 Review
Review

Affiliation:
1Programa de Pós-Graduação em Biologia Estrutural e Funcional, Escola Paulista de Medicina (EPM), Federal University of São Paulo (UNIFESP), São Paulo 04039-032, Brazil
2Genetics Laboratory, Instituto Butantan, São Paulo 05508-010, Brazil
ORCID: https://orcid.org/0000-0002-4043-3308
Affiliation:
3Laboratory of Ontogeny of Lymphocytes, Departamento de Microbiologia, Imunologia e Parasitologia, Federal University of São Paulo (UNIFESP), São Paulo 04039-032, Brazil
ORCID: https://orcid.org/0000-0002-0477-9600
Affiliation:
2Genetics Laboratory, Instituto Butantan, São Paulo 05508-010, Brazil
4Programa de Pós-Graduação em Endocrinologia e Metabologia, Escola Paulista de Medicina (EPM), Federal University of São Paulo (UNIFESP), São Paulo 04022-001, Brazil
ORCID: https://orcid.org/0000-0001-6694-9348
Affiliation:
2Genetics Laboratory, Instituto Butantan, São Paulo 05508-010, Brazil
4Programa de Pós-Graduação em Endocrinologia e Metabologia, Escola Paulista de Medicina (EPM), Federal University of São Paulo (UNIFESP), São Paulo 04022-001, Brazil
ORCID: https://orcid.org/0000-0003-3786-6169
Affiliation:
2Genetics Laboratory, Instituto Butantan, São Paulo 05508-010, Brazil
ORCID: https://orcid.org/0000-0002-8197-2260
Affiliation:
1Programa de Pós-Graduação em Biologia Estrutural e Funcional, Escola Paulista de Medicina (EPM), Federal University of São Paulo (UNIFESP), São Paulo 04039-032, Brazil
2Genetics Laboratory, Instituto Butantan, São Paulo 05508-010, Brazil
5BioDecision Analytics, São Paulo-SP 05713-510, Brazil
Email: rodrigo.pinheiro.araldi@gmail.com
ORCID: https://orcid.org/0000-0001-8288-2577
Explor Immunol. 2023;3:70–81 DOI: https://doi.org/10.37349/ei.2023.00090
Received: August 17, 2022 Accepted: January 12, 2023 Published: March 14, 2023
Academic Editor: Bernhard Ryffel, University of Orleans, France
Lung cancer is the leading cause of cancer-related deaths worldwide. The main risk factor for lung cancer is exposure to chemicals present in cigarettes and atmospheric pollutants, which, among other mechanisms, can increase the risk of cancer by inducing pulmonary inflammation. Among the complex features of inflammatory processes, the role of inflammasomes has attracted increasing attention due to their role in different stages of carcinogenesis. Inflammasomes are intracellular multiprotein complexes that when activated promote the maturation of interleukin-1beta (IL-1β) and IL-18, pro-inflammatory cytokines involved in the promotion, progression, epithelial-mesenchymal transition, metastasis, and resistance to therapy of lung cancer. In this way, this review summarizes the recent findings of inflammasome research in different stages of lung cancer, with a focus on non-small cell lung carcinoma (NSCLC), and highlights these multiprotein complexes as promising targets for cancer therapy.
Lung cancer is the second most common malignancy and the leading cause of cancer-related deaths in men and women worldwide [1–3]. According to epidemiologic data from global cancer observatory (GLOBOCAN), it registered nearly 2.2 million new cases of lung cancer and almost 1.8 million deaths by the disease in 2020 globally [1]. These numbers indicate that, despite the important advances in oncology, the 5-year survival rate after diagnosis remains low (10–20% in most countries) [1]. One of the reasons is that lung cancer is a silent disease, which is frequently diagnosed at advanced staging when the patients already have a local invasion or distant metastasis, which negatively impacts the survival rate [4, 5].
Lung cancer is classified into two types: (1) small cell lung carcinoma (SCLC), which represents around 10–15% of diagnostic cases, and (2) non-SCLC (NSCLC), which is the most frequent subtype, accounting for 80–85% of recorded cases. The NSCLC is sub-classified into three subtypes: (1) adenocarcinoma, which starts in the cells that normally secrete the mucus, and represents the most common histological type of lung cancer seen in people who don’t smoke; (2) lung squamous cell carcinoma (LUSC), which starts in squamous cells, being often linked to a history of smoking and tends to be found in the central part of lungs, near a main airway (bronchus); (3) large cell (undifferentiated) carcinoma, which can appear in any part of the lung, and tends to grow and spread quickly [4].
Although cancer is a multifactorial disease, studies show that about 20% of all human cancers are related to chronic inflammation [6–9]. However, the percentage of inflammation-related cancers can be higher when we focus on lung cancer. The reason is that the lung epithelium serves as a defensive barrier against foreign pathogens and particulates from environmental agents [6–8], making this tissue most susceptible to inflammatory processes. Furthermore, it is well established that exposure to chemicals present in a cigarette or atmospheric pollutants, such as benzene, benzopyrene, and asbestos, or even biological agents present as aerosols can cause pulmonary inflammation, increasing the risk for lung cancer [10, 11]. In this context, the role of inflammasomes in lung carcinogenesis has attracted the attention of scientists around the world. Mainly because these inflammasomes induce the secretion of pro-inflammatory cytokines which are closely related to cancer progression and metastasis, such as interleukin-1beta (IL-1β) and IL-18, as well as promote the activation of nuclear factor kappa B (NF-κB) signaling pathway [9, 12], that combined, contribute to different carcinogenesis steps [13, 14]. Based on that, this review aims to summarize the role of inflammasomes in lung cancer initiation, promotion, progression, and metastasis.
Inflammasomes are cytosolic complexes formed by many proteins, defining them as a high-molecular-weight complex. These complexes are assembled after sensing or binding specific stimuli [15, 16]. In general, the inflammatory stimuli came from the recognition of pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) by pattern-recognition receptors (PRRs). Different types of canonical inflammasomes are now described based on which PRR initiates the signaling. The most well-studied receptors in canonical activation are (1) nucleotide-binding oligomerization domain (NOD), (2) leucine-rich repeat (LRR)-containing protein [NOD-like receptor (NLR)/NOD-like] family members [(NLR protein 1) NLRP1, NLRP3 and NLR family CARD domain containing 4 (NLRC4)], (3) proteins absent in melanoma 2 (AIM2), and (4) pyrin. “Selection” of which receptor forms the complex relies on the stimulus nature. For example: (1) a specific toxin of anthrax activates NLRP1; (2) reactive oxygen species (ROS), potassium efflux out of the cell, pore-forming toxins, pathogens, translocation of NLRP3 to mitochondria, the release of mitochondrial DNA, release of cardiolipin, and release of cathepsins can activate NLRP3 inflammasomes; (3) certain types of bacterial proteins [(type III secretion system) T3SS and flagellin] activates NLRC4 inflammasomes and; (4) cytosolic double-strand DNA (dsDNA) binds and activates the AIM2 receptor [15, 17–19].
Once assembled, the inflammasomes can serve as a scaffold to recruit and activate caspase-1. In this sense, it was shown that the NLRP3 inflammasome assembly (which is one of the most studied inflammasomes) is controlled at several checkpoints. The first one is the priming step of NLRP3 inflammasome activation, which is associated with increased NLRP3 gene expression, followed by post-translational modifications that control NLRP3 levels and license the NLRP3 protein for inflammasome assembly [20]. After this step, the oligomerization of the NLRP3 serves as a scaffold to apoptosis-associated speck-like protein containing a CARD (ASC)-a crucial protein that permits pro caspase-1 and ASC interaction. When in proximity due to ASC filaments, the pro caspase-1 proteins are activated in caspase-1 by an autoproteolytic process [21]. This important step elicits the activation of pro-inflammatory cytokines IL-1β and IL-18. From that, combined with gasdermin D (GSDMD), can lead the cellular membrane pore formation in response to pyroptosis [22]. The other inflammasomes, such as NLRC4, AIM2, and pyrin also need the ASC speck to be activated. The difference between them is how the oligomerization of the receptor occurs. NLRC4 forms a scaffold with other proteins called NLR family apoptosis inhibitory protein (NAIP), which differ in specificity and number of isomers in mice and humans; and AIM2 inflammasome requires a dsDNA binding to be oligomerized. Importantly, NLRP1 can be activated despite ASC filaments [17, 19]. The classical mechanism of activation and assembly are summarized in Figure 1, using NLRP3 as a model.
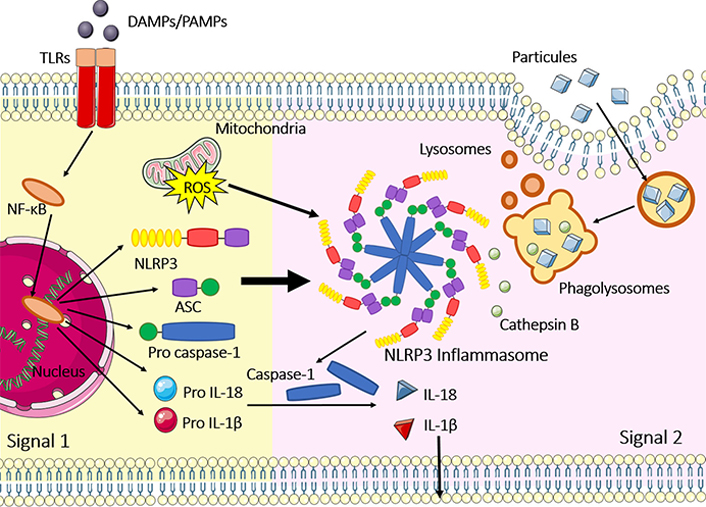
Classical NLRP3 inflammasome activation and assembly. Signal 1 represents the priming step, where the NF-κB signaling pathway is activated after toll-like receptors (TLRs)/PRR recognition of PAMPs and DAMPs. Priming is necessary to augment gene expression of NLRP3, ASC, pro caspase-1, pro IL-1β and pro IL-18; signal 2 occurs after several stimuli, such as phagolysosomal rupture, cathepsin B release, pore-forming toxins, mitochondrial DNA (mtDNA) release, potassium efflux and ROS generated at the mitochondria. After these 2 steps, inflammasomes are activated and assembled including NLRP3 oligomerization, recruitment of ASC specks and pro caspase-1 binding in the CARD domain of ASC. Caspase-1 is activated by an autoproteolytic process and IL-1β and IL-18 also assume their active forms, leading to inflammation; inflammasome activation can also result in pyroptosis, a type of cell death, where GSDMD is cleaved by caspase-1 and induces the formation of pores in the cellular membrane. The figure was partly generated using Servier Medical Art (https://smart.servier.com) provided by Servier, licensed under a Creative Commons Attribution 3.0 Unported License
Non-canonical inflammasomes are formed by caspase-11 (in mice) or caspase-4 and caspase-5 (in humans). These caspases are activated after interaction with lipopolysaccharide (LPS) and their activation leads to cleavage of GSDMD in a caspase-1-independent manner, leading to pyroptosis [23].
Besides IL-1β and IL-18, other pro-inflammatory cytokines, such as tumour necrosis factor alpha (TNF-α), IL-6, IL-8, and IL-17 are also involved in lung cancer initiation, progression, and metastasis. The main reason for it is that TNF-α induces the release of IL-1β and IL-6 [24], acting in the initiation and regulation of the cytokine signaling cascade.
The IL-1β also activates the cyclooxygenase 2 (COX-2) and the nitric oxide synthase [24, 25], serving as a chemoattractant cytokine to neutrophils in the inflammation process [26].
Similar to verified, IL-1β and TNF-α also stimulate the secretion of IL-6 by monocytes and macrophages. This action prevents apoptosis, contributing to cancer progression [27, 28]. For this reason, the levels of IL-6 within the tumor microenvironment (TME) also serve as a biomarker of poor prognosis.
Cancer initiation is characterized by irreversible genetic alterations (driver mutation) that lead to the gain of function of oncogenes and/or loss of tumor suppression genes [29]. In this sense, the airway epithelium is in constant contact with many mutagens and carcinogens present in the air. Among these molecules, it is known that asbestos can activate the NLRP3 inflammasome in macrophages and induces NLRP3 inflammasome activation in mesothelial cells, which line the pleural cavities [30]. The activation of these inflammasomes promotes pulmonary injury, contributing to the development of pulmonary fibrosis and inflammation [31]. However, other chemicals present in cigarette smoke, including nicotine and pollutants can also activate the NLRP3 inflammasome in human bronchial and alveolar epithelial cells [32–34].
The activation of these inflammasomes increases the production and secretion of IL-1β, which activates downstream proteins of the NF-κB signaling pathway, and the recruitment of immune cells to target sites [35]. Interestingly, alveolar macrophages show high expression of inflammasome complexes [36], so it is suggested that tumor-associated macrophages serve as the main producer of IL-1β to de TME [37].
The release of pro-inflammatory cytokines within the TME recruits neutrophils to the TME [38], leading to the generation of ROS and nitrogen species (NOS) [39]. The oxidative stress, in turn, can lead to single and/or double-strand DNA breaks [single-strand DNA breaks (SSBs) or double-strand DNA breaks (DSBs), respectively], microsatellite and chromosomal instability, suggesting that inflammasomes can indirectly be associated with the genomic instability in the pre-cancer and TME [40]. Thus, the inflammasomes contribute to both driver mutations (which are responsible for cancer initiation) and passenger mutations (which increase the TME heterogeneity, negatively impacting the patient’s survival rate) [41]. The role of activated inflammasomes in cancer initiation is summarized in Figure 2.
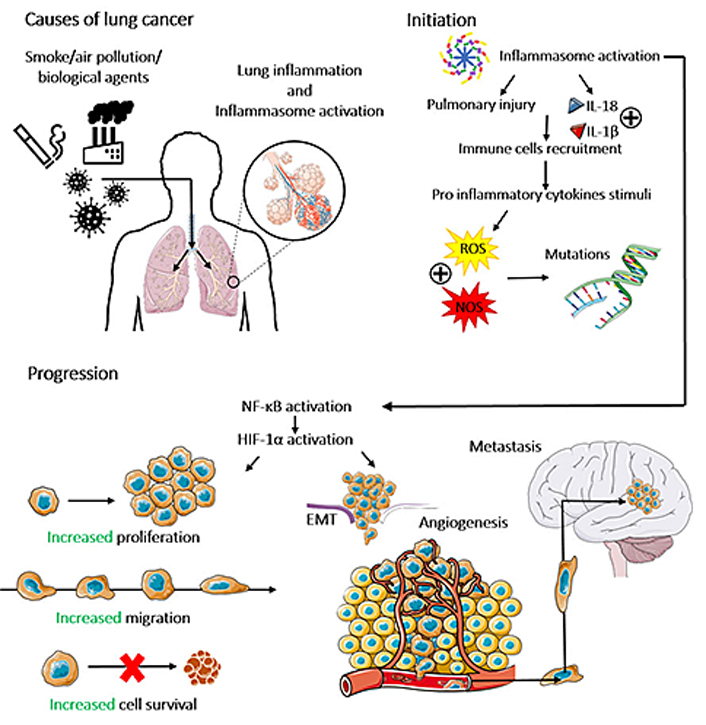
The role of inflammasomes in lung cancer initiation, progression, and metastasis. Different environmental stimuli, named cigarette smoke, air pollution, and biological agents can promote lung inflammation and inflammasome activation, which induce pulmonary injury and the secretion of the inflammatory cytokines IL-1β and IL-18, resulting in the recruitment of immune cells to the lung and leading to oxidative stress by the production of ROS and NOS. Which induces DNA breaks in lung cells, characterizing lung cancer initiation. Inflammasome activation and its consequent secretion of IL-1β and IL-18 activate the NF-κB/hypoxia inducible factor 1 alpha (HIF-1α) pathway, which modulates plenty of carcinogenic processes, such as the increase of proliferation, migration, survival, angiogenesis, epithelial-mesenchymal transition (EMT) and metastasis of lung cancer cells. The figure was partly generated using Servier Medical Art (https://smart.servier.com) provided by Servier, licensed under a Creative Commons Attribution 3.0 Unported License
Studies have shown that inflammasome components were upregulated in lung cancer when compared with adjacent tissue [42]. Indeed, IL-1β and IL-18 are pro-inflammatory cytokines commonly upregulated in lung cancers [43, 44] and the serum of NSCLC patients [44, 45]. Kong et al. [42] showed that inflammasome components are minimally expressed in adjacent normal tissues, but they have a heterogeneous expression in lung tissues (according to the lung cancer histological type). According to the authors, NACHT, LRR, and PYD domains-containing protein 1 (NALP1) and NLRC4 components were indetectable in lung cancer tissues, whereas AIM2 and NLRP3 were highly expressed in adenocarcinomas. By contrast, LUSCs exhibit a moderate expression of AIM2 and low expression of NLRP3, whereas, in SCLCs, AIM2 showed low expression, while NLRP3 was moderately expressed [42]. Interestingly, high-grade adenocarcinomas show higher levels of NLRP3 inflammasome [42]. Also, studies showed that the GSDMD levels were upregulated in lung adenocarcinoma (LUAD) and LUSC when compared to the matched adjacent normal tissues [46, 47]. NLRP3, AIM2, and GSDMD are the most inflammasome components studied in lung cancer. However, there are few studies describing the role of NLRC4 and NLRP1 in lung cancer.
Confirming that the inflammasomes are related to lung cancer progression, studies based on expression analysis of inflammasome-related genes have demonstrated a differential expression between cancer and non-cancer lung tissues [48]. In another independent study, Huang et al. [49] showed that NLRP3 deletion inhibits lung tumorigenesis in mouse models, whereas the activation of the NLRP3 inflammasome enhanced the proliferation and migration of lung cancer cells, by activation of mitogenic pathways, named phosphorylation of Akt, ERK1/2, and CREB [50]. Hillegass et al. [51] showed that the inhibition of NLRP3 inflammasome decreases the expression and release of IL-1β—pro-inflammatory cytokine which increases the proliferation and migration of lung cancer, as demonstrated in in vitro study [45]. Furthermore, Zhang et al. [52] showed that the AIM2 inflammasome serves as an oncogene in NSCLC, since its overexpression increased cell survival and migration, while the AIM2 knockdown significantly reduced cell viability. Reinforcing these data, Yu et al. [53] showed that the use of luteolin (a natural flavonoid used as an anti-inflammation in clinical treatments) decreased the proliferative, migratory, and invasive capabilities of lung cancer cells by downregulating AIM2 expression. Besides that, Gao et. al. [46] showed that higher GSDMD expression is associated with aggressive clinicopathological features, named larger tumor size, more advanced tumor-node-metastasis (TNM) stages in LUSC and LUAD, and less survival rate in LUAD patients, being a potential molecular marker for poor prognosis in LUAD. The same study showed that the knockdown of GSDMD decreases the expression of metalloproteinases and facilitates apoptosis in lung cancer cell lines. Furthermore, the knockdown of GSDMD impaired the proliferation by inhibition of the EGFR/Akt signaling pathway [46]. Altogether, these data provide strong evidence that inflammasomes are related to lung cancer promotion and progression (Figure 2).
One of the molecular mechanisms in literature connecting inflammasome activation and lung cancer development is the increase of IL-1β. It has been shown that IL-1β increases the expression of HIF-1α in lung cancer cells through an NF-κB-COX-2 pathway and by silencing miR-101 [45, 54]. It’s well known that NF-κB is a positive regulator of cell growth and proliferation in lung cells, through the upregulation of cyclin D1 and consequently enhanced transition from G1 to S phase [55, 56]. In addition, it has been shown that HIF-1α is essential for the proliferation of NSCLC cells [57], once HIF-1α overexpression is correlated with c-Myc, a transcriptional factor associated with proliferation, overexpression in lung cancer specimens [58].
Metastasis represents the end-products and the most dramatic cancer consequence. Metastasis occurs when cancer cells spread from a primary site to distant organs through a multi-step cell-biological process that involves local invasion, intravasation, transport, extravasation, and colonization [59]. These steps require a series of genetic, biochemical, and morphological deregulations that are present in an evolutionarily conserved developmental program known as the EMT [60–63]. In this sense, studies have shown that activation of NLRP3 inflammasome downregulates E-cadherin expression and upregulates snail expression, while the blockage of AIM2 inflammasome decreased vimentin, and matrix metalloproteinase 9 (MMP-9) expression and increased E-cadherin, indicating that the activation of NLRP3 and AIM2 inflammasome induce the EMT in lung cancer cells [50, 53]. Indeed, as mentioned above, IL-1β increases the expression of HIF-1α in lung cancer [45], in which the correlation of HIF-1α with EMT in cancer is well established in the literature (Figure 2). HIF-1α is a major regulator of transcription factors associated with the mesenchymal phenotype, such as Twist-related protein (TWIST), slug, snail, zinc finger E-box-binding homebox 1 (ZEB1), and SIP1 [64]. Moreover, it is known that the overexpression of IL-1β induces EMT in lung cancer [65, 66]. The outcome of cancer metastasis depends on the interactions between metastatic cells and the host microenvironment [67]. Wang et al. [68] showed that neutrophil extracellular traps (NETs) promote EMT in non-small cell lung cancer and metastasis by activation of NF-κB/NLRP3 inflammasome pathway, mediated by long non-coding RNA (lncRNA) MIR503HG suppression. Zhang et al. [69] demonstrated that circulating NETs are correlated with inflammatory cytokines (IL-1β, IL-6, IL-18, and TNF-α). Their work demonstrated that the pro-inflammatory co-activity of NETs and macrophages promotes the increase in migration and invasion of lung cancer cells in vitro [69].
Therapy resistance is the major challenge in cancer treatment [70] and could be innate or acquired. Innate resistance is a consequence of the genetics and biochemical changes that occur during cancer progression. These changes, combined with the passenger mutations, not only increase the cancer heterogeneity, but also contribute to the dedifferentiation from cancer cells to cancer stem cells (CSCs)—a small population of cells within the TME with capabilities of self-renewal, differentiation, and tumorigenicity when transplanted in animals. For sharing characteristics in common with stem cells, it is not surprising that CSCs also exhibit a higher capability to repair DNA damage than cancer cells, which confers radioresistance. In addition, CSCs also express high levels of ATP-binding cassette (ABC) transporters, which increase the efflux of chemotherapeutics, limiting the efficacy of chemo agents. Furthermore, therapeutic resistance can be acquired as a consequence of molecular changes and clonal selection observed after an extended period of treatment [71–75].
Interestingly, inflammation can mediate cancer resistance therapy, affecting the tumor response to chemo agents and immunotherapy [10]. In this context, studies show that inflammasomes are related to chemoresistance in different carcinomas [76–78], including lung cancer [42]. Besides that, few studies had showed a direct correlation between lung cancer therapy resistance and inflammasome activation. In the lung cancer context, there is a niche prone to inflammation because of external substances exposure (several particulates, allergens, and air-borne pathogens). Regarding this, inflammasomes represent key modulators in controlling cytokine production, since IL-1β and IL-18 can act in feedback with inflammasomes, activating these complexes, at the same time that these complexes, once activated, can increase the production of these cytokines [79].
In this sense, many agents could be used to inhibit inflammasomes in the priming or activation steps of the complexes, as reviewed by Dey Sarkar et al. [80]. However, priming involves the NF-κB pathway, for example, which includes a transcription factor expressed in many cells and is important to several processes. Thus, disrupting inflammasome activation and assembly may represent a better choice [80]. Importantly, it is necessary to develop a mechanism by which the inhibitor exclusively acts on the overactive inflammasome component in the tumor tissue, to avoid disruption of other important pathways in the host’s defense. Ahn et al. [81] reviewed recently the impact of several drugs in the priming or activation step, highlighting the importance to stimulate priming but inhibit the activation of inflammasomes.
In the context of inhibition of inflammasome activation, a randomized, double-blind, placebo-controlled Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) clinical trial evaluate the use of canakinumab, an IL-1β-neutralizing monoclonal antibody, in 10,061 patients with atherosclerosis who had a myocardial infarction. The study showed that the effects were dose-dependent and reduce 67% of lung cancer incidence and a reduction of 77% of lung cancer mortality. However, this study requires replication, because lung cancer was not a prespecified study endpoint, so replication in formal settings of cancer screening and treatment is necessary [82]. Other studies suggest of compounds associated with AIM2 and NLRP3 inflammasome inhibition, such as luteolin and polydatin, might be potential therapeutic candidates in the treatment of NSCLC [53, 83]. Studies have reported that the use of MCC950, an NLRP3 inhibitor, can delay tumorigenesis and improve the antitumor response in different models of cancer [84]. Despite that, preclinical and clinical reports studying MCC950 and other inflammasome inhibitors in cancer remain limited [85]. Moreover, a recent review summarized different ways to target NLRP3 inflammasome as an option to overcome cancers [86].
Considering that the inflammasomes can govern all steps of lung carcinogenesis, novel therapeutic approaches able to reduce the inflammatory response within the lung TME may be biotechnologically explored to improve the survival rate of lung cancer.
AIM2: absent in melanoma 2
ASC: apoptosis-associated speck-like protein containing a caspase activation and recruitment domains
CARD: caspase activation and recruitment domains
CSCs: cancer stem cells
EMT: epithelial-mesenchymal transition
GSDMD: gasdermin D
HIF-1α: hypoxia inducible factor 1 alpha
IL-1β: interleukin-1beta
LUAD: lung adenocarcinoma
LUSC: lung squamous cell carcinoma
NETs: neutrophil extracellular traps
NF-κB: nuclear factor kappa B
NLR: nucleotide-binding oligomerization domain-like receptor
NLRC4: nucleotide-binding oligomerization domain-like receptor family caspase activation and recruitment domains domain containing 4
NLRP1: nucleotide-binding oligomerization domain-like receptor protein 1
NSCLC: non-small cell lung carcinoma
PRRs: pattern-recognition receptors
ROS: reactive oxygen species
TME: tumor microenvironment
TNF-α: tumour necrosis factor alpha
VRDC: Conceptualization, Writing—original draft, Visualization. OFS: Conceptualization, Writing—original draft, Visualization. ALA: Conceptualization, Writing—original draft, Visualization. MRT: Conceptualization, Writing—original draft, Visualization. RPA: Conceptualization, Writing—original draft, Visualization, Supervision. HV: Visualization.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

