 Review
Review

Affiliation:
Laboratory of Inflammation Research, School of Life Science, Handong Global University, Pohang 37554, Republic of Korea
ORCID: https://orcid.org/0009-0000-1158-5647
Affiliation:
Laboratory of Inflammation Research, School of Life Science, Handong Global University, Pohang 37554, Republic of Korea
Email: jbaek@handong.edu
ORCID: https://orcid.org/0000-0003-4439-5545
Explor Immunol. 2025;5:1003211 DOI: https://doi.org/10.37349/ei.2025.1003211
Received: January 14, 2025 Accepted: July 23, 2025 Published: August 18, 2025
Academic Editor: Esma Isenovic, University of Belgrade, Serbia
Cardiac fibrosis, characterized by excessive extracellular matrix (ECM) deposition, plays a central role in the progression of heart diseases such as myocardial infarction, heart failure, and hypertensive cardiomyopathy. The dynamic interplay between fibroblasts and macrophages is pivotal in regulating ECM remodeling and the fibrotic response. Fibroblasts, as primary ECM producers, undergo phenotypic changes during pathological conditions, transitioning into myofibroblasts that exacerbate fibrosis. Macrophages, both resident and non-resident, contribute to cardiac fibrosis by influencing fibroblast activation through cytokine secretion and direct cell interactions. Emerging evidence from preclinical studies highlights the transformation of macrophages into myofibroblast-like cells, known as macrophage-to-myofibroblast transformation (MMT), a key mechanism linking chronic inflammation to fibrosis. During MMT, macrophages acquire characteristics like myofibroblasts. This process is driven by signaling pathways such as TGF-β/Smad3, ALKBH5, and mineralocorticoid receptor (MR)/connective tissue growth factor (CTGF) pathways. Recent single-cell transcriptomics and lineage-tracing studies have provided deeper insights into the molecular regulation of MMT and its contribution to myocardial remodeling. Additionally, the balance between resident cardiac macrophages and monocyte-derived macrophages plays a crucial role in determining the fibrotic outcome following cardiac injury. This review discusses the cellular composition of the heart, the interactions between macrophages and fibroblasts, and the mechanisms driving MMT. By synthesizing these insights, we aim to evaluate MMT as a therapeutic target for mitigating cardiac fibrosis and improving clinical outcomes in cardiovascular diseases.
Fibrosis is characterized by the excessive deposition of extracellular matrix (ECM) components, such as collagen and fibronectin. This process leads to tissue stiffness, organ dysfunction, and eventually failure [1, 2]. Fibrosis contributes to nearly 45% of deaths in industrialized countries. It is not a disease itself, but rather a dysregulated tissue repair response that becomes chronic due to repetitive injury or inflammation [2].
Cardiac fibrosis is a hallmark of cardiac diseases such as myocardial infarction (MI), hypertensive heart disease, and chronic kidney disease-induced cardiorenal syndrome. Myocardial remodeling due to fibrosis results in reduced compliance, impaired systolic and diastolic function, and arrhythmias [3, 4]. The initial fibrotic response plays a reparative role. It helps maintain structural integrity and prevent ventricular wall rupture. However, the exaggerated fibrotic response can lead to ECM deposition and reactive fibrosis [5]. This maladaptive response leads to ventricular stiffening, impaired cardiac function, and the progression to heart failure (HF) [6].
The central role of fibroblasts and macrophages in cardiac fibrosis is increasingly recognized. Fibroblasts serve as the primary producers of ECM. Macrophages support their activation by secreting cytokines and growth factors, such as TGF-β [7–10]. RCMs are a critical component of the heart’s immune microenvironment. They make up approximately 5–10% of the non-myocyte population in the murine myocardium during steady-state conditions [11, 12]. These macrophages originate mainly from embryonic progenitors, such as yolk sac and fetal liver-derived cells. They maintain their population through local proliferation rather than monocyte recruitment [11, 12]. Both resident and monocyte-derived macrophages interact with fibroblasts directly or via paracrine mechanisms, facilitating the fibrotic process [7, 9]. These interactions highlight the importance of studying both macrophage subpopulations and their roles in fibrosis progression.
One emerging concept in cardiac fibrosis is macrophage-to-myofibroblast transformation (MMT). Recent studies have demonstrated that macrophages can transdifferentiate into myofibroblast-like cells [1, 13–15]. MMT is a crucial mechanism connecting chronic inflammation to tissue fibrosis. Inflammation is widely recognized as a key driver of fibrotic progression. Human studies and experimental models have consistently demonstrated the pivotal role of MMT in advancing fibrotic conditions [16–20]. Myofibroblasts represent the primary cell type responsible for producing collagen during the development of tissue fibrosis [21]. CD206+ M2 macrophages can be differentiated into myofibroblasts [22, 23]. MMT is characterized by the co-expression of F4/80 (CD68) and α-SMA. The trans-differentiation process is mediated by TGF-β/Smad3 signaling [21, 24].
In this review, we summarize the cellular dynamics of cardiac fibrosis. We focus on the interactions between macrophages and fibroblasts and their roles in ECM remodeling. Through current insights into macrophage plasticity and fibroblast activation, we aim to elucidate potential therapeutic strategies for mitigating cardiac fibrosis and improving cardiac function.
Changes in the number and function of various cardiac cells significantly impact myocardial structure and function. These cells include cardiomyocytes, fibroblasts, endothelial cells, and macrophages. Such cells contribute to the progression of HF [25]. Cardiomyocytes dominate the myocardial mass, providing contractile force, but their function relies on structural and immune support from other cell types. Fibroblasts regulate and remodel the ECM, and endothelial cells maintain vascular function [26]. Macrophages, another key component, are involved in immune modulation and tissue repair [27].
Single-cell and single-nucleus RNA sequencing (scRNA-seq and snRNA-seq) allow the identification of specific cell populations, including rare and disease-specific cell states, by analyzing transcriptional signatures [28]. Prior to the advent of these tools, histology and flow cytometry were commonly used but lacked the capacity to resolve cellular heterogeneity at the single-cell level [29]. In combination with spatial transcriptomics and multiplex imaging, they provide detailed insights into cellular interactions and their spatial organization, offering a comprehensive understanding of cardiac dynamics in both health and disease.
In HF, fibroblasts undergo significant phenotypic changes, transitioning into myofibroblasts with enhanced ECM production. This shift contributes to excessive fibrosis, disrupting cardiac structure and impairing ventricular compliance. scRNA-seq has revealed diverse fibroblast subpopulations and disease-specific phenotypes. This finding emphasizes the importance of targeting pathways, such as TGF-β and Wnt signaling, to mitigate fibrosis progression [30]. In hearts affected by pathogenic variants, fibroblasts shift toward a secretory phenotype characterized by increased release of inflammatory mediators and growth factors. This phenotype, despite limited proliferative capacity, perpetuates ECM remodeling and chronic inflammation, contributing to cardiomyopathy [31].
Under physiological conditions, fibroblasts maintain ECM homeostasis and structural integrity, comprising a substantial proportion of non-myocyte cells in the heart. However, pathological stimuli such as myocardial injury activate fibroblasts, leading to excessive ECM production and fibrosis. Chronic activation has been closely linked to ventricular dysfunction and disease progression. Fibroblast subpopulations not only regulate ECM dynamics but also interact with other cardiac cells, underscoring their role as key mediators in the cardiac cellular network [32, 33]. In the non-failing human heart, fibroblasts exhibit a wide range of transcriptomic profiles, with certain subtypes specializing in ECM regulation and fibrotic processes. Beyond maintaining structural stability, these fibroblasts respond actively to mechanical and pathological stress, further highlighting their importance as therapeutic targets for cardiac fibrosis [34].
Cardiac macrophages are essential for the immune response and tissue repair following MI. Initially, they mediate inflammation by recruiting neutrophils and clearing necrotic tissue through ECM degradation [35]. As healing progresses, macrophages shift toward anti-inflammatory and reparative phenotypes, promoting fibroblast activation and ECM remodeling. However, prolonged or excessive activity can lead to maladaptive ventricular remodeling and fibrosis, contributing to HF. The polarization of macrophages into pro-inflammatory (M1) or anti-inflammatory (M2) phenotypes is dynamically regulated by epigenetic mechanisms. These mechanisms include DNA methylation and histone modifications [36]. In cardiovascular diseases, this polarization is critical. Ly6Chi macrophages (M1) intensify inflammation and tissue damage, while Ly6Clow macrophages (M2) facilitate repair and fibrosis by secreting cytokines like TGF-β1. Dysregulated polarization often exacerbates cardiac fibrosis [37].
Molecular regulators such as YAP, TAZ, and Lgr4 further influence macrophage behavior. YAP and TAZ deficiencies enhance reparative macrophage polarization, improving infarct healing and reducing fibrosis [38]. Similarly, the absence of Lgr4 in macrophages has been shown to increase reparative populations, decrease inflammation, and improve healing, highlighting its potential as a therapeutic target [39]. Additionally, macrophages contribute to cardiac repair by producing VEGF-C during efferocytosis, promoting lymphangiogenesis and reducing inflammation. Deficiency in VEGF-C production disrupts healing and exacerbates fibrosis [40].
In pathological conditions, macrophages exhibit context-dependent roles. For instance, in COVID-19-associated cardiac injury, macrophages release cytokines like IL-6 and TNF-α, exacerbating cardiomyocyte apoptosis and fibrosis [41]. In immune checkpoint inhibitor-induced myocarditis, macrophages interact with T-cells to amplify inflammatory responses, leading to myocardial damage and remodeling [42]. These findings indicate the dual role of macrophages in both repair and pathological processes, highlighting their significance in cardiac health and disease.
RCMs are a specialized population of immune cells derived from embryonic progenitors. They are uniquely adapted to maintain cardiac homeostasis and facilitate tissue repair under both normal and pathological conditions [12, 43]. These macrophages self-renew locally within the myocardium and exhibit anti-inflammatory properties through the expression of CCR2– markers, enabling them to suppress excessive inflammation and promote vascular remodeling following cardiac injury [44, 45]. Notably, RCMs play an active role in preserving cardiac structure and function by phagocytosing apoptotic cardiomyocytes, clearing cellular debris, and releasing cytokines that modulate fibroblast activation and ECM remodeling [43, 46, 47].
Under stress conditions such as hypertension or MI, RCMs undergo phenotypic changes, upregulating reparative programs that include the secretion of insulin-like growth factor-1 (IGF-1), a critical mediator for adaptive cardiac growth [45]. This function of RCMs is essential to counteract the fibrotic response, as it prevents excessive ECM deposition and maintains myocardial compliance [12, 45]. However, in conditions where RCM populations are depleted or impaired, their protective effects diminish, leading to exacerbated fibrosis and functional decline [46].
Non-resident macrophages (NRMs) are monocyte-derived and are actively recruited into the heart during injury or inflammation, where they adopt a predominantly pro-inflammatory phenotype characterized by CCR2+ markers [48, 49]. These macrophages amplify the inflammatory response by releasing high levels of inflammatory cytokines, including TNF-α, IL-6, IL-1β, and MCP-1, and pro-fibrotic factors, such as TGF-β, PDGF, and FGF, which stimulate fibroblast activation and drive the transition to pathological fibrosis [50, 51]. In acute MI, NRMs infiltrate the infarcted myocardium in large numbers, replacing a portion of the RCM population, and dominate the inflammatory phase by promoting ECM deposition and exacerbating cardiac remodeling [44]. Additionally, macrophages contribute to cardiac fibrosis through the actions of angiotensin II (Ang II) and aldosterone, which promote their polarization toward a fibrogenic phenotype [52].
The interaction between RCMs and NRMs is critical in shaping the progression and resolution of cardiac injury. RCMs are involved in orchestrating the transition from inflammation to repair by modulating NRM activity, thereby facilitating a controlled reparative response [43, 46]. However, when the recruitment of NRMs is excessive, their pro-inflammatory activity can overwhelm the reparative capacity of RCMs, resulting in maladaptive remodeling characterized by stiffened myocardium and impaired cardiac function [27, 44].
Emerging technologies, including single-cell transcriptomics and advanced imaging techniques, have provided deeper insights into the distinct roles and gene expression profiles of RCMs and NRMs [12]. These studies reveal that RCMs exhibit transcriptional programs geared toward cardiac repair and homeostasis, while NRMs display a more inflammatory signature associated with fibrosis and tissue damage [53, 54]. Understanding the precise mechanisms governing the behavior of these macrophage populations opens new avenues for targeted therapies aimed at reducing fibrosis and enhancing cardiac repair (Figure 1).
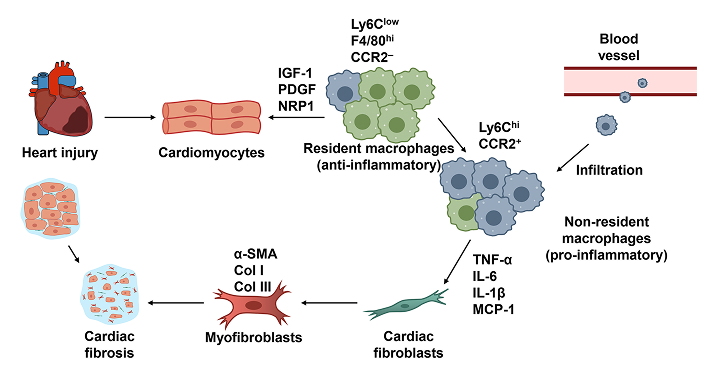
The mechanism of resident macrophages and non-resident macrophages in cardiac fibrosis. Resident cardiac macrophages with a Ly6Clow F4/80hi CCR2– phenotype support cardiac function and attenuate excessive fibroblast activation by secreting growth factors that promote tissue repair. In contrast, infiltrating Ly6Chi CCR2+ non-resident macrophages produce higher levels of inflammatory cytokines and pro-fibrotic mediators, such as TNF-α, IL-6, IL-1β, and MCP-1. These factors stimulate the differentiation of fibroblasts into myofibroblasts, which are characterized by elevated expression of α-SMA, Col I, and Col III
Macrophages and fibroblasts are central to the processes of cardiac repair and fibrosis. They engage in dynamic interactions that modulate each other’s behavior. These interactions are influenced by various signaling pathways, cytokines, and growth factors, which can either promote or suppress fibrosis depending on the context. Macrophages recruit fibroblasts by releasing growth factors and cytokines [55]. In addition, macrophages express coagulation factor XIIIa and provide extracellular fibrin matrix to fibroblasts [56, 57]. Myofibroblasts, derived from fibroblast differentiation under the influence of cytokines such as TGF-β, are key players in ECM remodeling and scar formation during cardiac repair [58, 59]. TGF-β signaling also promotes the secretion of matrix proteins [60, 61]. Persistent activation of myofibroblasts, along with their interactions with macrophages, leads to excessive ECM deposition, contributing to pathological fibrosis [58, 62]. Myofibroblasts secrete factors such as IL-6 and COX-2-derived prostaglandins (PGE2 and PGD2), which enhance the anti-inflammatory phenotype of macrophages. In turn, alternatively activated macrophages downregulate myofibroblast activity, including cell migration and cytokine output, creating a feedback loop that can limit fibrosis [63]. After MI, macrophages infiltrate the infarcted myocardium and release cytokines and growth factors that recruit and activate fibroblasts. This recruitment is crucial for ECM deposition and scar formation but can also result in adverse remodeling if not properly regulated [64, 65]. Macrophages secrete MCP-1 and TGF-β, which are essential for initiating fibroblast activation but also drive fibrosis when overexpressed [60, 66].
Fibroblasts and macrophages reciprocally regulate each other through the colony-stimulating factor 1 (CSF-1) and its receptor (CSF-1R). Fibroblasts secrete CSF-1 to sustain macrophage populations, while macrophages, in turn, influence fibroblast survival and activity through the secretion of cytokines, such as PDGFs [62]. This signaling axis is critical in fibrotic conditions, as CSF-1R blockade has been shown to attenuate fibrosis by limiting macrophage recruitment and reducing fibroblast activation [67, 68]. Matrix metalloproteinases (MMPs), secreted by both macrophages and fibroblasts, play a pivotal role in ECM degradation and remodeling. Dysregulation in MMP activity contributes to persistent ECM deposition, reinforcing tissue stiffness and fibrotic progression [62, 69]. This feedback loop highlights the need for strategies targeting MMP regulation to mitigate fibrotic remodeling. Studies on macrophage-derived factors, such as low-density lipoprotein receptor-related protein 1 (LRP1), have revealed their role in rejuvenating fibroblast functions. Young macrophages secrete factors that suppress fibrotic signaling, while aged macrophages show impaired ability to regulate fibroblasts, leading to exacerbated fibrosis [70].
Cardiac injury triggers the release of damage-associated molecular patterns (DAMPs) from necrotic myocardial cells, which act through toll-like receptor 4 (TLR4) to activate fibroblasts. This activation increases fibroblast proliferation, motility, and expression of ECM components such as collagen. DAMPs, including HMGB1 and S100 proteins, are pivotal in this process, as demonstrated by their ability to induce myocardial inflammation and fibrosis when injected into the heart [71]. In inflammatory dilated cardiomyopathy (DCM), fibroblasts respond to IL-17A by producing granulocyte/macrophage colony-stimulating factor (GM-CSF). This cytokine attracts pro-inflammatory macrophages, which further exacerbate cardiac fibrosis through a feed-forward loop. The IL-17A-GM-CSF axis demonstrates how cytokine-mediated interactions between fibroblasts and macrophages contribute to the progression of cardiac inflammation and fibrosis [72, 73]. Macrophage migration inhibitory factor (MIF), secreted by macrophages, exhibits antifibrotic effects by inhibiting fibroblast proliferation and matrix deposition via CD74/AMPK signaling. While primarily studied in liver fibrosis, similar mechanisms are likely applicable to the heart, where MIF may regulate macrophage-fibroblast interactions to suppress excessive ECM production [74].
MMT is defined as the trans-differentiation of macrophages into myofibroblasts. This process is characterized by the co-expression of macrophage-specific markers, such as CD68, alongside myofibroblast markers like α-SMA. MMT contributes significantly to ECM accumulation and fibrosis progression [1, 75, 76]. Recent studies have identified MMT in a range of organ systems, indicating its involvement in numerous fibrotic conditions and suggesting it as a potential therapeutic target for mitigating the progression of fibrotic diseases [77]. The tissue-specific plasticity of macrophages underscores the significance of MMT. A study identifies 12,743 macrophage-specific enhancers and demonstrates that tissue-resident macrophages possess distinct enhancer landscapes shaped by tissue- and lineage-specific transcription factors. The findings highlight the critical role of the microenvironment in reprogramming chromatin landscapes. This reprogramming occurs even in differentiated macrophages, underscoring their plasticity and adaptability [78]. Bone marrow-derived macrophages (BMDMs) play a key role in the transition of macrophages to myofibroblasts, especially M2 macrophages [21, 79]. Pro-fibrotic alternative (M2) macrophages can produce TGF-β1, a cytokine whose effects can shift between promoting healing and inducing fibrosis [80]. BMDMs (M2) can undergo a proliferation-dependent phenotypic switch in inflamed tissues, losing their suppressive and reparative functions and contributing to fibrosis through increased expression of pro-inflammatory cytokines such as TNF-α and IL-6. Blocking M-CSF signaling prevents this switch [81]. This plasticity enables macrophages to transition into myofibroblasts (MMT).
Lineage-tracing experiments have provided critical evidence for MMT in fibrotic diseases. In a renal fibrosis model, LysM-Cre/Rosa26-tdTomato mice demonstrated that macrophages expressing F4/80 transitioned into α-SMA-positive myofibroblasts through the TGF-β/Smad3 signaling pathway, directly linking macrophages to the production of collagen I [22]. Computational modeling and experimental validation have further reinforced the role of MMT in fibrosis. In granuloma-associated fibrosis, macrophages were shown to transition into myofibroblasts following sequential inflammatory and anti-inflammatory signaling [82]. Similarly, experiments in kidney fibrosis models have identified macrophage-derived myofibroblasts as a significant source of collagen deposition. In preclinical studies, blocking MR activation reduced MMT, attenuating renal fibrosis [79, 83]. These results provide a compelling basis for hypothesizing that similar mechanisms could be applicable to cardiac tissue, suggesting potential therapeutic strategies for heart fibrosis. Moreover, extracellular vesicle-derived microRNAs, such as miR-21, were identified as mediators of macrophage plasticity, enhancing their capacity for transformation into myofibroblasts [84, 85]. Another study showed that the MMT process is regulated by transcription factors, such as Pou4f1, driving ECM deposition and tissue remodeling [86].
MMT significantly contributes to myocardial fibrosis (MF). In scRNA-seq and lineage-tracing experiments, macrophages derived from circulating monocytes were shown to preferentially undergo MMT. In a myocardial ischemia-reperfusion (MIR) injury model, macrophages producing high levels of TGF-β were identified as key contributors to fibrotic remodeling. Notably, S100a9hi macrophages, a subset identified through scRNA-seq, activated the Myd88/NFκB/NLRP3 and TGF-β/Smad3 signaling pathways. This dual activation facilitated both macrophage-to-myofibroblast transition and the transformation of cardiac fibroblasts into myofibroblasts [87]. A study [85] showed that this transition was driven by an increased ALKBH5 expression, which mediated m6A demethylation of IL-11 mRNA, enhancing its stability and protein expression. Mice with macrophage-specific deletion of ALKBH5 exhibited reduced MMT, attenuated cardiac fibrosis, and improved cardiac function. Notably, targeted siRNA delivery against ALKBH5 or IL-11 receptor α in macrophages effectively mitigated fibrosis in mice [85]. In cardiac fibrosis post-MI, macrophages were observed to acquire fibrotic markers such as fibroblast activation protein-α (FAP) and collagen type I during the reparative phase. The IKKε-p38 signaling axis plays a pivotal role in regulating macrophage responses to inflammation and tissue repair. In MI models using IKKε-deficient mice, reduced phosphorylation of p38 in macrophages lacking IKKε has been associated with impaired resolution of inflammation and an increased transition of MMT, which exacerbates fibrosis. This highlights the significance of p38 signaling in macrophage plasticity and its potential impact on fibrotic progression [88].
Integrated single-cell transcriptomics revealed that macrophages release profibrotic signals, including FN1, SPP1, and THBS1. In MI and DCM, macrophages expressing elevated levels of M1 polarization markers and myofibroblast markers directly facilitate MF progression. Transcription factors such as FOSB in DCM and MI and KLF6 in HF were identified as key regulators of this transformation. Moreover, the study highlights that macrophages undergoing MMT exhibit significant activation of toll-like receptors, ERK signaling, and the HIF-1 pathway, which are critical for promoting fibrosis in response to myocardial injury [89]. Treatment with eplerenone, an MR antagonist, was shown to inhibit MMT transformation by reducing the expression of intercellular adhesion molecule-1 (ICAM-1) and MCP-1, thereby limiting macrophage-driven inflammatory injury. The findings reveal the MR/SGK-1 pathway as a key regulator of MMT and underscore its role in the pathogenesis of unilateral ureteral obstruction (UUO)-induced cardiac fibrosis [90]. In another study using a rat model of UUO, significant cardiac fibrosis and MMT were observed, processes that were effectively suppressed by the MR antagonist, eplerenone. In vitro studies further demonstrated that aldosterone-activated MR promoted MMT and increased the secretion of connective tissue growth factor (CTGF) from macrophages. Blocking MR or CTGF inhibited these effects, suggesting that the MR/CTGF pathway plays a pivotal role in MMT-driven cardiac fibrosis [91, 92].
Erythropoietin receptor (EPOR) signaling has emerged as a significant regulator of MMT in fibrosis. Under TGF-β1 stimulation, EPOR activation in BMDMs enhances the expression of both F4/80 and α-SMA, indicating an increased MMT process [93]. In an MI model, macrophages undergoing MMT can express fibroblast markers, such as type I collagen (COL1A1), FSP1, and FAP, contributing to cardiac fibrosis (Figure 2). Depletion of macrophages reduces these collagen-producing cells, indicating that macrophages are key drivers of fibrotic remodeling in the heart [13, 94, 95]. Research on the MMT’s role in cardiac fibrosis remains limited. However, the emerging concept of MMT offers valuable insights into the mechanisms driving cardiac fibrosis. It also highlights MMT’s potential as a therapeutic target. Targeting MMT pathways could pave the way for novel strategies to treat cardiovascular diseases associated with cardiac fibrosis, providing hope for more effective interventions and improved clinical outcomes.
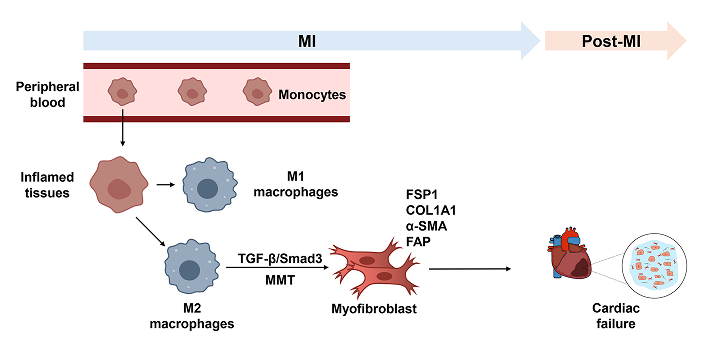
Diagram of MMT in myocardial infarction. Upon MI, circulating monocytes infiltrate the damaged site and primarily differentiate into an M2 pro-fibrotic phenotype rather than an M1 pro-inflammatory phenotype. The TGF-β/Smad3 pathway mediates MMT, leading these cells to express fibroblast-associated markers such as α-SMA, COL1A1, FSP1, and FAP. MI: myocardial infarction; MMT: macrophage-to-myofibroblast transformation
Cardiac fibrosis is a critical factor in the progression of MI, HF, and hypertensive cardiomyopathy. This pathological process is driven by excessive deposition of ECM components, such as collagen type I and type III. These components are primarily produced by activated fibroblasts and myofibroblasts. The interplay between fibroblasts and macrophages plays a pivotal role in this process. Macrophages regulate fibroblast activation through cytokines and growth factors, including TGF-β, PDGF, and IL-6.
A significant breakthrough in understanding fibrosis is the recognition of MMT. During MMT, macrophages express dual markers such as CD68, F4/80 (macrophage markers), and α-SMA or collagen type I (myofibroblast markers). This process is primarily regulated by the TGF-β/Smad3 signaling pathway and influenced by factors like CTGF and MR activation. Inhibition of pathways like MR/CTGF with agents such as eplerenone has shown potential in suppressing MMT and reducing fibrosis.
Other regulatory mechanisms of MMT include epigenetic modifications like ALKBH5-mediated m6A RNA demethylation, which impacts macrophage plasticity and differentiation. Furthermore, macrophage subsets such as CD206+ M2 macrophages are more prone to MMT, contributing significantly to collagen deposition and scar formation. Depleting macrophages or inhibiting specific pathways can reduce MMT-derived myofibroblasts, highlighting therapeutic potential.
Despite these insights, research on MMT in cardiac fibrosis remains incomplete. The specific contributions of RCMs versus NRMs during different stages of fibrosis need further clarification. Additionally, the environmental cues such as hypoxia, mechanical stress, and inflammatory signals that drive MMT require more detailed investigation. Understanding how these factors interact with signaling pathways like TGF-β/Smad3, MR/CTGF, and ALKBH5 could uncover new intervention points. However, potential effects on macrophage-mediated tissue repair and immune regulation should be considered.
This review underscores the importance of MMT in cardiac fibrosis and its potential as a therapeutic target. By identifying key markers and pathways involved in macrophage-fibroblast interactions, this work lays the foundation for developing targeted therapies. Future studies integrating advanced techniques like scRNA-seq, spatial transcriptomics, and lineage tracing will be crucial for mapping the precise roles of macrophage subsets in fibrosis. Translating these insights into clinical applications could lead to innovative treatments for fibrotic heart diseases. This progress offers new hope for patients with MI, HF, and related conditions.
In conclusion, targeting MMT and its regulatory pathways holds promise for mitigating cardiac fibrosis. Addressing current knowledge gaps and pursuing these research avenues may transform the treatment landscape for cardiovascular diseases associated with fibrosis.
BMDMs: bone marrow-derived macrophages
CSF-1: colony-stimulating factor 1
CSF-1R: colony-stimulating factor 1 receptor
CTGF: connective tissue growth factor
DAMPs: damage-associated molecular patterns
DCM: dilated cardiomyopathy
ECM: extracellular matrix
EPOR: erythropoietin receptor
FAP: fibroblast activation protein-α
GM-CSF: granulocyte/macrophage colony-stimulating factor
HF: heart failure
MF: myocardial fibrosis
MI: myocardial infarction
MIF: macrophage migration inhibitory factor
MMPs: matrix metalloproteinases
MMT: macrophage-to-myofibroblast transformation
MR: mineralocorticoid receptor
NRMs: non-resident macrophages
RCMs: resident cardiac macrophages
scRNA-seq: single-cell RNA sequencing
UUO: unilateral ureteral obstruction
DK: Investigation, Writing—original draft. JHB: Conceptualization, Validation, Writing—review & editing, Supervision. Both authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by the National Research Foundation of Korea (NRF) through the Ministry of Education [2021R1I1A3059820] (to Jea-Hyun Baek). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

