 Review
Review

Affiliation:
1Health Science Center, State University of Ceará, Fortaleza 60714-903, Brazil
2Drug Research and Development Center (NPDM/UFC), Faculty of Medicine, Federal University of Ceará, Fortaleza 60714-903, Brazil
Email: gislei.frota@uece.br
ORCID: https://orcid.org/0000-0002-0689-8371
Affiliation:
1Health Science Center, State University of Ceará, Fortaleza 60714-903, Brazil
ORCID: https://orcid.org/0000-0002-4254-7210
Affiliation:
1Health Science Center, State University of Ceará, Fortaleza 60714-903, Brazil
ORCID: https://orcid.org/0000-0001-6700-5533
Affiliation:
1Health Science Center, State University of Ceará, Fortaleza 60714-903, Brazil
ORCID: https://orcid.org/0000-0001-7692-8372
Affiliation:
1Health Science Center, State University of Ceará, Fortaleza 60714-903, Brazil
ORCID: https://orcid.org/0000-0002-8346-1384
Affiliation:
1Health Science Center, State University of Ceará, Fortaleza 60714-903, Brazil
ORCID: https://orcid.org/0000-0002-7396-4252
Affiliation:
1Health Science Center, State University of Ceará, Fortaleza 60714-903, Brazil
ORCID: https://orcid.org/0000-0003-4689-564X
Explor Immunol. 2023;3:56–69 DOI: https://doi.org/10.37349/ei.2023.00089
Received: October 11, 2022 Accepted: December 12, 2022 Published: February 28, 2023
Academic Editor: Bernhard Ryffel, University of Orleans, France
The infection of COVID-19 is directly linked to the destruction of lung epithelial cells, and the cyclic guanosine monophosphate-adenosine monophosphate synthase-stimulator of interferon genes (cGAS-STING) system has been implicated in the pathology of respiratory infections. This study aimed to systematize the relationship between the pathophysiology of COVID-19 and the cGAS-STING system’s activation in the lungs. Severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) is an RNA virus that belongs to the Coronaviridae family whose genetic material is produced by a single positive RNA molecule (RNA+). The cGAS-STING signaling pathway has emerged as a key mediator of injury caused by infection and cellular or tissue stress. The cGAS-STING cyclic pathway is part of innate immunity and is activated from cytosolic DNA responses present in newly formed syncytia, by cell-to-cell fusion, in target of angiotensin-converting enzyme 2 (ACE2) expression and SARS-CoV-2 Spike protein. Although this pathway is canonically understood to be responsive to both pathogen-derived and host-derived DNA, it has been demonstrated to cross-communicate with the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs). cGAS-STING activation is significant to interferon production, mainly type-I interferons (IFN-I), in a SARS-CoV-2 infection scenario, indicating a major antiviral role of the cGAS-STING pathway. It was identified that in SARS-CoV-2 the cGAS-STING axis is activated, but the inflammatory response could be specific for nuclear factor-κB (NF-κB) in infected cells, and that this axis is potentiated by a cytokine storm produced by the immune system’s cells.
Severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) caused the coronavirus disease 2019 (COVID-19) pandemic with origin in Wuhan, China [1]. With confirmed 522 million infections and death of 6.27 million until May 2022, COVID-19 has affected people from all countries and all ages, with a significant impact on morbidity and mortality, especially in older people [2]. The pathobiology of COVID-19 is directly linked to the destruction of lung epithelial cells, and in a significant proportion of infections, severe symptoms are developed by individuals and that can lead to long-lasting lung damage or death [3, 4]. The infection is characterized by low levels of type-I interferons (IFN-I) in addition to an overproduction of inflammatory cytokines or chemokines such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) [5–7].
In the SARS-CoV-2 infection, the inflammatory and innate immune response are initiated by sensing the pathogenic components via pattern-recognition receptors (PRRs) such as toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)/stimulator of interferon genes (STING) axis [8, 9]. Through the cGAS viral DNA and RNA are recognized, which activates the STING signaling. The interferon gene stimulator STING, when activated in response to the recognition of cytosolic DNA including viral DNA, plays a role in controlling the induction of innate immune genes. Thus, STING activation exerts protection on the host against DNA pathogens, although its prolonged activation can lead to inflammatory complications that can be lethal for the patient [10].
It has been demonstrated that a nuclear factor-κB (NF-κB) response was mediated by activation of cGAS-STING, this response being attenuated by several STING-targeting drugs [11]. Both cGAS and STING have additional functions, regardless of the interferon response. These involve non-catalytic roles of cGAS in the regulation of DNA repairing and signaling via STING to NF-κB and mitogen-activated protein kinase (MAPK), as well as induction of STING-mediated autophagy and lysosome-dependent cell death [12]. The cGAS is a cyclic DNA detector to induce alternate runner cyclic guanosine monophosphate-adenosine monophosphate (cyclic GMP-AMP, or cGAMP) for the operation of the crucial signaling appendage STING, which later initiates the NF-κB and interferon regulatory factor 3 (IRF3) driven pathways to promote seditious cytokines and the interferon-β (IFN-β) product [13]. The cGAS/STING axis has been linked to aging and age-associated respiratory infections due to its critical role in connecting natural cellular stress to pathogenic signals [14].
The SARS-CoV-2 virus is a weak inducer of the IFN-I response, being a characteristic of coronavirus infections that seem to have developed strategies to neutralize IFN-I’s production [15]. While the infection is progressing, viral spike protein (S protein) is produced on the cell surface and infected cells fuse with neighboring cells, leading to the formation of micronuclei that, when detected by cGAS, activating STING [3]. As an important signaling molecule, STING detects pathogen and virus derivatives, especially double-stranded DNA (dsDNA) [10]. The dsDNA source pathogens are endocytosed by phagocytosis and digested. Extracellular micronuclei formed during the mitotic process are sources of cytosolic dsDNA, which in the mitotic process of normal cells can be exposed to the cytoplasm, but hardly activate cGAS [16]. The combination of these occurrences results in the generation of innate immune response genes via the IRF3 and NF-κB pathways [10]. In this process, the IFN-I response is of paramount importance for inducing the transcription of hundreds of interferon-stimulated genes (ISGs) that promote an immune response against the viral infection, inducing an antiviral state in humans [17]. In view of the importance of activating the cGAS-STING pathway in IFN-I signaling, this study sought to systematize the relationship between the pathophysiology of COVID-19 and the activation of the cGAS-STING system in the lung. It started with the question: How does the cGAS-STING pathway correlate with lung damage caused by COVID-19?
The signaling cascade that activates the two major arms of the innate immune response (the type I/III interferon response and the inflammatory cytokine response) is induced by RNA or DNA sensors, which include cytoplasmic DNA sensor cGAS-STING. This system not only recognizes dsDNA from DNA viruses, but also plays an important role in the infection of the RNA virus. In addition, the cytosolic sensor protein, cGAS is a key protein molecule in immune responses to viral infections by acting to regulate basal expression levels of cell-intrinsic immune genes in cells [14].
It is known that IFN-I constitute an important antiviral defense and play a critical role in activating the adaptive immune system. Although there are many cytoplasmic DNA sensors related to the induction of IFN-I transcription, cGAS is thought to be the most relevant DNA PRR, as the induction of IFN-I responses was shown to be paramount, regardless of the type of cell or DNA sequence. Activation of this antiviral pathway is essential for controlling viral replication, although viruses develop abilities to escape the IFN system at different stages of the pathway. Several RNA viruses from different families, including human coronaviruses, can interact and block STING signaling [11].
When the molecule cGAS recognizes nucleic acid damage or foreign DNA, STING is stimulated to recruit the enzyme TNF receptor-associated factor (TRAF) family member associated NF-κB activator TANK-binding kinase 1 (TBK1). The STING signaling pathway is important in the induction of type I and type III IFN, being a key mediator of the host’s antiviral defense, in addition to inducing other antiviral proteins. After its activation, STING recruits the TBK1 enzyme that phosphorylates it, forming a STING-TBK1 complex that is translocated to perinuclear lysosomal compartments [18, 19].
Also, the activated STING can cause inhibitor of NF-κB (IκB) kinase α (IKKα) and IKKαβ phosphorylation which activates NF-κB, mediated by TRAF3 and TRAF6, respectively. TRAF6 may be recruited to the signaling complexes with STING and TBK1, which in turn activates the canonical NF-κB p65 signaling pathway through the IKKαβ activation loop. IFN-β production mediated by dsDNA could be associated with NF-κB p65 activation STING-mediated. However, STING also has the ability to activate the non-canonical NF-κB signaling pathway via the TRAF3-IKKα axis, which leads to a dsDNA-mediated modulation of the canonical NF-κB signaling pathway. Then, IFNs transcription is induced by the translocation of IRF3 and NF-κB, both already activated into the nucleus, for the formation of IFN and cytokines [10]. These pathways are described in diagram form in Figure 1.
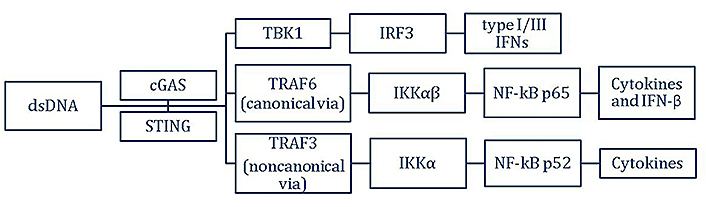
Cell signaling pathway for the production of type I/III IFN, and inflammatory cytokines caused by activation of the cGAS-STING upon dsDNA
IFN is secreted and binds to the interferon-α/β receptor (IFNAR) on the cell surface. This binding induces the expression of ISG and interferon-induced proteins with tetratricopeptide repeats (IFIT), which produces an “antiviral state” in host cells. This mediator can be divided into three types: type I includes IFN-α, IFN-β, IFN-κ, and IFN-ω; type II includes only IFN-γ; and type III includes IFN-λ1, IFN-λ2, and IFN-λ3 [20]. Type I and type III IFNs are vital to the cell’s intrinsic antiviral response and are directly involved in protecting it against the spread of the virus to neighboring cells [14].
The production of pro-inflammatory cytokines is a key aspect of the innate immune response, which is necessary to attract specialized immune cells to the primary site of infection and support the initiation of an adaptive immune response [14]. The secondary messenger cGAMP, synthesized by cGAS, binds to STING and this complex cGAMP-STING migrates from the endoplasmic reticulum (ER) to the Golgi apparatus. During this migration, there is the downstream activation of NF-κB and IRF3. Infected lung epithelial cells produce an unbalanced immune response that results in a polarized NF-κB response, to the detriment of the classic antiviral immune response (NF-κB, IRF3/7, and IFN signaling) [14, 21].
The levels of released cytokines and the various stimuli and changes in the intracellular environment are sufficient to trigger the activation of downstream immune cells such as macrophages and neutrophils [22]. Furthermore, this pro-inflammatory response leads to the recruitment of these immune cells, which are associated with cell death and lung pathology. Macrophages are activated and polarized into different phenotypes, it is believed that this polarization can be significantly regulated by the cGAS-STING pathway [14, 23].
The main site of infection is the lung epithelial cells, which are therefore responsible for initiating immune responses to infection by the SARS-CoV-2 virus. The low IFN response is often observed in COVID-19, especially in the initial phase, which can be explained by the impairment in the IFN expression [24]. That observation can be associated with a deficient activation of the cGAS-STING pathway, since this pathway is involved in the IFN expression [16].
At the early infection stage, it is verified that the virus effectively suppresses the production of interferons when there are low amounts of viral components [25]. Plus-strand RNA viruses developed numerous ways to limit or block the cellular immune pathways. The activation of bystander cells rather than the initial infection leads to IFN activation, which may explain the fact that interferon production by the SARS-CoV-2 virus occurs only hours after infection. Another factor that explains this interference in IFN production is due to the virus-encoded proteins actively targeting and antagonizing multiple steps of the immune activation pathway, leading to a robust blockade in the IRF3-mediated IFN response [15].
Some viral proteins can inhibit the cGAS-STING pathway activation, more likely at the early stages of the pathway, impairing antiviral activity. The 3C-like protease (3CL) from SARS-CoV-2 was able to inhibit immune responses induced by the RLR and cGAS-STING pathways. Furthermore, structural protein N has shown to have little effect on cGAS-STING-mediated innate immune activation, but with a strong ability to antagonize RLR innate immune activation. In contrast, a conserved coronavirus protein that is involved in virus replication and release, the SARS-CoV-2 protein open reading frame 3a (ORF3a), specifically antagonizes the immune activation induced by cGAS-STING, without detectable effect on RLR-mediated immune responses [26].
Thus, the 3CL and ORF3a proteins act, probably by inhibiting NF-κB signaling. ORF3a acted by blocking the NF-κB accumulation caused by the cGAS-STING activation. About 3CL, this protease inhibited the modification of STING by K63-ubiquitin, which impaired the NF-κB signaling, and also impeded the downstream recruitment of TBK1 and IKKβ by STING [22, 26]. SARS-CoV-2 open reading frame 10 (ORF10), another encode viral protein, interacts with STING, attenuates the STING–TBK1 association, and impairs STING oligomerization, STING-mediated aggregation, and autophagy; ORF10 also prevents STING trafficking from the ER to the Golgi by anchoring the STING in the ER. Finally, ORF10 suppresses STING-mediated IRF3 activation and IFN induction [27].
SARS-CoV-2 has been shown to inhibit the RIG-I-mitochondrial antiviral-signaling protein (MAVS)-TBK1 pathway [19, 28], which makes the IFN response induced by the cGAS-STING activation, through the syncytium formation, key for an antiviral defense [19]. RLRs, including RIG-I, play an important role in triggering antiviral and inflammatory responses to control viral replication, in the response to specific RNA structures of the cytoplasmic virus. RLRs detect distinct types of viruses through a common adapter protein known as MAVS. Upon viral RNA recognition, RIG-I recruits the MAVS, which leads to a signaling cascade, including kinases TBK1/IKKε and the IKK complex, which coordinates the induction of type I interferons, as well as a large variety of antiviral ISGs [29].
In endothelial cells infected by SARS-CoV-2, the IFN-I response was not affected when it was a knockdown of MAVS. Still, about the endothelial cells, there were high levels of IFN-β secretion after infection, but this production was stopped when H-151, a STING inhibitor, was administered. It was also verified that macrophages could contribute to the resultant IFN-I response on the vascular side, in a way that it was dependent on cGAS in SARS-CoV-2 infection. Confirming the STING involvement, perinuclear foci of phosphorylated stimulator of interferon genes (p-STING) were found in the endothelial cell after infection [30]. Therefore, cGAS-STING activation is significant to IFNs production in a SARS-CoV-2 infection scenario, indicating an important antiviral role of the cGAS-STING pathway [31].
Syncytia are evolutionarily conserved cellular structures formed by the multiple cell fusions of mononuclear cells. As the infection progresses and Spike protein is produced on the cell surface, there is a cell-cell fusion to form multinucleated syncytia that promote the formation of micronuclei that contains DNA damage [19, 25]. This syncytium formation occurs in cells that express the angiotensin-converting enzyme 2 (ACE2) and SARS-CoV-2 spike protein, resulting in an average of approximately 4 micronuclei (> 93%) per syncytium. Notably, these micronuclei showed high levels of DNA damage response activation and cGAS-STING signaling through translocation of the phosphorylated form of the H2A histone family member X (γH2Ax) and cGAS micronuclei and upregulation of their respective target genes [25].
According to recent studies, the micronuclei are detected by cGAS, which activates STING, triggering the phosphorylation of IRF3 and the generation of IFNs [25, 31]. The complex phosphorylates IRF3, which is a critical downstream effector for IFN expression, is present in SARS-CoV-2-induced micronuclei. IFN is directly related to the cytokine storm present in patients with COVID-19, being an essential mediator of innate immunity in the disease, and a marker of host antiviral immunity. The study by Guo et al. [20], demonstrates that IFN-β1 and IFIT1 genes are higher in patients with moderate and severe COVID-19 than in healthy patients, but there is no difference between the two clinical types. Soon after SARS-CoV-2 infection, IFNs are protective, but in the long term, they become pathological, which suggests that the initial IFN response is essential to block the infection, spread, and pathogenesis of the virus [18].
These statements are supported by the upregulation of IFN expression in cells that formed syncytium, followed by the increase in the expression and phosphorylation of cGAS, STING, and IRF3. Also, when cGAS was genetically ablated by ready-to-use cells engineered to stably express the ACE2 receptor in HeLa cells (HeLa-ACE2), suppression of IFN-β induction and IRF3 phosphorylation was observed [31]. Interestingly, STING molecules of bats presented deficiency IFN-I induction, which has been associated with coronavirus infection persistence in bats [26].
There is growing evidence that patients with severe COVID-19 show a strong response to interferon type I (IFN-I), as opposed to the delayed and perhaps suppressed interferon response seen early in the infection. A single-cell RNA sequencing analysis was performed on patients with COVID-19 and unique hyperinflammatory signatures were observed across all types of immune cells, particularly the upregulation of TNF-α and IL-1-driven inflammatory responses. In patients with severe COVID-19, IFN-I responses co-occurred with inflammatory responses induced by TNF-α and IL-1 in classical monocytes, these responses were not observed in patients with mild COVID-19. This suggests that IFN-I play an important role in the progression to severe COVID-19 with the exacerbation of inflammation induced by TNF-α and IL-1 [32]. It is also worth mentioning that lethal SARS is associated with a greater release of pro-inflammatory cytokines, especially IFN-α and IFN-γ. Cytokines with increased levels in fatal SARS are IL-6, IL-1β, IFN, and C-X-C motif chemokine ligand 10 (CXCL10) [33].
Some research has also shown that the activation of the cGAS-STING pathway at a late stage of infection promotes an exacerbated inflammatory response [25, 30]. The effect of SARS-CoV-2 infection on host defense is not yet fully understood. A transcriptomic analysis of patients who died from acute respiratory distress syndrome (ARDS) during the first outbreak of SARS-CoV showed that IFNs and IRF3 were upregulated, suggesting activation of the cGAS-STING pathway signaling [34].
An overproduction of inflammatory cytokines, such as IL-6 and TNF-α, and low levels of IFNs are often seen in severe COVID-19 patients, which might be the cause of sustained viral replication and disease progression [35]. Those findings can be correlated with the NF-κB-specific inflammatory response in infected cells, selectively induced by cGAS-STING axis activation, with impairment of IRF3 and IFN signaling, demonstrated by Neufeldt et al. [11]. This association is likely because various cytokines, such as IL-1, IL-6, and TNF-α, important proinflammatory cytokines, are expressed with NF-κB activation [36]. To support the involvement of NF-κB activation at a severe stage of COVID-19, it was found that the genes upregulated in critical patients were mostly products of the NF-κB pathway. A type I IFN response impairment was also verified in those patients [37], which is consistent with the low activation of IFN signaling found by Neufeldt et al. [11]. Regarding the cGAS-STING correlation, since exacerbated activation of the NF-κB pathway can be caused by excessive innate immune sensor activation [37], the progressive syncytium formation can trigger a high activation of cGAS-STING.
However, it was not clear why STING would selectively activate the NF-κB pathway. It is known that cell survival and proliferation are as well favored by NF-κB activation [38], which can be beneficial for sustained viral replication and dissemination [14]; maybe this hypothesis explains the reason for the activation of the NF-κB pathway being strongly expressed. Thereby, the inhibition of NF-κB signaling, a downstream of cGAS-STING pathway, is considered a therapeutic target against inflammatory response to SARS-CoV-2 infection [14, 39]. A summary view of what was covered in this topic is shown in Figure 2.
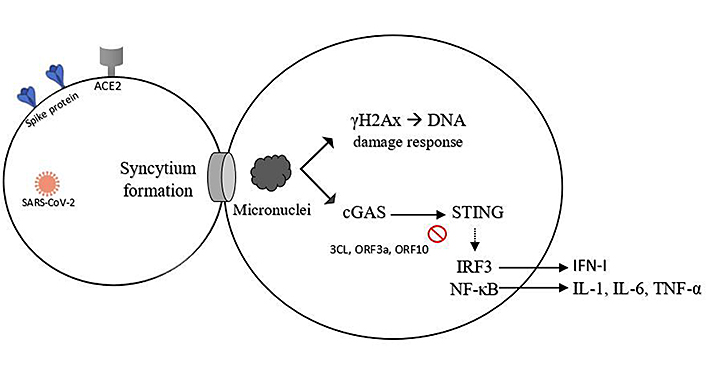
The syncytium is formed by cell-cell fusion in late-stage SARS-CoV-2 infection. Micronuclei formed are recognized for the DNA damage and cause the activation of many factors, such as phosphorylation of the histone variant H2AX, producing γH2Ax, and phosphorylation of IRF3 and NF-κB, via cGAS-STING, with overproduction of inflammatory cytokines. 3CL, ORF3a, and ORF10 are cGAS-STING inhibitors
Recently, infectious agents have emerged as a decisive cause to develop pulmonary fibrosis (PF). In this context, SARS-CoV-2 is associated with the development of ARDS and PF. A significant number of patients with COVID-19 (40%) develop ARDS, which has PF as a sequel, and the pathological feature of ARDS is diffuse alveolar damage (DAD) that begins with an acute inflammatory phase, followed by a fibrotic phase [40]. Although patients infected with SARS-CoV-2 can be cured, in most cases, the development of these pulmonary fibrotic lesions cannot be prevented; moreover, it remains unclear whether these lesions are stable or progressive fibrotic events [41].
Endothelium and epithelium injuries rise as a general mechanism for the onset of fibrosis [42]. More recently, it has become increasingly clear that viral infections such as SARS-CoV-2 can also cause severe lung damage, leading to endothelial and epithelial damage that serves as a starting point for an extensive inflammatory and fibrotic processes. Macrophages and neutrophils activations release profibrotic mediators that promote an accumulation of myofibroblasts. These are specific types of cells that are capable of producing extracellular matrices and have enhanced contractility. However, the exact role of viral and bacterial infections in the development and transmission of PF remains controversial [40, 43].
Risk factors associated with the predisposition to greater severity of COVID-19 may contribute to the occurrence of PF. These risk factors are advanced age, comorbidities, prolonged intensive care unit (ICU), time and duration of mechanical ventilation, smoking, and alcoholism. Laboratory findings such as lymphopenia, leukocytosis, and elevated lactate dehydrogenase (LDH) are associated with greater disease severity, with serum LDH being an indicator of lung tissue destruction, associated with a risk of mortality [25, 44]. Prolonged duration of mechanical ventilation confers an additional risk of ventilation-induced lung injury (VILI), since abnormalities in volume and pressure settings lead to the release of pro-inflammatory modulators and increase PF in patients [41].
Patients with severe COVID-19 can have severe lung damage, this is due much more to a hyperactivated immune system than inadequate viral clearance. Regarding the association between the cGAS-STING pathway and those lung lesions, as already discussed, at SARS-CoV-2 infection, there is micronuclei formation, which activates the cGAS-STING pathway. This constant activation itself promotes syncytium death, which contributes to inflammation and tissue damage in patients with severe COVID-19 or at a critical stage [19]. It has also been shown that in SARS-CoV-2 infection, the cGAS-STING axis is activated, but generating an NF-κB-specific inflammatory response in cells infected with the virus. This response is probably potentialized by immune cells producing the COVID-19 cytokine storm. The macrophages and neutrophils recruited by this inflammatory state are associated with cell death and pulmonary pathology [14]. It was found that NF-κB potentially triggers a viral replication and a higher influx of innate immune cells, such as neutrophils and monocytes, within the lungs, which causes tissue damage and maybe an auto-amplification inflammatory loop [37].
Studies show that interferons can disrupt the lung epithelial barrier and impair lung epithelial regeneration during viral infection, highlighting the importance of timely administration of interferon for the treatment of viral lung diseases [8]. It is also suggested that type III IFN (IFN-III) act as the first line of defense in the lung epithelium in the early stages of viral diseases and the role of IFN-I offers greater protection in later stages. The mechanisms of antiviral action of IFN-III are still being elucidated, but these mediators could have therapeutic potential against viruses that preferentially infect epithelial cells [17]. Activation of the cGAS pathway increased IFN expression to promote local and systemic inflammation, which was further enhanced by syncytial lysis. Furthermore, the intrinsic action-response participation of IFN in SARS-CoV-2 infection resonates with the identification of different profiles from immature secretory cells to ciliated cells with a distinctly strong IFN-γ response structure, suggesting their stimulation by the host immune system [45].
Host ACE2 receptor and spike protein (S protein) viral are known to be the initial portal targets of SARS-CoV-2 in the upper respiratory tract mucosa [45]. This binding is facilitated by specific proteases such as transmembrane serine protease 2 (atp) or Furin 28-32. Notably, ACE2 expression levels with one or both of the S-priming proteases were found to be increased three-fold in secretory cells in COVID-19 patients compared to uninfected controls. A greater representation of ACE2 of the immune cells was detected in the epithelium of the infected respiratory tract of analysis. In addition, there was a high correlation between ACE2 expression and IFN response, once the activity of signal transducer and activator of transcription 1 (STAT1), a central transcription factor for the IFN response, which was among the greatest predictors of ACE2 expression, was detected [45]. It is worth remembering that ACE2 and S protein are expressed on the surface of the cells that form the syncytium capable of activating the cGAS-STING pathway [19]. The syncytial cells give rise to the formation of nuclear membrane vesicles, accompanied by subsequent budding to form cytoplasmic chromatin. In some cases, cGAS is recruited to the cytoplasmic chromatin after nuclear membrane vesicles have developed from the nuclei. In other cases, cGAS is recruited to intranuclear chromatin, which is in the process of transport from the nucleus to the cytoplasm, and transported to the cytoplasm [31].
Recent evidence suggests that the cGAS-STING pathway, in addition to its well-established and previously described role in cytosolic DNA detection, is also involved in the attenuation of RNA viral infections, suggesting that there is an interaction between innate detection of cytosolic DNA and RNA. Indeed, cGAS or STING deficiency in cells or mice facilitates the replication of various RNA viruses, demonstrating the importance of cGAS in viral control. One of the suggested mechanisms would be that RNA viruses cause damage to mitochondria and release of mitochondrial DNA (mtDNA) into the cytosol, which would then activate the cGAS-STING pathway to potentiate host defense responses [46]. In the cytosol of syncytia formation, dsDNA of any source activates cGAS. This converts adenosine triphosphate (ATP) and guanosine triphosphate to form a second messenger cGAMP, which binds to STING, and induces its dimerization and translocation to perinuclear proteins. Subsequently, in late stages of COVID-19, it is suggested to activate TRAF6 and STING phosphorylates TBK1, which in turn activates NF-κB p65 leading to the synthesis of IFN-I and cytokines [31, 34] (Figure 3).
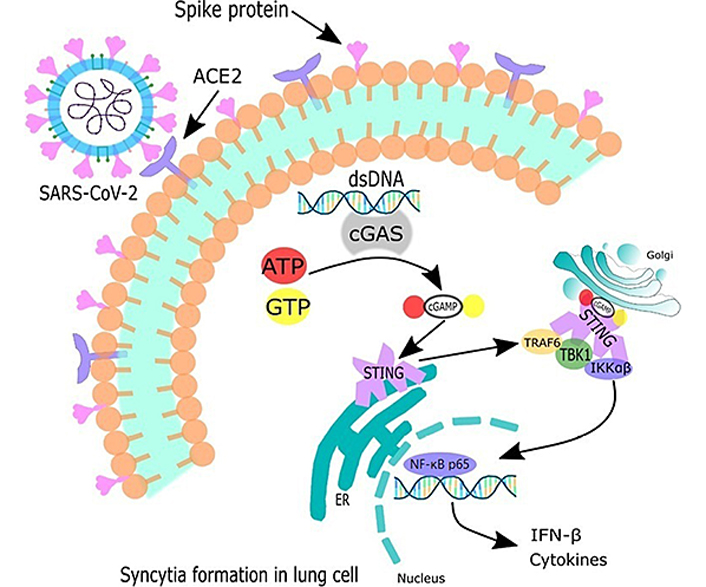
Syncytia formation in lung cells activates cGAS-STING signaling pathway canonical in late phase of SARS-CoV-2 infection. Therefore, dsDNA activates cGAS, which goes on to synthesize cGAMP. This second messenger activates the STING. Thus, STING translocates from the ER to the Golgi apparatus, starting to recruit kinases such as TBK1 and IKK. TRAF6 is recruited to the signaling complexes with STING and TBK1, which in turn activates the canonical NF-κB p65 signaling pathway to activate the transcription of IFN-β and cytokines encoding genes
From another perspective, p-STING is known to be a selective marker for activated STING observed in perivascular macrophages and in endothelial cells in COVID-19 lesions. Histopathological studies indicate that lung specimens expressing p-STING were from patients with rapidly lethal disease (death within 10 days of disease onset) characterized by early signs of DAD with an extensive transparent film [30]. On the other hand, samples without p-STING are most often from patients with a longer disease course (death within 14 days of onset) and show fibrotic changes more characteristic of advanced stages of DAD. Furthermore, samples with hallmarks for early DAD, but not late DAD, showed an IFN-I response. Together, these analyses link tissue damage produced by SARS-CoV-2 infection in the lung to activation of the cGAS-STING pathway and IFN-I signaling and postulate that endothelial cells, in addition to macrophages, are implicated in this antiviral response. Atypical inflammation and poor clinical outcome are associated with a sustained increase in IFN-I levels in the late phase of infection [30, 35, 40].
cGAS has been shown to be stimulated into endothelial cells through damage to mitochondrial homeostasis and associated accumulation of mtDNA, driving expression of IFN-I, cell activation, and finally cell death. Moreover, the study also lists macrophages as mediators of an IFN-I response on the vascular side in a cGAS-dependent pathway. In conclusion, it is evident that endothelial dysfunction and IFN-I production are the main virus-induced cellular mechanisms regulated by the cGAS-STING pathway in COVID-19 [30]. This regulation of IFN production by cGAS-STING can be strongly associated with pulmonary pathology since it was found that abundant or prolonged IFN production can impair the regeneration of lung epithelium [14].
The initiation of the response in the lung is followed by an inflammatory process that leads to massive activation of the resident immune cells. Lung myofibroblasts can come from different sources and their fate is apoptosis to complete the healing process [47]. Unfortunately, during fibrosis, the termination of extracellular matrix (ECM) production by these cells is compromised and increased rigidity leads to further cell damage and further activation of myofibroblasts. When this happens, a self-amplifying activation circuit is established and the fibrotic process becomes irreversible [48]. Regardless of origin, endothelial damage causes the release of profibrotic factors and cytokines, such as transforming growth factor-β (TGF-β), connective tissue growth factor/CCN family member 2 (CTGF/CCN2), and plasminogen activator inhibitor-1 (PAI-1) that support all stages of fibrotic processes. In this scenario, aging also emerged as a critical actor [42]. Damaged aged cells are unable to regulate genes such as nitric oxide synthase 3 (Nos3), which encodes the enzyme endothelial nitric oxide synthase (eNOS), and is a key protein involved in the resolution of PF [40, 49].
COVID-19 survivors developed post-acute sequelae covid (PASC), and animal studies have shown that histologic evaluation identified subpleural lesions containing collagen, fibroblast proliferation, and chronic inflammation with tertiary lymphoid structures 15–120 days after the viral spread. The longitudinal spatial transcription profile identifies global repair pathways dysregulated and fibrosis in diseased regions, similar to human COVID-19. Alveolar intermediate cell populations, as well as local regulation of profibrotic markers, were found in persistently diseased areas [50].
Some drugs have been investigated, and diamidobenzimidazole (diABZI), a STING agonist, has been the most studied. The studies on diABZI demonstrated an inhibition of the viral infection [8, 18, 31, 35] and a reduction of viral replication and lung inflammation, as well, played by diABZI treatment against SARS-CoV-2 infection [8]. Considering that diABZI is a STING-TBK1-IRF3 signaling pathway activator, those results show the importance of STING activation against SARS-CoV-2 infection [8].
The STING-TBK1-IRF3 signaling pathway activation by diABZI was observed by Li et al. [8] through human respiratory Calu-3 epithelial cells, treated with that agonist. After 2 h of treatment, besides phosphorylated STING, there was an increase of TBK1, IRF3 phosphorylation, and also NF-κB signaling. In addition, IFNs, ISGs type I and III, and cytokines, which help with neutrophil recruitment, were induced by the treatment. Zhou et al. [31] also concluded that diABZI had a positive regulation upon IFNs, which was correlated to a lower viral replication. The amount of viral replication reduction was observed to be similar to the amount caused by Remdesivir, a SARS-CoV-2 RNA replication inhibitor [8]. Those results reveal a potential use of diABZI as a treatment for SARS-CoV-2 infection.
However, in the treatment with mice, with H-151, a STING antagonist, as shown by Domizio et al. [30], reduces the pathology caused by SARS-CoV-2, preventing weight loss and death after infection. To those findings, STING was associated with malefic inflammation in the late phases of the infection, pointing to the H-151 as a possible treatment and prevention from a progression of the infection to serious clinical manifestations.
Neufeldt et al. [11], by identifying the triggering effect of the inflammatory response from the activation of the cGAS-STING axis in COVID-19 infection, sought to test the pharmacological inhibition of the pathway, more precisely, of the STING molecule. To that end, he incubated SARS-CoV-2 infected human lung cells with H-151, VS-X4, and TBK1 inhibitor amlexanox (AMX), all STING inhibitors. After 24 h of infection, it was possible to visualize a significant reduction in the levels of TNF messenger RNAs (mRNAs) in cells treated with H-151 and VS-X4, compared to those treated with AMX and dimethyl sulfoxide (DMSO). Moreover, in this same specific population of cells, a reduction in p65/v-rel avian reticuloendotheliosis viral oncogene homolog A (RELA) nuclear accumulation was observed, and, regarding the effects of VS-X4, an important reduction in virus-induced upregulation of IL-6 and interferon gamma-induced protein 10 (IP-10) was noted. Therefore, such evidence made it possible to conclude the efficacy of STING inhibitors in the pro-inflammatory response induced by activation of the cGAS-STING pathway in cells infected by SARS-CoV-2.
Furthermore, from another study perspective, Liu et al. [25] evaluated that the role of proteolytic activation of the viral S protein in syncytial generation has a direct implication in the pathogenesis of COVID-19, since the action strengthens the infectivity of cell-free virions and increases lymphocytic clearance by multinucleated pneumocytes. Thus, based on such evidence, their study assumes the therapeutic potential of neutralizing antibodies, drugs, and protease inhibitors in the disruption of syncytial formation, reproducing greater efficacy against the late or critical state of the infection.
Regarding the use of IFNs in COVID-19, the effectiveness is still questionable. The use of IFNs during the inflammatory and severe stages causes immunopathology and long-lasting damage to patients. After the viral peak, the late use of IFNs fails to establish a protective effect, in addition to aggravating the SARS-CoV-1 infection, causing inflammation and severe pneumonia. Therefore, it is of fundamental importance to consider the best time window for the administration of IFNs, since these are double-edged immunomodulatory agents; they may have a protective effect when administered in the initial phases of the disease before the viral peak (early use of IFN leads to a protective effect), while the deleterious effects are observed when administered in the inflammatory phase. More human experiments are required to find the best time window to administer IFN-I to patients with various modalities of COVID-19 [51].
Briefly, the expression of the spike protein by SARS-CoV-2 induces the fusion of the viral membrane with the plasma membrane, culminating in the release of the viral genome, and this process is accompanied by syncytial formation, which activates the cytosolic protein sensor cGAS and stimulates the adapter molecule STING to activate downstream signaling pathways that eventually trigger expression of antiviral genes such as IFNs and ISGs. In the early stages of coronavirus infection, there is a suppression in the production of interferon, especially IFN-I. One of the explanations is due to the SARS-CoV-2 inhibitory viral proteins 3CL, ORF3a, and ORF10 that inhibit the RLR or cGAS-STING pathways that cause the inhibition of the NF-κB and TBK1 signaling cascade. After the formation of syncytial cells, there is an increase in the inflammatory response that potentiates the activation of the cGAS-STING pathway with a selective increase in NF-κB activation and consequent increase in inflammatory cytokines, such as IL-1, IL-6, and TNF-α. Thus, NF-κB is considered the main determining factor in the exacerbation of the disease.
IFN is directly related to the cytokine storm present in patients with COVID-19, but it is still unclear at what exact moment this increase occurs, due to the various forms of the disease. It was observed that in the early stages of the disease, the production of IFNs is contained by the virus, and hours later there is an increase in its concentration. This increase is related to the protective action of IFNs and infection blocking, but in the long term and in cases of severe COVID-19, this increase becomes pathological, which together with the accumulation of other cytokines and chemokines, tends to worsen the prognosis of the disease.
3CL: 3C-like protease
ACE2: angiotensin-converting enzyme 2
ARDS: acute respiratory distress syndrome
cGAMP: cyclic guanosine monophosphate-adenosine monophosphate
cGAS: cyclic guanosine monophosphate-adenosine monophosphate synthase
COVID-19: coronavirus disease 2019
DAD: diffuse alveolar damage
diABZI: diamidobenzimidazole
dsDNA: double-stranded DNA
ER: endoplasmic reticulum
IFN-I: type-I interferons
IFN-β: interferon-β
IKK: inhibitor of nuclear factor-κB kinase
IL-6: interleukin-6
IRF3: interferon regulatory factor 3
ISGs: interferon-stimulated genes
MAVS: mitochondrial antiviral-signaling protein
NF-κB: nuclear factor-κB
ORF3a: open reading frame 3a
ORF10: open reading frame 10
PF: pulmonary fibrosis
p-STING: phosphorylated stimulator of interferon genes
RIG-I: retinoic acid-inducible gene I
RLR: retinoic acid-inducible gene I-like receptor
SARS-CoV-2: severe acute respiratory syndrome coronavirus type 2
STING: stimulator of interferon genes
TBK1: TANK-binding kinase 1
TNF-α: tumor necrosis factor-α
TRAF: tumor necrosis factor receptor-associated factor
GFA:Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Supervision, Visualization, Writing—original draft preparation, Writing—review & editing. SGF: Data curation, Formal analysis, Investigation, Visualization, Writing—original draft preparation, Writing—review & editing. HNV: Data curation, Formal analysis, Investigation, Visualization, Writing—original draft preparation, Writing—review & editing. CGAPLF: Data curation, Formal analysis, Investigation, Visualization, Writing—original draft preparation. KSA: Data curation, Formal analysis, Investigation, Visualization, Writing—riginal draft preparation. LMA: Data curation, Formal analysis, Investigation, Visualization, Writing—original draft preparation. SLMT: Data curation, Formal analysis, Investigation, Visualization, Writing—original draft preparation.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 4469
Download: 46
Times Cited: 0
