Review
Review

Affiliation:
1Department of Pharmacology, Seacom Pharmacy College, Howrah 711302, West Bengal, India
Email: souravpal2525@gmail.com
ORCID: https://orcid.org/0009-0006-1532-4069
Affiliation:
2Seacom Pharmacy College, Howrah 711302, West Bengal, India
Affiliation:
1Department of Pharmacology, Seacom Pharmacy College, Howrah 711302, West Bengal, India
ORCID: https://orcid.org/0009-0007-0610-3595
Affiliation:
3Department of Pharmacy, Seacom Pharmacy College, Howrah 711302, West Bengal, India
Explor Immunol. 2025;5:1003213 DOI: https://doi.org/10.37349/ei.2025.1003213
Received: May 10, 2025 Accepted: August 05, 2025 Published: August 26, 2025
Academic Editor: Sunil K. Arora, Postgraduate Institute of Medical Education & Research, India
Mpox, caused by the monkeypox virus (MPXV), has re-emerged as a global health concern due to recent outbreaks and the emergence of new variants. Current antiviral options are limited, prompting the search for alternative therapeutic strategies. This review explores the therapeutic potential of marine-derived bioactive compounds as antiviral agents against MPXV, focusing on their mechanisms of action and clinical relevance. Marine phytoconstituents, including mycosporine-like amino acids, carrageenan, fucoidans, and griffithsin, exhibit diverse antiviral, immunomodulatory, and anti-inflammatory properties. Understanding their role may offer innovative solutions for mpox management and address gaps in current treatment approaches. A comprehensive literature search was performed across PubMed, Scopus, and Web of Science to identify peer-reviewed articles published between 2010 and June 2024 using keywords such as “mpox”, “monkeypox virus”, “marine-derived antivirals”, and “orthopoxvirus”. Emphasis was placed on studies from 2021–2024 to capture recent developments in mpox pathogenesis and marine-based therapeutics. Eligible sources included original research, systematic reviews, meta-analyses, and official health reports published in English. Marine-derived compounds demonstrate promising antiviral and immunomodulatory effects against MPXV in preclinical models. While further research is needed to confirm their clinical efficacy and address issues of scalability and safety, these agents represent a valuable adjunct or alternative for future mpox therapeutics.
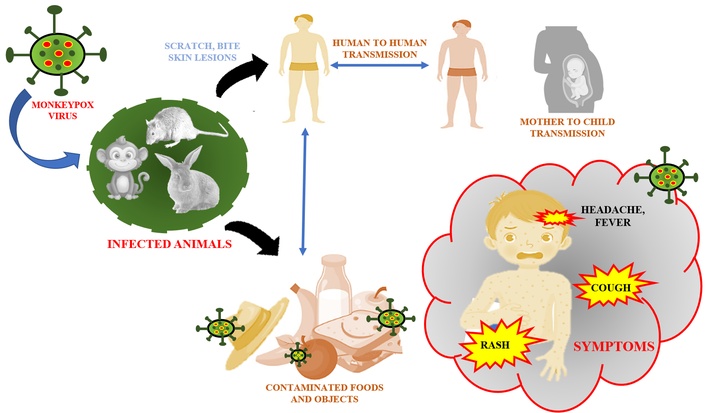
Mpox, formerly known as monkeypox, is a viral disease caused by the monkeypox virus (MPXV), a member of the Orthopoxvirus genus, alongside the variola virus, which causes smallpox [1]. While historically confined to central and western Africa, mpox has recently garnered global attention due to outbreaks occurring beyond these endemic regions. There is concern that mpox could pose a pandemic threat. The transmission of MPXV occurs through various mechanisms, including direct human-to-human contact, zoonotic transmission from animals to humans, and contact with contaminated objects [2]. Human-to-human transmission primarily occurs through direct contact with lesions, respiratory droplets, or bodily fluids from an infected individual. Zoonotic transmission typically results from contact with infected animals (Figure 1) [3]. Additionally, contaminated objects, such as bedding or clothing used by an infected person, can facilitate the spread of the virus. Symptoms typically appear within 1 to 21 days after exposure, including fever, chills, headache, muscle aches, back pain, fatigue, and swollen lymph nodes. A hallmark of mpox is a rash, which progresses through the stages of macules, papules, vesicles, pustules, and scabs. It often starts on the face and spreads to other parts of the body, including the palms, soles, and genital areas [4]. Recent findings highlight atypical presentations, such as milder symptoms or fewer lesions, complicating diagnosis. Severe complications include bacterial superinfections, pneumonia, encephalitis, myocarditis, and gastrointestinal symptoms like vomiting and diarrhea. The contagious period lasts from symptom onset until all lesions heal completely. Vulnerable populations include children, pregnant individuals, and immunocompromised persons (e.g., those with untreated HIV), all of whom are at higher risk for severe disease outcomes. As mpox continues to spread globally in 2025, with new variants like Clade Ib emerging in Africa, timely diagnosis and intervention are critical.
In 2022, a significant mpox outbreak in Africa resulted in 6,884 reported cases across eight African Union member states, with a case fatality rate (CFR) of approximately 2.5% [5]. The World Health Organization declared this outbreak a public health emergency of international concern on July 23, 2022 [6]. By 2025, the situation has escalated dramatically, with approximately 20,000 total cases reported across 15 African countries, including the Democratic Republic of the Congo (DRC), which remains the epicenter with over 15,411 cases and a rising CFR of 3–6% due to the emergence of Clade Ib [7].
The spread of mpox has extended beyond Africa to Europe, Asia, and the Americas, prompting renewed global concern (Table 1). The lack of specific antiviral treatments for mpox has led researchers to explore alternative therapies, particularly marine-derived compounds sourced from sponges, corals, and seaweeds. These unique compounds have demonstrated potential antiviral properties against orthopoxviruses in preclinical studies. However, challenges remain regarding the scaling of production and ensuring safety for human use. The ongoing outbreak underscores critical needs for targeted interventions, including equitable vaccine distribution for high-risk populations and strengthening surveillance systems to address underreporting.
Reported mpox cases and deaths by region [8]
| Region | Total reported cases | Total deaths reported |
|---|---|---|
| DRC | 15,411 | 43 |
| Burundi | 3,586 | 1 |
| Uganda | 2,949 | 21 |
| Kenya | 37 | 1 |
| Central African Republic | 263 | 1–10 |
| Republic of the Congo | 158 | 1 |
| Ivory Coast | 28 | 1 |
| South Africa | 24 | 3 |
| Nigeria | 1,484 | 0 |
| United States | 4,259 | 11–25 |
| Canada | 365 | 0 |
| United Kingdom | 2,137 | 0 |
| Australia | 1,680 | 0 |
DRC: Democratic Republic of the Congo
MPXV is an enveloped, double-stranded DNA virus belonging to the Orthopoxvirus genus, with a genome size of approximately 197 kilobases encoding nearly 190 proteins [9]. Structurally, MPXV consists of a lipoprotein envelope, a viral core, and two lateral bodies that house essential enzymes and transcription factors required for replication within the cytoplasm. The virus’s central genomic regions, which exhibit a 96.3% similarity to the vaccinia virus (VACV), encode key structural proteins, while most variable areas contribute to virulence [10]. MPXV demonstrates intricate immune evasion mechanisms, enabling it to establish infection and persist within the host. Proteins like MOPICE suppress the complement pathway, while B16 inhibits type I interferon (IFN) signaling, impairing the host’s antiviral defenses [11]. Additionally, the virus interferes with major histocompatibility complex (MHC)-I presentation through proteins such as B10 and N3R, reducing cytotoxic T cell recognition. Unlike other orthopoxviruses, MPXV does not downregulate MHC-I molecules but selectively modulates T cell activation, facilitating localized immune suppression while maintaining a viral reservoir [12]. These strategies contribute to the virus’s capacity to evade the host immune response effectively. Recent structural insights into MPXV proteins, such as the VP39 methyltransferase adopting a Rossmann fold, reveal its role in RNA cap modification, a critical step in viral replication and immune evasion. The virus’s ability to produce both intracellular mature virus (IMV) and extracellular enveloped virus (EEV) further supports its adaptability and transmission. Genetic variability across Clades I and II, along with the emergence of Clade Ib in 2023, underscores MPXV’s capacity for rapid evolution. Clade-specific mutations, particularly in immune evasion genes like A10 and A42R, influence virulence and adaptation to human hosts [13].
Clade I of the MPXV, primarily found in Central Africa, particularly the DRC, is associated with more severe disease and higher mortality rates, ranging from 4% to 11% in some studies [14]. This Clade has been recognized for a longer period and has historically caused the majority of endemic cases in Africa. Recent findings indicate that Clade I exhibits more efficient human-to-human transmission compared to Clade II, particularly in settings where close contact occurs, such as healthcare or familial environments. The emergence of new lineages within Clade I, such as Clade Ib, has raised concerns regarding their potential for increased transmissibility and virulence. Genomic analyses have shown that Clade Ib is distinct from previously circulating strains and has been linked to sexual transmission routes, indicating a shift in transmission dynamics. Surveillance data from 2023 to early 2024 revealed that a significant proportion of cases involved younger populations, with many being sex workers, suggesting that behavioral factors may play a role in the spread of this Clade [15].
In contrast, Clade II, predominantly found in West Africa, encompasses subclades such as IIa and IIb, with cases reported in countries like Nigeria and Cameroon. This Clade is generally associated with milder disease and lower mortality rates, typically around 1% to 3%. Although it is less efficient in human-to-human transmission compared to Clade I, outbreaks have still occurred, particularly during the global epidemic that began in 2022. Clade IIb gained significant attention due to its rapid spread outside endemic regions during the recent global outbreak, with genomic analyses revealing mutations indicative of adaptation to human hosts. The B.1 lineage of Clade IIb was responsible for a substantial number of cases globally, demonstrating a shift toward increased transmissibility through sexual contact [16].
The transmission dynamics of MPXV in 2025 reflect evolving viral adaptations and socio-behavioral factors. Genomic and epidemiological analyses reveal that Clade IIb (lineage B.1), responsible for the 2022–2023 global outbreak, spread predominantly through heavy-tailed sexual contact networks among men who have sex with men (MSM), as observed in New York City [16]. Phylogeographic models indicate that early declines in transmission (effective reproduction number Re < 1 by mid-July 2022) were driven by immunity among highly connected individuals in these networks, with secondary attack rates of 0.1–0.4 [17]. This rapid saturation of “super-spreader” nodes reduced cluster sizes even before widespread vaccination, highlighting the role of network structure in curbing outbreaks. Molecular insights into MPXV underscore Clade-specific differences: Clade Ib, emerging in Central Africa (2023), exhibits enhanced transmissibility via sexual and non-sexual contact, though with lower mortality (CFR 3–6%) compared to Clade Ia [18]. Viral proteins like VP39 (RNA methyltransferase) and immune-evasion factors (e.g., A47R, B13R) enable host immune suppression, facilitating prolonged viral shedding [19]. Structural adaptations in Clade Ib’s A35R protein further enhance zoonotic spillover potential. Recent data emphasize demographic shifts, with 50% of pediatric cases in endemic regions linked to household transmission [13].
Direct contact with infected animals remains a major risk factor in mpox transmission. Recent findings highlight the fire-footed rope squirrel (Funisciurus pyrropus) as a potential reservoir, capable of asymptomatic viral shedding [19]. Evidence from a 2023 mangabey outbreak supports cross-species transmission, while pouched rats and other rodents may also contribute to a broader reservoir network. Transmission occurs through direct handling, exposure to bodily fluids, bites, or indirect contact, such as consuming undercooked bushmeat [20]. A meta-analysis reports a 5.61-fold increased infection risk among individuals exposed to infected animals [21]. Activities like hunting, trapping, and skinning are particularly hazardous. However, identifying true reservoir hosts remains challenging due to limited surveillance and the complex ecology of mpox in endemic regions.
Mpox infection elicits strong immune activation characterized by an increase in cluster of differentiation 38 (CD38)+, HLA-DR+ CD8+ T cells, which are essential for viral control but may also contribute to tissue damage [22]. Alterations in CD4+ T cell subsets, particularly rise in effector and transitional memory cells, reflect adaptive immune engagement. Elevated plasma levels of regulated upon activation, normal T cell expressed and secreted (RANTES), chemokine C-C motif ligand 3 (CCL3), C-X-C motif chemokine ligand 10 (CXCL10), and interleukin-2 receptor α (IL-2Rα) indicate a robust inflammatory response, while interleukin-6 (IL-6) shows variable expression. B cell responses include a shift from IgA to IgG isotypes in plasmablasts, signaling adaptive humoral immunity [23]. People living with HIV (PLHIV), especially those with advanced immunosuppression, are at significantly higher risk for mpox infection and severe disease outcomes [24]. A 2024 meta-analysis found that PLHIV are 4.46 times more likely to contract mpox than HIV-negative individuals, with the risk increasing markedly in those with CD4 counts below 200 cells/mm3 [21]. These individuals often experience severe clinical manifestations, including extensive lesions and systemic complications, leading to elevated mortality. Hospitalization risk is also heightened, with a 56.6% increase compared to HIV-negative patients [25]. During the 2022–2023 outbreak, PLHIV accounted for 38–50% of global cases, underscoring the public health burden. Despite many having well-controlled HIV, those with advanced disease faced worse outcomes. Immunologically, impaired cellular responses in advanced HIV hinder viral control, contributing to mpox severity [26].
Recent systematic reviews and meta-analyses have confirmed that pre-existing sexually transmitted infections (STIs) significantly increase the risk of acquiring mpox, particularly during recent outbreaks marked by sexual transmission among MSM [27]. A pooled OR of 1.76 (95% CI: 1.42–2.91) demonstrates a 76% increased risk of mpox in individuals with prior STIs such as syphilis, gonorrhea, chlamydia, hepatitis B/C, and varicella-zoster virus. Moderate heterogeneity (I2 ≈ 40%) is reported, with results remaining statistically significant (P < 0.0001) [28]. Additionally, comorbidities, including STIs, elevate mpox risk (OR = 1.58, 95% CI: 1.31–1.91), and multiple sexual partners further compound susceptibility (OR ≈ 1.6–1.8) [21]. Biological plausibility supports this association, as STI-related mucosal disruption and immune suppression increase mpox susceptibility. Mpox lesions also facilitate STI transmission, creating a bidirectional risk [28]. The 2022 outbreak strains (Clade IIb, lineage B.1) show rapid evolution driven by apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3 (APOBEC3)-mediated mutations (e.g., GA>AA, TC>TT), with 66–86 nucleotide and 28–39 amino acid substitutions, particularly in immune-related genes such as B21R, F13L, and A18R [29]. Mpox employs multiple immune evasion mechanisms, including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway inhibition via ankyrin-like and B cell lymphoma 2 (BCL-2)-like proteins, T cell suppression by B21R, and mucosal barrier disruption through genital/rectal lesions [30]. Co-infections, especially with HIV, intensify disease severity through immune compromise (e.g., CD4+ T cell depletion), increased pathogen load, and delayed viral clearance, factors that may elevate the risk of tecovirimat resistance.
Recent data reinforce sexual behavior as a key driver of mpox transmission in non-endemic regions. A case-control study in California (2022–2023) revealed a significantly elevated infection risk from sexual contact with asymptomatic or suspected mpox partners (OR = 7.7) [31]. Risk increased with the number of partners: individuals with ≥ 4 partners had nearly fourfold higher odds of infection (OR = 3.8) [21]. Site-specific sexual practices correlated with lesion location, linking insertive and receptive anal sex to penile and anorectal lesions, respectively [31]. A 2023 Chinese study found that safer sexual behaviors, such as avoiding group or commercial sex and using condoms, significantly reduced mpox risk. Meta-analyses further support these findings, identifying multiple partners (OR = 1.61), unprotected sex (OR = 1.53), HIV (OR = 4.46), and other STIs (OR = 1.76) as key risk factors [21]. These insights highlight the critical role of sexual networks and behavior in the spread and the importance of behavioral interventions in outbreak control.
MPXV transmission involves multiple routes, with recent outbreaks emphasizing human-to-human spread, particularly through sexual contact. Zoonotic transmission persists in endemic regions via rodent reservoirs, such as Funisciurus spp. and Cricetomys gambianus, with viral shedding through saliva, lesions, and excreta [32]. Bushmeat handling and consumption remain relevant risk factors. Human-to-human transmission is now primarily driven by sexual exposure, with culturable virus detected in semen up to 21 days post-symptom onset and anogenital lesions present in 60–70% of cases, facilitating mucosal entry [33]. While respiratory droplet transmission occurs during prolonged close contact, no evidence supports aerosol spread outside healthcare settings. Fomite transmission is substantiated by MPXV persistence on surfaces for over 15 days. Vertical transmission is rare but confirmed, with placental infection linked to high fetal mortality [34]. At the cellular level, MPXV entry is mediated by mature virion (MV) proteins (A26, A27, H3, D8) binding to glycosaminoglycans and EV proteins (B5, A33), promoting actin-driven intercellular spread [35]. Fusion is pH-independent, involving 11–12 transmembrane proteins and host actin remodeling. Early immune evasion involves the suppression of NF-κB by ankyrin-repeat proteins (J3L, D1L) and the inhibition of apoptosis via BCL-2-like protein (A47R) [36]. Following mucosal entry, the virus disseminates via lymphatic spread, leading to viremia and infection of distant organs, including skin, liver, testes, and placenta. Genomic adaptations, notably APOBEC3-mediated mutations and gene duplications in immunomodulatory loci, enhance host adaptation and immune evasion [37].
The replication of MPXV follows a complex cytoplasmic life cycle distinct from most DNA viruses [38]. MPXV initiates infection by binding to host cell surface molecules such as glycosaminoglycans (heparan sulfate, chondroitin sulfate) and integrins via viral surface proteins like hemagglutinin [39]. Entry occurs either through direct membrane fusion or clathrin-mediated endocytosis. Upon entry, the viral core is uncoated, and the genome is released directly into the cytoplasm, bypassing the nucleus [40]. MPXV gene expression proceeds in a tightly regulated cascade where early genes are expressed immediately post-entry and encode proteins for immune evasion and replication initiation; intermediate genes trigger viral DNA replication; late genes encode structural proteins required for virion assembly. Replication occurs in cytoplasmic viral factories called Guarnieri bodies [41]. Viral DNA replication produces concatemeric intermediates that are processed into individual genomes. These are packaged into IMVs, which mature into stable intracellular MVs. Some MVs acquire additional membranes from the Golgi or endosomal system, forming EEVs, which enhance cell-to-cell spread and immune evasion. Viral egress involves lysis for MVs and exocytosis for EEVs. MPXV virions carry multiple immunomodulatory proteins that inhibit host inflammatory responses, apoptosis, and complement activation [42].
MPXV has evolved complex mechanisms to evade host immune responses, facilitating persistent infection and effective transmission (Figure 2). These strategies target innate and adaptive immune systems, ensuring viral survival despite host defenses. In the innate immune response, MPXV suppresses type I IFNs, critical for antiviral defense, by inhibiting pattern recognition receptors (PRRs) such as retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated protein 5 (MDA5), and Toll-like receptors (TLRs), which would normally activate antiviral transcription factors like IFN regulatory factor 3 (IRF3) and NF-κB. Viral proteins like E3L and K3L block key antiviral pathways, including the protein kinase R (PKR) and 2',5'-oligoadenylate synthetase (OAS)/ribonuclease L (RNase L) systems, preventing antiviral protein synthesis and RNA degradation [43]. MPXV also evades type II IFN (IFN-γ) signaling, impeding the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathway [44]. The virus further inhibits the complement system, preventing the formation of the membrane attack complex (MAC) and protecting infected cells from lysis [45]. In the adaptive immune response, MPXV modulates T cell activity by interfering with MHC expression and TCR signaling, thereby reducing CD8+ T cell activation and differentiation [46]. It also blocks apoptosis in infected cells, allowing prolonged viral replication. Furthermore, MPXV evades neutralizing antibodies by utilizing cell-associated viremia, where infected monocytes shield the virus from antibodies, and by expressing Fc-binding proteins that prevent antibody-dependent cellular cytotoxicity [47]. MPXV also alters the cytokine environment by suppressing pro-inflammatory cytokines like tumor necrosis factor alpha (TNF-α) and IL-1β and sequestering chemokines such as CCL5 and CXCL10, impairing immune cell recruitment [48]. These immune evasion mechanisms contribute to MPXV’s ability to persist in the host, evade immune surveillance, and enhance transmission [13].
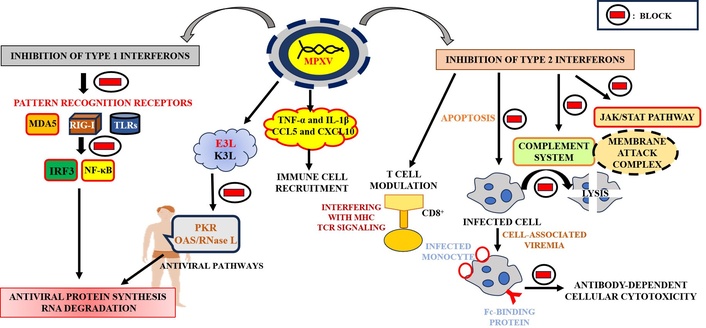
Viral immune evasion cascades in MPXV infection. MPXV: monkeypox virus; MDA5: melanoma differentiation-associated protein 5; RIG-I: retinoic acid-inducible gene I; TLRs: Toll-like receptors; IRF3: interferon regulatory factor 3; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; TNF-α: tumor necrosis factor alpha; IL-1β: interleukin-1 beta; CCL5: chemokine C-C motif ligand 5; CXCL10: C-X-C motif chemokine ligand 10; PKR: protein kinase R; OAS: 2',5'-oligoadenylate synthetase; RNase L: ribonuclease L; MHC: major histocompatibility complex; CD8: cluster of differentiation 8; JAK: Janus kinase; STAT: signal transducers and activators of transcription
A critical mechanism involves inhibition of the complement cascade via the viral MOPICE protein, a homolog of the VACV complement control protein. MOPICE mimics host regulators, binds C3b/C4b, and acts as a decay-accelerating factor to block convertase formation, thereby reducing MAC assembly and inflammatory signaling [49]. Additionally, MPXV suppresses IFN signaling through the B16 protein, which sequesters IFN-α/β extracellularly, preventing JAK1/tyrosine kinase 2 (TYK2) activation and downstream STAT1/2-mediated transcription of IFN-stimulated genes (ISGs) [50]. This significantly impairs early antiviral responses. In the adaptive immune arm, MPXV interferes with MHC-I antigen presentation through B10 and N3R proteins. B10 retains MHC-I in the endoplasmic reticulum, while N3R disrupts peptide loading, both resulting in impaired cytotoxic T lymphocyte recognition [51]. The virus also dampens inflammatory pathways by targeting NF-κB activation. Ankyrin repeat-containing and BCL-2-like proteins bind host IKK complexes and TNF receptor-associated factor 6 (TRAF6) adaptors, respectively, inhibiting nuclear translocation of NF-κB and reducing cytokine production [52]. Furthermore, the B21R glycoprotein directly suppresses T cell activation by modulating TCR signaling, lowering IL-2 and IFN-γ expression, and impairing CD4+/CD8+ T cell function [53]. Together, these mechanisms highlight MPXV’s ability to evade immune detection at multiple molecular checkpoints.
MPXV employs several mechanisms to suppress the host’s immune response, enabling effective immune evasion and prolonged viral replication. A crucial strategy is disrupting the JAK-STAT pathway, a vital signaling cascade that governs immune responses, cell growth, and apoptosis [54]. This pathway is typically activated by cytokine binding to their receptors, triggering JAK-mediated phosphorylation of STAT proteins, which then regulate the transcription of antiviral and inflammatory genes [55]. MPXV manipulates this pathway at multiple points to hinder the host’s immune defense. One of the primary mechanisms MPXV utilizes is the inhibition of type I IFN signaling. Viral proteins such as E3L and K3L play key roles in preventing the activation of RIG-I-like receptors (RLRs) and TLRs, crucial for the initiation of IFN production [56]. E3L inhibits the activation of IRF3 and NF-κB, essential transcription factors for IFN production, while K3L mimics the α subunit of eukaryotic initiation factor 2 (eIF2α), preventing PKR-mediated phosphorylation of eIF2α, which would otherwise halt viral protein synthesis [57]. Additionally, MPXV modulates suppressors of cytokine signaling (SOCS) proteins (SOCS1 and SOCS3) to suppress JAK-STAT signaling by promoting the degradation of activated JAKs and STATs, reducing immune response intensity [58]. MPXV also interferes with the activation and nuclear translocation of STAT1 and STAT2, blocking their ability to initiate the transcription of ISGs that are crucial for antiviral immunity [59]. The suppression of the JAK-STAT pathway has significant pathophysiological implications [60]. It allows MPXV to evade innate and adaptive immune responses by impairing the activation of natural killer (NK) cells and CD8+ T cells and ensures prolonged viral replication by preventing apoptosis [61]. This contributes to a state of chronic inflammation and increased susceptibility to secondary infections, highlighting the virus’s ability to persist within the host and complicating therapeutic interventions.
MPXV encodes a specialized nuclease, poxin, which plays a critical role in evading host innate immunity by targeting the cyclic GMP-AMP synthase-stimulator of IFN genes (cGAS-STING) signaling axis (Figure 3). Under normal conditions, cGAS detects cytosolic double-stranded DNA and catalyzes the synthesis of cyclic GMP-AMP (cGAMP), a second messenger that activates STING [62]. STING engagement triggers the phosphorylation of TANK-binding kinase 1 (TBK1), IRF3, and NF-κB, ultimately driving the expression of IFNs and pro-inflammatory cytokines such as IL-6 and TNF-α [63]. Poxin acts by specifically hydrolyzing 2',3'-cGAMP, thereby abolishing STING activation and downstream signal transduction [64]. Structural and enzymatic analyses have identified key residues His17, Tyr138, and Lys142 as essential for catalysis, forming a conserved triad within the active site. Unlike conventional nucleases, poxin does not require divalent metal ions for activity, instead utilizing a metal-independent mechanism likely based on acid-base catalysis. Moreover, the enzyme exhibits stringent substrate specificity, selectively degrading 2',3'-cGAMP while sparing bacterial cyclic dinucleotides such as 3',3'-cGAMP and c-di-GMP [63]. Through this mechanism, MPXV poxin effectively suppresses the host’s early antiviral response, impairing the activation of TBK1 and IRF3 and limiting the production of IFNs and inflammatory mediators [65]. This enables the virus to evade detection during the initial phase of infection and facilitates viral replication and spread, particularly in epithelial barriers where nucleic acid sensing is pronounced. Poxin thus represents a highly refined viral adaptation for immune modulation, underscoring its significance in MPXV pathogenesis and its potential as a therapeutic target.
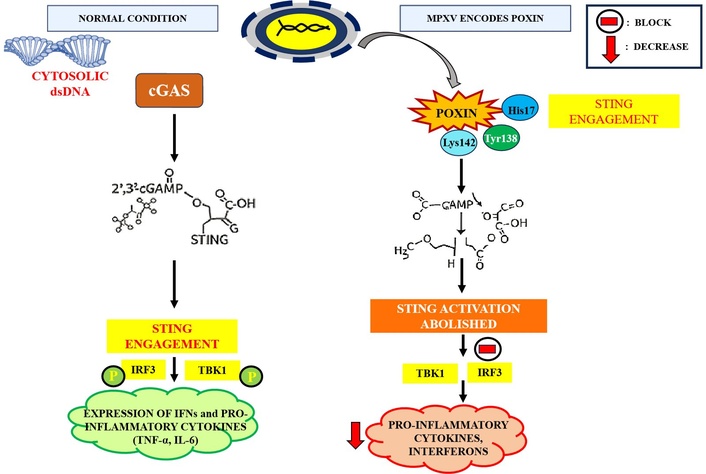
Modulation of the cGAS-STING antiviral response by MPXV. cGAS-STING: cyclic GMP-AMP synthase-stimulator of interferon genes; MPXV: monkeypox virus; cGAMP: cyclic GMP-AMP; IRF3: interferon regulatory factor 3; TBK1: TANK-binding kinase 1; IFNs: interferons; TNF-α: tumor necrosis factor alpha; IL-6: interleukin-6; P: phosphorylation
MPXV utilizes a strategic viral protein, F3, to evade host innate immune response pattern recognition pathways (PPs), primarily through interference with double-stranded RNA (dsRNA)-mediated antiviral signaling [66]. F3 is a homolog of the VACV E3 protein and retains a conserved C-terminal dsRNA-binding domain essential for immune suppression [67]. Although the N-terminal Z-DNA-binding domain present in VACV E3 is absent in MPXV F3, this truncated form remains functionally competent in binding viral dsRNA generated during replication [68]. The immunoevasive function of F3 is executed through dsRNA sequestration, effectively concealing this viral pathogen-associated molecular pattern (PAMP) from host PRRs such as RIG-I, MDA5, and PKR. By inhibiting PRR activation, F3 impedes the initiation of critical signaling cascades that would normally induce IFN production and pro-inflammatory responses [69]. In particular, F3 prevents PKR-mediated phosphorylation of the eIF2α, which would otherwise halt host and viral protein synthesis as part of the antiviral response [70]. This allows MPXV to maintain active translation machinery and sustain viral replication. Additionally, by blocking RIG-I and MDA5 signaling, F3 suppresses transcriptional activation of IFN-α/β and ISGs, further undermining host antiviral immunity [71]. Recent comparative studies have shown that MPXV F3 exhibits greater potency in dampening these innate responses than VACV E3 mutants, underscoring its evolutionary refinement as a key immune modulator [67].
Once MPXV enters the human body, it replicates initially at the entry site, typically leading to local infection [13]. This is commonly manifested as the characteristic skin lesions, an important clinical indicator of the virus. Early systemic symptoms often include fever, headache, malaise, and lymphadenopathy, which reflect the virus’s systemic spread [72]. The virus then disseminates via the lymphatic system to regional lymph nodes, where it further amplifies [12]. It subsequently enters the bloodstream, leading to viremia, which allows the virus to spread to various organs, including the skin, lungs, liver, spleen, and kidneys [73]. The presence of systemic involvement and widespread rash is a key hallmark of MPXV infection. In certain cases, MPXV can invade the CNS, leading to severe neurological complications such as encephalitis [74]. This may result from direct viral infection of the brain and spinal cord tissues, although such instances are relatively rare. Additionally, MPXV can target immune cells, particularly lymphoid tissues, and modulate immune responses, facilitating viral persistence and replication [11]. This immune evasion is critical for the virus as it allows the establishment of an infection that can evade the host’s immune defenses for extended periods. One notable aspect of MPXV infection is its potential for viral persistence. After the acute phase of infection, the virus can remain in certain tissues for extended periods, possibly leading to latent or subclinical infections. This persistence may result in individuals continuing to shed the virus even after clinical symptoms have subsided, which poses a significant risk for ongoing transmission [75]. These asymptomatic or subclinical carriers may harbor the virus in tissues such as lymphoid tissues, skin, and possibly even the genital tract, maintaining a reservoir of infection in the population [11].
MPXV employs a variety of immune evasion mechanisms to facilitate persistence and systemic spread. These mechanisms include the inhibition of IFN production, modulation of T cell function, suppression of apoptosis, and other strategies to dampen antiviral immune responses [44]. This allows MPXV to circumvent the host’s immune surveillance, replicate efficiently, and potentially lead to chronic or persistent infection, prolonging transmission risk. The hallmark clinical feature of MPXV infection is the development of skin lesions, which evolve from macules to papules, vesicles, pustules, and eventually scabs [76]. These lesions typically appear all over the body, particularly on the face, palms, soles of the feet, and genital areas. The progression of these lesions provides essential diagnostic information for identifying the virus. While most cases of MPXV infection resolve with supportive care, more severe cases can occur, particularly in immunocompromised individuals, leading to complications such as secondary bacterial infections or systemic organ failure. In rare instances, these severe infections can be fatal. The risk of severe disease highlights the importance of early diagnosis, clinical management, and preventive measures, especially in at-risk populations.
Marine environments, among the most virus-saturated ecosystems on Earth, harbor viral loads exceeding 107 particles per milliliter of seawater. This intense and persistent viral pressure has driven marine organisms, especially those lacking adaptive immunity, such as sponges and corals, to evolve diverse chemical defenses [77]. Marine phytoconstituents, including mycosporine-like amino acids (MAAs), shinorine, porphyra-334, omega-3 fatty acids, carrageenan, fucans, and squalene, offer potential therapeutic benefits for mpox (Table 2). These compounds modulate immune responses [78], reduce oxidative stress [79], and inhibit viral entry [80], suggesting their role in enhancing immune function and mitigating viral replication, thereby supporting mpox treatment. These bioactive secondary metabolites often target viral entry, replication, or propagation, forming an ecological arsenal against infection. Dense microbial populations and fierce resource competition further select for antiviral compounds as tools for survival and ecological advantage [81]. Additionally, environmental stressors like UV radiation and salinity shifts have favored the evolution of multifunctional molecules with antiviral, antioxidant, and photoprotective roles.
Proposed mechanisms and potential clinical benefits of marine-derived phytoconstituents in mpox management
| Marine-derived phytoconstituent | Experimental models | Potential mechanism | Potential clinical outcomes | References |
|---|---|---|---|---|
| MAAs | In vitro [cultured cyanobacteria, algae, fungi; HPLC/LC-MS; DPPH, ABTS assays; model protein (hen egg white lysozyme) glycation inhibition] | MAAs scavenge ROS, may activate the Nrf2 pathway, inhibit COX-2, iNOS, and NF-κB, modulate cytokine responses, and activate FAK/MAPK signaling—enhancing antioxidant defenses, reducing inflammation, balancing immunity, and promoting keratinocyte migration, proliferation, and wound healing | Reduces oxidative damage, protects tissues, and may limit viral spread. Decreases inflammation and pain, aiding recovery. Balances the immune response, lowering cytokine storm. Speeds healing, improves skin repair, and may prevent scarring and secondary infections | [82, 83] |
| Shinorine | In vitro (human THP-1 and THP-1-blue cell lines; LPS treatment of cell cultures; reporter assay in THP-1-blue cells; Kyn/Trp quantification) | Activates NF-κB in resting cells but suppresses it during inflammation. Reduces IDO-1 activity (↓ Kyn/Trp). Scavenges ROS | Balances immune response to reduce inflammation and tissue damage. May enhance T cell activity and viral clearance by lowering immune tolerance. Protects against oxidative stress, aiding tissue preservation and recovery in mpox | [84] |
| Porphyra-334 (P-334) | In vitro (human skin fibroblasts, DCF-DA fluorescence assay; HaCaT cells; ABTS, DPPH assay) | Suppresses T cell activation, NF-κB, and IFN signaling. Inhibits IFN-induced JAK-STAT signaling, potentially promoting viral persistence. Enhances cell-associated viremia through monocytes/macrophages, aiding viral spread and immune evasion | Antioxidant and immunomodulatory effects may restore immune activation, counteracting viral suppression. Reduces oxidative stress, supports JAK-STAT activation, enhances ISG expression, and promotes innate immune clearance while reducing inflammation and supporting adaptive immunity | [84, 85] |
| Omega-3 fatty acids (EPA & DHA) | In vitro (human THP-1 monocyte/macrophage cell line; HEK293T-ACE2; Vero E6-ACE2); in vivo (rodent model, ARDS models) | Alter membrane properties and enhance immune receptor signaling. Generate resolvins, protectins, and maresins to reduce inflammation. Suppress NLRP3 inflammasome, enhance IFN signaling, promote M2 macrophages, regulate T/B cell activation, and reduce oxidative stress and inflammation | Improve immune recognition and reduce viral immune evasion. Resolve excessive inflammation and balance antiviral defense with tissue protection. Strengthen antiviral responses, inhibit replication, enhance clearance, limit spread, and improve systemic control while preserving barriers and reducing dissemination | [86] |
| Carrageenan | In vitro (NCM460 cell line, human colonic mucosa, HCT-8 human colonic adenocarcinoma cells); in vivo (guinea pig, TLR5–/– mouse, IL-10–/– mouse, B16-F10 melanoma model, 4T1 mammary carcinoma model) | Macrophage toxicity suppresses T cell-dependent immunity, while TLR4-mediated NF-κB activation induces inflammation. λ-Carrageenan can promote immune cell infiltration (M1 macrophages, dendritic cells, T cells) and cytokine production, potentially enhancing antiviral immunity and limiting systemic spread, depending on its type and context | Carrageenan may impair antiviral macrophage function and dysregulate IFN responses, aiding immune evasion. Its adjuvant effects could enhance antiviral signaling, potentially improving vaccine efficacy and systemic immunity while balancing immune suppression and response | [87–89] |
| Squalene | In vivo (mouse C57BL/6, BALB/c, KO; ferret; NHP-macaques, human Covid-19 patient); ex vivo (lymph node) | Squalene promotes M2 anti-inflammatory responses (↑ IL-10, IL-4, IL-13), inhibits NF-κB-driven pro-inflammatory mediators, activates Keap1-Nrf2-ARE antioxidant pathways, enhances tissue-repair factors (TIMP-2, GM-CSF), and preserves mitochondrial and ER function, supporting immune balance and recovery in mpox | By balancing immune activation and preventing excessive inflammation, squalene may reduce immune evasion by the virus, support effective JAK-STAT antiviral signaling, and limit systemic viral dissemination | [90, 91] |
| Fucans | In vivo (rodents, zebrafish embryo); in vitro (RAW 264.7 murine macrophages, HaCaT human keratinocytes, Caco-2/RAW 264.7 co-culture) | TLR4 activation induces ROS, NO, and cytokines, inhibiting immune cell migration. It reduces IL-6, TNF-α, and IL-1β levels, modulates redox balance, enhances phagocytosis, increases antioxidant enzymes, and reduces oxidative stress and tissue damage | It reduces inflammation, enhances immune clearance, and protects against oxidative stress, helping resolve lesions, limit viral spread, and prevent severe complications | [92, 93] |
MAAs: mycosporine-like amino acids; ROS: reactive oxygen species; Nrf2: nuclear factor erythroid 2-related factor 2; COX-2: cyclooxygenase-2; iNOS: inducible nitric oxide synthase; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; FAK: focal adhesion kinase; MAPK: mitogen-activated protein kinase; IDO-1: indoleamine 2,3-dioxygenase 1; IFN: interferon; JAK: Janus kinase; STAT: signal transducers and activators of transcription; ISG: IFN-stimulated gene; EPA: eicosapentaenoic acid; DHA: docosahexaenoic acid; NLRP3: NOD-like receptor family, pyrin domain containing 3; TLR5: Toll-like receptor 5; IL-10: interleukin-10; Keap1: Kelch-like ECH-associated protein 1; ARE: antioxidant response element; TIMP-2: tissue inhibitor of metalloproteinases-2; GM-CSF: granulocyte-macrophage colony-stimulating factor; ER: endoplasmic reticulum; TNF-α: tumor necrosis factor alpha; Kyn: kynurenine; Trp: tryptophan; HPLC: high performance liquid chromatography; LC-MS: liquid chromatography-mass spectrometry; DPPH: 2,2-diphenyl-1-picrylhydrazyl; ABTS: 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid); THP-1: human acute monocytic leukemia cell line; LPS: lipopolysaccharide; DCF-DA: 2',7'-dichlorofluorescein diacetate; HaCaT: human adult keratinocyte cell line; ACE2: angiotensin-converting enzyme 2; ARDS: acute respiratory distress syndrome; HCT-8: human colorectal adenocarcinoma cell line; NHP: non-human primates
Marine-derived compounds such as griffithsin, carrageenan, and fucoidans have demonstrated potent antiviral activity against MPXV and related orthopoxviruses through distinct molecular mechanisms. Griffithsin, a red algal lectin, binds with high affinity to high-mannose glycans on the envelope glycoproteins of various enveloped viruses, suggesting it could similarly block MPXV attachment and fusion [94]. While direct evidence for MPXV A33 and B5 is pending, its broad-spectrum mechanism makes it a compelling candidate for further testing. This glycan-specific interaction sterically inhibits viral entry without cytotoxic effects, making griffithsin a promising entry inhibitor. In parallel, carrageenan and fucoidans, sulfated polysaccharides from red and brown algae, act as polyanionic inhibitors by forming electrostatic interactions with positively charged viral surface proteins [95]. These interactions mimic host glycosaminoglycans and prevent viral adsorption to cell surface receptors. Beyond direct antiviral effects, these polysaccharides modulate host immune responses by targeting the TLR4 signaling pathway. By attenuating TLR4-mediated myeloid differentiation primary response 88 (MyD88)/NF-κB activation, they suppress the overproduction of pro-inflammatory cytokines such as IL-6 and TNF-α, thus reducing the cytokine storm associated with severe MPXV infection [96]. Additionally, fucoidans enhance type I IFN responses, contributing to host antiviral defense [97]. Together, these marine compounds offer a multifaceted antiviral strategy, combining viral entry blockade with immunomodulation, highlighting their therapeutic potential against MPXV and other emerging zoonotic viruses.
Marine organisms have adapted to virus-rich environments by evolving a diverse arsenal of antiviral compounds, including peptides, terpenoids, and alkaloids. These molecules often serve multifunctional roles, such as UV protection and cellular resilience, alongside antiviral defense [98]. Their structural complexity, ecological specificity, and broad-spectrum bioactivity position them as promising leads in antiviral drug discovery. Advances in marine biotechnology and genomics now allow targeted exploration and biosynthetic manipulation of these compounds. The unique chemical diversity found in marine ecosystems represents a frontier for developing novel antivirals to address emerging viral threats and overcome limitations of existing therapeutic agents. Importantly, many of these compounds originate from symbiotic microorganisms associated with marine hosts, adding complexity and diversity to marine chemical ecology [99]. Collectively, these evolutionary pressures have fostered the development of structurally unique and potent antiviral agents, positioning marine ecosystems as a rich and underexplored resource for novel therapeutic discovery. Marine-derived compounds exhibit multifunctional bioactivities that make them highly promising in antiviral drug development. Beyond directly inhibiting viral replication, agents like fucoidans, carrageenan, griffithsin, and MAAs possess potent antioxidant, anti-inflammatory, and UV-protective properties. These functions collectively target key aspects of viral pathogenesis, including oxidative stress, cytokine storms, and barrier dysfunction. Recent findings highlight their efficacy against viruses such as influenza, HIV, and SARS-CoV-2, and support their therapeutic potential in both systemic and topical applications.
Marine natural products (MNPs) hold immense potential for antiviral drug discovery, yet their therapeutic development faces significant hurdles. A major limitation is supply and scalability; many bioactive compounds occur in minute quantities [100]; and wild harvesting (e.g., Bugula neritina for bryostatins) is ecologically unsustainable [101]. Cultivation techniques and metagenomic approaches are being developed to alleviate this bottleneck [102]. The structural complexity of marine compounds often complicates chemical synthesis. While total synthesis has succeeded for a few agents (e.g., eribulin), it remains labor-intensive. Advances in artificial intelligence (AI)-assisted retrosynthesis and biosynthetic pathway engineering are helping streamline this process [103]. Reproducibility and standardization remain difficult due to environmental influences on metabolite profiles. Integrated omics approaches and chemometric tools are enhancing quality control, though regulatory acceptance still requires consistent reproducibility.
To address the challenges associated with the development of MNPs, several advanced biotechnological strategies have emerged. Synthetic biology now enables the heterologous expression of marine biosynthetic gene clusters in engineered microbial systems such as Escherichia coli, Streptomyces, and microalgae. This approach offers scalable, reproducible, and environmentally sustainable production of complex marine metabolites, mitigating the limitations of wild harvesting and low natural yields [104]. Concurrently, aquaculture and mariculture provide viable, controlled cultivation methods for bioresource-rich marine organisms, especially those producing high-value polysaccharides such as carrageenan and fucoidans. These methods support the continuous and sustainable supply of bioactive compounds without compromising marine biodiversity. For instance, modified carrageenan derivatives, such as iota-carrageenan nasal sprays, have shown safety and efficacy in human trials against respiratory viruses, including SARS-CoV-2, supporting their potential for mucosal delivery platforms [105]. Likewise, fucoidan nanoparticles have demonstrated improved oral bioavailability and antiviral effects in murine models of influenza [106]. Derivatization of native molecules, such as fucoidans, has led to improved antiviral potency, reduced cytotoxicity, and enhanced bioavailability, thereby increasing their therapeutic potential [107]. Furthermore, the integration of high-throughput screening technologies, powered by robotics, microfluidics, and AI, has significantly accelerated the identification of lead compounds [108]. These platforms enable rapid screening of complex marine extract libraries against a range of viral targets, enhancing hit discovery and optimization processes. Collectively, these interdisciplinary advances spanning synthetic biology, sustainable cultivation, medicinal chemistry, and automated screening are overcoming traditional barriers in marine drug development. They are transforming MNPs from ecologically derived chemical entities into viable candidates for clinical antiviral therapeutics, bridging the gap between marine chemical ecology and pharmaceutical innovation.
The global resurgence of mpox underscores the critical need for effective and accessible therapeutic interventions. Current treatment options remain limited, prompting growing interest in alternative strategies, including marine-derived phytoconstituents. These compounds, such as MAAs, omega-3 fatty acids, and carrageenan, have demonstrated notable antiviral, anti-inflammatory, and immunomodulatory properties. Emerging evidence suggests their potential to modulate host immune responses and inhibit key viral entry and replication stages. Despite these promising findings, further research is required to elucidate their specific mechanisms of action against the MPXV, evaluate their efficacy in preclinical and clinical models, and address scalability, formulation, and bioavailability challenges. Advancing our understanding of these marine-derived agents may contribute significantly to developing novel therapeutics for mpox and improving preparedness for future viral outbreaks.
AI: artificial intelligence
APOBEC3: apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3
BCL-2: B cell lymphoma 2
CCL3: chemokine C-C motif ligand 3
CD38: cluster of differentiation 38
CFR: case fatality rate
cGAMP: cyclic GMP-AMP
cGAS: cyclic GMP-AMP synthase
CXCL10: C-X-C motif chemokine ligand 10
DRC: Democratic Republic of the Congo
dsRNA: double-stranded RNA
EEV: extracellular enveloped virus
eIF2α: α subunit of eukaryotic initiation factor 2
IFN: interferon
IL-6: interleukin-6
IMV: intracellular mature virus
IRF3: interferon regulatory factor 3
ISGs: interferon-stimulated genes
JAK: Janus kinase
MAAs: mycosporine-like amino acids
MAC: membrane attack complex
MDA5: melanoma differentiation-associated protein 5
MHC: major histocompatibility complex
MNPs: marine natural products
MPXV: monkeypox virus
MSM: men who have sex with men
MV: mature virion
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
PKR: protein kinase R
PLHIV: people living with HIV
PRRs: pattern recognition receptors
RIG-I: retinoic acid-inducible gene I
SOCS: suppressors of cytokine signaling
STAT: signal transducers and activators of transcription
STING: stimulator of interferon genes
STIs: sexually transmitted infections
TBK1: TANK-binding kinase 1
TLRs: Toll-like receptors
TNF-α: tumor necrosis factor alpha
VACV: vaccinia virus
SP: Conceptualization, Investigation, Writing—original draft. SS: Writing—review & editing. BB: Writing—review & editing. KB: Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.