagy. Similarly, active AMPKα could activate the autophagy via the inhibition of the mTORC1 complex [63], which might be involved in physiological ageing [36, 37, 55] (Figure 3).
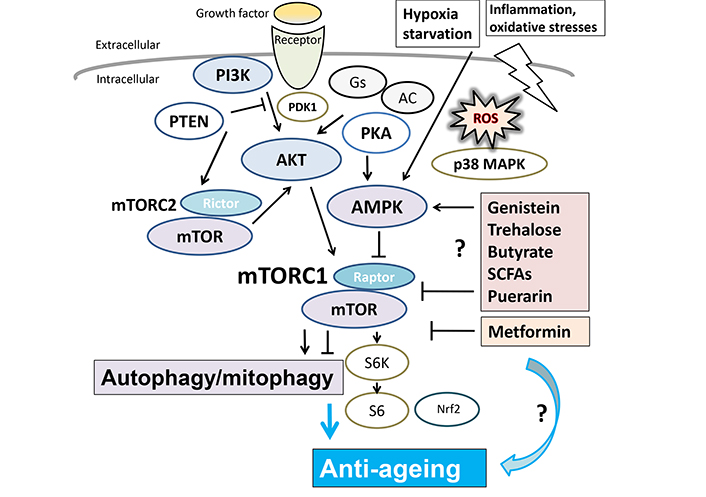
Several modulator molecules linked to the PI3K/AKT/AMPK/mTOR/mTORC1 signaling pathway for autophagy/mitophagy and/or anti-ageing are demonstrated. Example compounds known to act on the AMPK/mTOR pathway and/or autophagy/mitophagy are also shown on the right side. Arrowhead means stimulation whereas hammerhead represents inhibition. Note that some critical events such as antioxidants feedback pathway have been omitted for clarity. PI3K: phosphoinositide-3 kinase; Gs: G protein stimulatory subunit; AC: adenylate cyclase; PTEN: phosphatase and tensin homologue deleted on chromosome 10; PDK1: phosphoinositide-dependent kinase-1; PKA: protein kinase A; AKT: protein kinase B; MAPK: mitogen-activated protein kinase; mTOR: mammalian/mechanistic target of rapamycin; SCFAs: short chain fatty acids; S6: ribosomal protein S6; S6K: S6 kinase; Nrf2: nuclear factor erythroid 2-related factor 2
Mitochondria are like energy plants in a cell. In addition, mitochondria are the main metabolic hubs, which are also involved in signaling pathways for health and/or disease conditions through alterations in these metabolites. Most ROS might be produced in mitochondria, which could work both as signaling and/or detrimental molecules [64]. The resultant oxidative stresses may be also existing in physiological processes for ageing, where they could lead to genomic instability with potential onset of various pathologies. Hence, autophagy/mitophagy might be mandatory in the maintenance of genomic integrity as well as healthy homeostasis [65]. A basal autophagy/mitophagy impairment may lead to inefficient cellular responses to oxidative stresses and increased susceptibility of cells, which could increase cellular vulnerability to external and/or internal stresses [66]. Autophagy/mitophagy might be also mandatory for blocking and/or delaying cell death as well as ageing. As a prosurvival mechanism, autophagy could be encouraged by different types of cellular stresses including DNA damage, ROS, nutrient deprivation, and/or hypoxia, promoting adaptation of cells to the harmful environment [67]. PARP1 might be involved in the regulation of autophagy/mitophagy with PARylation of autophagy/mitophagy-related proteins, where the SIRT1 could be a key regulator of autophagy/mitophagy [68]. Remarkably, several PARPs and PARylation could also modify the function of mitochondria from the nucleus through depletion of NAD+ molecules and/or epigenetic regulation of nuclear genes [69]. Activated PARP1, for example, could regulate some pathological pathways including mitophagy and/or mitochondrial dysfunction [68].
Mitophagy is an intracellular process through which damaged mitochondria are removed [70]. Mitophagy is a kind of autophagy. Under excess oxidative conditions, massive DNA damage might be recognized by PARP1, triggering the initiation of mitophagy. Again, impaired regulation of autophagy/mitophagy could cause cellular functional decline and/or cell death, bringing about several human diseases [71]. Therefore, autophagy/mitophagy pathways might play crucial roles in keeping the cellular homeostasis [60, 71]. Activated PARP1 may be also accompanied by PARylation-induced depolarization of mitochondrial membrane [72]. In fact, PARP1 could localize to mitochondria, where it poly(ADP)ribosylates (PARylates) electron transport chain proteins [73]. Similar to nuclear PARP1, surplus activation of mitochondrial PARP1 could impede the biogenesis of mitochondria, leading to cell death, which is mitochondria-related apoptosis [74]. DNA damage-dependent mitochondria signaling may be due to the accumulation of PAR [75]. Active PARP1 could synthesize the PAR polymer from NAD+ to mark the site of DNA damage [75]. PARylation is a reversible protein modification carried out by the concerted actions of PARPs and PAR, which could disrupt the mitochondrial energy mechanisms linking the release of mitochondrial proapoptotic factors [76]. It has been shown that PARP1 might play a key role in autophagy/mitophagy by inhibiting SIRT1 [77]. Notably, the SIRT1 might be repressed by the activation of PARP1 [78]. Furthermore, NAD+ precursors could rescue the mitochondrial defects and extend lifespan [78], which may suggest a potential therapeutic strategy for several progeroid disorders [78]. As for a key role in longevity, SIRT1 might be involved in autophagy/mitophagy in physiological and/or pathological ageing. Interestingly, it has been revealed that PARP1-induced defective mitophagy may be also a key mechanism in peripheral neuropathic injuries [79].
The involvement of impaired mitophagy, NAD+ depletion, and accumulation of damaged mitochondria have been suggested in the metabolic dysfunction of Werner syndrome [80]. Interestingly, renovation of the mitophagy could improve the disease phenotypes of Werner syndrome in both Drosophila melanogaster and Caenorhabditis elegans models as well as in primary cells isolated from patients of Werner syndrome, suggesting that an impairment of the NAD+-mitophagy axis might play a critical role in the development of Werner syndrome [80]. The Bloom syndrome DNA helicase (BLM) is a recombination factor in maintaining genome stability, and BLM-deficient cells may also exhibit increased mitochondrial mass, higher ATP levels, and/or increased respiratory spare capacity [81]. In Cockayne syndrome, cells may exhibit mitochondrial fragmentation and/or excessive fission, which could be rescued by considerably stimulating mitophagy via the overexpression of Parkin [82]. RECQL4 is associated with Rothmund-Thomson syndrome, which could be found in mitochondria [83]. The loss of RECQL4 could also alter the mitochondrial integrity in cells with Rothmund-Thomson syndrome [83]. Remarkably, accumulated evidence indicates that RECQL4 is important not only in cancer development but also in the accelerated ageing process [84]. A basal activation of mitophagy in cells of Hutchinson-Gilford progeria syndrome has been also shown [85]. Interestingly, all of the markers of mitophagy could be reversed in response to the treatment of Hutchinson-Gilford progeria syndrome cells with the specific inhibitor of chromosomal region maintenance 1 (CRM1) [85]. Consistently, the increase of defective mitochondria caused by impaired mitophagy has been also observed in the progeroid syndromes Xeroderma pigmentosum and/or Cockayne [78, 86]. In general, PARP proteins are usually involved in the modification of proteins and/or nucleic acids through mono(ADP-ribosyl)ation or PARylation, which may decrease the activity of AMPK and/or mitochondrial turnover. Lastly in this section, therefore, comprehension of mitochondrial profiles of PARylation, mono(ADP-ribosyl)ation, and the details of AMPK function in the mitochondrial dynamics should be explored in future studies of progeroid disorders.
Ageing refers to the phenomenon that the physical and psychological adaptability of the body to the environment gradually declines. The increasing proportion of ageing people represents a swelling economic burden, because of age-associated various diseases. Finding potential interventions to age-associated conditions might have widespread implications. A number of genetically defined accelerated ageing diseases have been characterized, which seem to be associated with defects in the maintenance of genomic integrity. As shown here, a key factor that might connect the association between several progeroid disorders and natural ageing may be simultaneously the function of PARPs, oxidative stresses, and/or mitochondrial function. In particular, PARP1 and mitophagy might be important. Emerging evidence has linked mitochondria to physiological ageing and found mitochondrial dysfunction in accelerated ageing disorders [6, 87], which may indicate that an accumulation of damage to the mitochondria could partly underlie the process of ageing. The mitochondrial alterations may be caused by activation of PARP1, which implies that modulation of PARP1 could potentially become a target of therapeutic strategy for treating accelerated ageing disorders and/or age-associated diseases.
Excessive activation of PARP1 may accelerate ageing in mice model [88]. In line with this finding, PARP1 inhibitor could improve the function of senescent cells by increasing NAD+ levels and/or SIRT1 activity [89]. Actually, NAD+ supplementation in the treatment with nicotinamide riboside or nicotinamide mononucleotide, both of which are NAD+ precursors, has significantly protective effect against the process of ageing [90], as similarly does SIRT1 [91]. Interestingly, it has been shown that both p53 and PARP1 might be responsible for the telomere-shortening [92]. The p53 could control cell cycle, cell apoptosis, and/or senescence instigated by the telomere-shortening [93]. In the telomere-related ageing, PARP1 might be also crucial for the p53 activity [94]. Besides, PARP1 could also prevent for the abnormal telomere shortening as well as the physiological shortening [95]. Therefore, PARP1 inhibition could occasionally induce telomere lengthening [96]. PARP1 may be a pleiotropic molecule in the regulation of ageing. On one hand, PARP1 could protect cells from senescence. On the other hand, PARP1 could also promote cell death in senescent conditions. Increased activity of PARP1 may help to extend lifespan [97]. Importantly, PARP1 has a substantial role in DNA damage-repair, and thereby might contribute to the extension of lifespan in animal models. PARP1 is abundantly expressed in the primary cell cultures derived from long-lived rat species [98]. Consequently, these cells might possess high efficiency in DNA repair ability. Taken together, PARP1 might be a kind of longevity-guarantee factor. It is possible that PARP activity may correlate with maximum lifespan.
Ageing is a natural process, characterized by progressive loss of biological integrity, impaired function, and increased susceptibility to death. This natural and physiological ageing may be associated with excessive ROS production and/or resulting DNA damage [99]. In this meaning, several antioxidants may have more or less anti-ageing effects. Up to the present, however, studies have focused on the role of mitochondrial dysfunction in ageing and/or ageing-related degenerative diseases, suggesting that restoring mitochondrial biogenesis may exert favourable effects in extending lifespan and/or supporting healthy ageing [100]. Moreover, decreasing levels of autophagy and/or NAD+ have been suggested as further features of ageing or age-related disease [101]. It has been revealed that enhancing autophagy/mitophagy and/or intracellular NAD+ pools could endorse the extension of lifespan [102]. The NAD+ replacement has been shown to alleviate cellular function by enhancing autophagy/mitophagy [103]. Clearance of impaired mitochondria via the mitophagy has been demonstrated to be linked to the longevity [104]. Conversely, impairment of autophagy/mitophagy has been revealed to trigger NAD+ reduction following apoptotic cell death [105]. In other words, both autophagy/mitophagy and NAD+ pools mutually depend on each other for ideal cellular function and/or longevity [106]. Elimination of impaired mitochondria by selective autophagy/mitophagy might be a longevity relevant key process throughout ageing.
Longevity of lifespan has attracted growing attention as with the social development. In other words, life satisfaction is an imperative provider to the broad construct of subjective well-being both in physical and/or social levels. Public awareness has been elevated about the beneficial potential of specific natural chemical species in the human diet, which should be further focused on the exploration of the precise mechanisms in order to counter the effect of ageing for the longevity. It has been shown that some composition of diet might be one of the major determining factors for longevity of lifespan [107]. In addition, the dietary regulation may contain important, cost-effective, and/or harmless factors to counteract the effect and/or adjust the autophagy/mitophagy. A certain diet could control the autophagy/mitophagy [108], which may also extend the lifespan. For example, it has been reported that downregulation of PARP1 by the puerarin supplementation may be promising for improving the longevity of Drosophila melanogaster by activating autophagy [109]. In mammals, not only has autophagy been associated with the profits of lifespan-extending interventions, but the activation of autophagy may be essentially sufficient to prolong the lifespan in mice [110]. Furthermore, sodium butyrate is known to improve several age-related pathologies and prolongs survival time in mice model [111], which is one of SCFAs detected in several foods. Interestingly, butyrate could promote mitochondrial biogenesis [112]. The anti-senescence activity of genistein is associated with inducing autophagy, in which SIRT1/AMPK pathway might be involved in accelerating autophagy and/or mitigating senescence [113] (Figure 3). Furthermore, an association between longevity and trehalose, a glucose disaccharide, has been recognized [114]. Trehalose could enhance the removal of protein aggregates through the process of autophagy in mammalian cell culture [115]. Disaccharide trehalose might deliver structure-specific effects on cellular energy production with mitochondria [108] (Figure 3). Metformin, a biguanide drug for the treatment of type II diabetes, may have beneficial effects on the lifespan by stimulating autophagy [116] (Figure 3). Additionally, it has been known in a variety of organisms including from yeast to mammals that autophagy might be involved in the regulation of longevity. As a result of the removal of dysfunctional mitochondria, mitophagy could contribute to increased longevity [117]. It seems that prolonged longevity may require autophagy/mitophagy. Consequently, autophagy/mitophagy-stimulating situations provoked by nutritional and/or pharmacologic interventions might contribute to the longevity of lifespan. Remarkably, enhanced autophagy/mitophagy could also counteract ageing of specific organs [118].
As shown here, quality control of mitochondria has a crucial role in counteracting the process of ageing [119]. Therefore, many cellular components that impact autophagy/mitophagy might be linked to longevity-related molecules such as members of sirtuin family proteins [119]. SIRT1 is the most noticeable member of the sirtuin family, which is a group of class III histone deacetylases implicated in longevity [120]. Some deacetylation genes by SIRT1 in the situation of caloric restriction may impact the longevity of lifespan [121]. For example, nuclear SIRT1 works to deacetylate the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α), thereby enabling the expression of genes required in energy-exhausted cells [122]. This modification of histone acetylation might play critical roles in epigenetic gene regulation, which might contribute to the alteration in intracellular metabolism for the longevity of lifespan [122]. Possible causes of ageing might be associated with mitochondria. ROS could lead to damaged DNAs following the accumulation of dysfunctional mitochondria thus increasing autophagy/mitophagy. Therefore, autophagy/mitophagy could be regulated by ROS signaling. As mentioned in the introduction section, NAD+ levels, PARP1 activation, mitochondrial dysfunction, neurodegenerative diseases, neuron senescence, and/or ageing may construct a vicious cycle, whose propagation may drive pathologies. Here, we have highlighted the relationship between PARP1 and autophagy/mitophagy for the deceleration of ageing, which may provide potential therapeutic targets for struggling with age-related diseases and/or promoting longevity.
ADP: adenosine diphosphate
AMPKα: adenosine monophosphate-activated protein kinase alpha
ATP: adenosine triphosphate
C: carboxy
CSB: Cockayne syndrome protein B
LMNA: lamin A/C
mTORC1: mammalian/mechanistic target of rapamycin complex 1
N: amino
NAD+: nicotinamide adenine dinucleotide
NF-κB: nuclear factor-kappa B
PAR: poly(adenosine diphosphate-ribose)
PARP1: poly(adenosine diphosphate-ribose) polymerase 1
PARylation: poly(adenosine diphosphate-ribosyl)ation
RECQL4: RecQ like helicase 4 gene
ROS: reactive oxygen species
SIRT1: sirtuin 1
WGR: tryptophan glycine and arginine rich
WRN: Werner syndrome protein
NS: Conceptualization, Writing—original draft. YI: Conceptualization, Writing—original draft, Visualization. SY: Writing—original draft. SM: Conceptualization, Writing—original draft, Visualization, Supervision. Each author (NS, YI, SY, and SM) has participated sufficiently in this work of drafting the article and/or revising the article for the important rational content. Then, all authors gave final approval of the version to be submitted. Finally, all authors have read and agreed to the published version of the manuscript.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Prasert Sobhon ... Sawaek Weerakiet
Peter Martin ... Leonard W. Poon
Renad I. Zhdanov ... Alexey S. Sozinov
Freda Gonot-Schoupinsky
