 Review
Review

Affiliation:
1School of Dentistry, International Medical University, Kuala Lumpur 57000, Malaysia
Email: spoorthiravi@imu.edu.my
ORCID: http://orcid.org/0000-0002-3902-6836
Affiliation:
2School of Postgraduate Studies, International Medical University, Kuala Lumpur 57000, Malaysia
ORCID: https://orcid.org/0000-0001-5659-8660
Affiliation:
3School of Health Sciences, International Medical University, Kuala Lumpur 57000, Malaysia
ORCID: https://orcid.org/0000-0001-7297-5948
Affiliation:
1School of Dentistry, International Medical University, Kuala Lumpur 57000, Malaysia
ORCID: https://orcid.org/0000-0003-0434-3492
Explor Immunol. 2024;4:129–151 DOI: https://doi.org/10.37349/ei.2024.00133
Received: September 14, 2023 Accepted: January 11, 2024 Published: February 29, 2024
Academic Editor: Yongjian Geng, the University of Texas, USA
Periodontitis is a ubiquitous chronic inflammatory worldwide disease. The multiplicity of gram-negative microbiomes and their endotoxins, such as lipopolysaccharides (LPS), play a crucial role in its pathogenesis. The detection and consequent effects of LPS occur either via membrane-based cluster of differentiation 14 (CD14)/myeloid differentiation factor 2 (MD2)/Toll-like receptor (TLR)-4 complex activation or through intracellular cytosolic LPS detection that further cascades its effects, resulting in a variety of cell death processes, including apoptosis, pyroptosis, necroptosis, NETosis, and their crosstalk. Irrespective of the detection of LPS, the cellular response is for protecting and resolving the inflammation. However, chronic and exaggerated responses in periodontitis result in the destruction of periodontal structures. This review summarizes the extracellular and cytosolic detection of LPS and its further consequences. Then, it sheds light on methods reported to mitigate the adverse effects of LPS.
Periodontitis is a chronic multifactorial inflammatory disease characterized by progressive destruction of the teeth-supporting apparatus, resulting in tooth mobility and eventual loss [1]. This process involves complex dynamic interactions among specific bacterial pathogens, destructive host immune responses, and environmental factors [2]. The subgingival biofilm of specific gram-negative, anaerobic microorganisms, termed “red complex” [Porphyromonas gingivalis (P. gingivalis), Tannerella forsythia, and Treponema denticola] [3, 4], and their toxins such as lipopolysaccharides (LPS) along with other virulence factors stimulate the host cells such as macrophages, monocytes, neutrophils, and other constituent cells such as fibroblasts, osteocytes, keratinocytes, and B cells [5]. This process leads to the production of a range of pro-inflammatory cytokines like interleukin (IL)-1β, IL-18, tumour necrosis factor (TNF)-α, prostaglandin E2 (PGE2), IL-6, and IL-8 further resulting in the production of matrix metalloproteinases (MMPs) by a variety of cells including fibroblasts, keratinocytes in the junctional epithelium, macrophages, and neutrophils [2, 6–9]. The resulting MMPs break down the collagen fibres in the periodontal ligament (Figure 1).
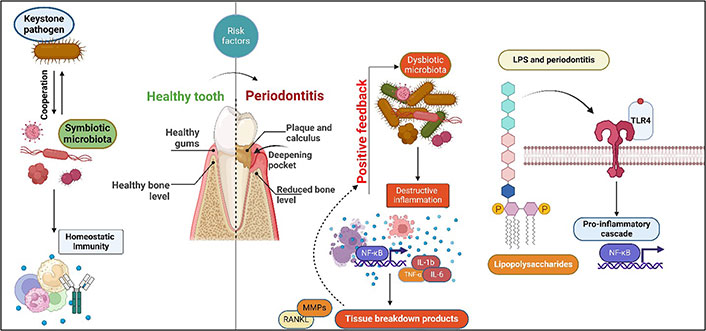
Schematic illustration of a link between microbial dysbiosis resulting in endotoxin overload and initiation and progression of periodontitis. NF-κB: nuclear factor-kappa B; RANKL: receptor activator of NF-κB ligand; TLR-4: Toll-like receptor-4. Created with BioRender.com
Additionally, the release of pro-inflammatory cytokines results in the expression of RANKL, a receptor activator of NF-κB (RANK) in the osteoblasts and T helper cells. The interaction of these cells with the RANK on osteoclast precursors leads to the formation and maturation of osteoclasts, which mediate the destruction of the alveolar bone [2, 10]. Such periodontitis and tissue destruction occur when there is an imbalance between the biofilm and the host immune response (Figure 1). The condition’s aetiology needs to be more comprehended, as do the agents responsible for the alternating periods of aggravation and remission. In this regard, LPS is considered a pivotal structure and a prototypical microbial toxin, triggering an immunological response from macrophages, which are crucial regulators of the host immune response. Repeated exposure has been shown to cause a state of inactivity called endotoxin tolerance, which is believed to be a protective mechanism. However, this condition may also provide protective advantages to prospective infections. P. gingivalis, a keystone periodontal pathogen, can alter the bio-reactive lipid A component of LPS, affecting the immune response [11].
LPS is a vital, inherent component of the outer membrane of gram-negative bacteria [12]. They are important for maintaining the structural integrity of bacteria and provide a construct that interacts with other surfaces [13]. The LPS molecule is a large, thermostable amphipathic glycoconjugate with a hydrophobic lipid domain linked to a central oligosaccharide and a distal polysaccharide. An LPS molecule consists of a hydrophobic component called lipid A, a hydrophilic distal polysaccharide part called O-antigen, and the core polysaccharide [14].
The lipid A component is the most bioactive among the three, and it varies from organism to organism [15], which imparts pathogenic, endotoxic characteristics. LPS generates a strong, pleiotropic pro-inflammatory stimulus upon interaction with the immune system, inducing an innate immune response. The immune system recognizes the lipid A component of LPS, which is released in soluble form from dividing cells, from lysed bacterial cells upon autolysis, complement activation, phagocytosis, or the action of antibiotics. The primary response is the recognition and binding of the lipid A component through the host cell’s TLRs [14]. Due to the compositional variability of lipid A, different bacterial strains mount varying degrees of immune response [16]. As a strategy, certain bacteria are reported to be able to modify their lipid A structure to adapt to the host environment. Even in bacterial lysis, the lipid A moiety or its fragments can lead to severe ill effects when released into the bloodstream. Zaric et al. [17] aimed to explore the clinical relevance of structural variations in salivary lipid A isoforms as potential biomarkers for chronic periodontitis. Salivary lipid A was isolated and analyzed using mass spectrometry. Their results showed a predominance of under-acylated and under-phosphorylated lipid A isoforms in chronic periodontitis patients. The LPS extracted from these patients exhibited lower inflammatory potential than healthy individuals. This observation suggests that key periodontal pathogens can evade the host immune system by producing low-potency LPS, allowing them to thrive in subgingival areas [17].
The O-antigen of the LPS is the most variable part that imparts specific antigenicity to bacteria that can lead to the production of antibodies. Variability in O-antigen’s chain length can inhibit complement-mediated bacterial control and killing [18]. The O-antigen also contributes to pathogen evasion of immune cell phagocytosis. The composition or its size indicates the virulence of a particular bacterial strain. The O-antigen can cause attachment and comes in direct contact with the external environment [14, 19]. This component can help bacterial colonization, develop drug resistance, and evade the host defense processes. A compromised immune system contributes to the endotoxic effects of LPS during the infection phase. If left unchecked, it might result in severe effects such as septic shock or hypotension [14]. The LPS molecules containing only lipid A and core oligosaccharides are referred to as “rough” or lipooligosaccharides, whereas the entire LPS topped with O-antigen is referred to as “smooth” [20].
Effectively, LPS acts as a permeability barrier against substances potentially harmful to a bacterial cell [14]. Listeria monocytogenes is the only gram-positive bacteria with a genuine LPS [21]. It has been demonstrated that most commensal and pathogenic gram-negative bacteria form biofilms. These biofilms stabilize the bacterial cell populations and resist various drugs and antibiotics. Palmitoylation of LPS is one of the strategies that lead to the formation of stable biofilms. Palmitic acid increases the hydrophobicity of LPS, a characteristic of biofilms forming on biotic and abiotic surfaces.
This review summarizes the different ways of extracellular and intracellular/cytosolic detection of LPS and its further consequences. Then, it sheds light on methods reported to mitigate the adverse effects of LPS.
The LPS molecule is classically described to be detected by various cells through structures on their cell membranes. Specialized proteins termed pattern recognition receptors (PRRs) on the innate immune cells facilitate the detection of LPS even at picomolar concentrations. TLR-4, a receptor for bacterial LPS, is among the most-studied PRRs for LPS. Bruce Beutler received the 2011 Nobel Prize for establishing the role of TLR-4 as the LPS receptor [22]. Dysregulation of TLR-4 promotes aberrant cytokine production in bacterial sepsis. Therefore, understanding the TLR-4 signalling pathway will help address the basic mechanisms and underlying processes of LPS-induced inflammation.
TLR-4 encounters LPS in the extracellular space via contact with intact bacteria or exposure to soluble LPS aggregates. This process is mediated by lipid-binding proteins (LBPs) and LPS-binding proteins, which are carriers of LPS in the blood. Physiologically, they can transfer LPS to the macrophages or the serum carrier lipoproteins. LPS initially binds to LBP in the serum, which then transfers it to the cluster of differentiation 14 (CD14) receptor on the cell membrane of immune cells. The CD14 [glycosylphosphatidylinositol (GPI) anchored protein] transfers the LPS information to the myeloid differentiation factor 2 (MD2) (an associated factor of TLR-4 and an unanchored protein), which interacts with TLR-4. Thus, LPS binds to the CD14/TLR-4/MD2 receptor complex found on numerous types of host cells, including monocytes, dendritic cells, macrophages, and B cells [23]. LPS-binding stimulates TLR-4/MD2 dimerization, a prerequisite for the subsequent Myddosome- and Toll-IL-1 receptor domain-containing adaptor inducing interferon-β (TRIF)-dependent responses and endocytic activity of TLR-4 [24].
Upon detection of LPS, TLR-4 rapidly causes the construction of a supramolecular organizing center (SMOC) termed a “Myddosome” [25–28]. The Myddosome comprises two adapter proteins, myeloid differentiation factor 88 (MyD88) and Toll-IL-1 receptor domain containing adaptor protein (TIRAP), and many another serine threonine [IL-1 receptor (IL-1R)-associated kinase] (IRAK) groups. Thus, this SMOC triggers the TLR-4 signalling pathways.
TLRs are composed of two key domains: extracellular and intracellular regions. The extracellular region has leucine-rich repeats, an intracellular domain belonging to the IL-1R family [29]. Hence, TLRs are believed to utilize the same signalling components as IL-1R. When molecules such as LPS, IL-1. IL-1-related cytokine, IL-18, macrophage-activating lipopeptide (MALP) binds to the cell-surface receptor, and the adaptor molecule MyD88 is recruited to the receptor [30, 31]. An IRAK is recruited, phosphorylated, dissociated from the receptor complex, and interacts with [TNF receptor (TNFR)-associated factor] (TRAF) 6 [32]. Thus, two distinct pathways are activated involving the c-Jun N-terminal kinase (JNK)/p38 mitogen-activated protein kinase (MAPK) family and the Rel family transcription factor NF-κB. In support of this, few studies have shown no production of IL-1, TNF-α, and IL-6 in response to LPS and MALP-2 in macrophages lacking MyD88. However, LPS stimulation of MyD88-deficient macrophages activates NF-κB and JNK/p38, albeit slower than in wild-type macrophages [33]. In contrast, NF-κB activation by MALP-2 stimulation is eliminated in macrophages lacking MyD88 [34]. These data support the existence of multiple mechanisms in the activation of NF-κB and JNK/p38 pathways in TLR-4 signalling.
Two signalling pathways (Figure 2) are triggered by the activation of TLR-4 by LPS: the MyD88-dependent pathway and the MyD88-independent pathway. These signalling pathways activate numerous transcription factors, including NF-κB and [interferon (IFN)-regulatory factors] (IRFs), and induce the production of pro-inflammatory cytokines and IFNs, respectively.
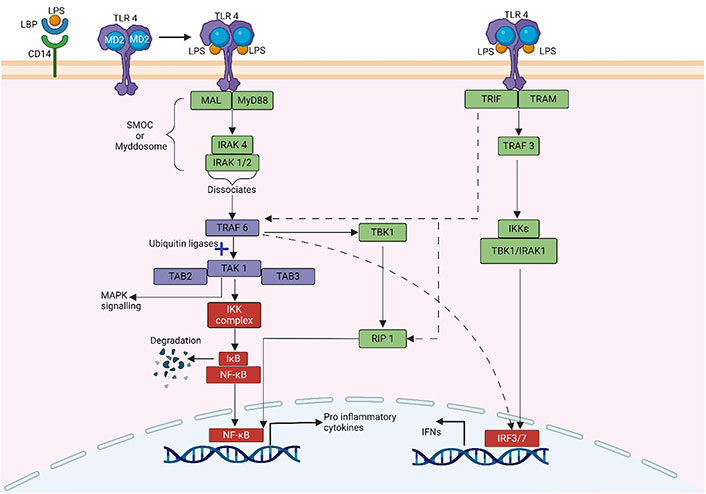
Schematic illustration of MyD88 dependent and MyD88 independent pathway of NF-κB activation. The dotted line indicates the interaction between both pathways. MAL: MyD88-adapter-like protein; TAK: transforming growth factor β-activated kinase; TAB: TAK-binding protein; IκB: inhibitor κB; IKK: IκB kinase; TBK: TANK binding kinase; RIP: receptor-interacting protein; TRAM: TRIF-related adaptor molecule. Created with BioRender.com
MyD88, an adaptor protein containing a death domain, also referred to as Toll/IL-1 receptor (TIR) domain-containing, links the cytoplasmic TIR domain of TLR-4 to MyD88. The death domain of MyD88 then binds to the IRAK4, activating either IRAK1 or IRAK2 [35, 36]. The MyD88-IRAKs family complex, known as the “Myddosome” is particularly important for inflammation and the host immune system. After dissociating from the Myddosome, the IRAK complex interacts with TRAF6. TRAF6 combines with TAK1, TAB1, and TAB2 to form a complex. The complex then attaches to ubiquitin ligases, such as ubiquitin-conjugating enzyme 13 and ubiquitin-conjugating enzyme variant 1A. TAK1 then causes inhibitory B (IB) phosphorylation by activating the complex of IKKα/IKKβ/IKKγ (also known as IKK1, IKK2, and NF-κB essential modulator (NEMO), respectively). IB dissociates from the complex and is directly targeted for ubiquitination and proteasomal destruction, activating the transcription factor NF-κB. The released NF-κB translocates into the nucleus and regulates the expression of several genes encoding pro-inflammatory cytokines [37, 38]. TAK1 can activate MAPK signalling pathways, such as the extracellular signal-regulated kinase (ERK) pathway, the JNK, and the p38 pathway, in addition to the IKK complex [39]. Transcriptional factor activator protein-1 (AP-1) can be activated by MAPK signalling pathways. NF-κB and AP-1 activation contributes to the generation of pro-inflammatory cytokines including IL-6, IL-1, and TNF-α [40].
The MyD88-independent mechanism, also known as the TRIF-dependent pathway or the TIR-containing adapter molecule-1-dependent pathway, can activate both IRF and NF-κB, resulting in the synthesis of IFNs and pro-inflammatory cytokines, respectively. TRAM and TRIF activate the MyD88-independent pathway. Localized on the cytosolic surface of the plasma membrane, TRAM is a specific bridging adapter protein in the TLR-4-mediated MyD88-independent pathway [41]. TRIF interacts with TRAF family member-associated TBK1 and IKK-ε to phosphorylate the transcription factor IRF3 [42]. Moreover, TRIF can interact with IRAK1 and IKK-ε to activate the transcription factor IRF7. IRFs that have been activated then translocate into the nucleus, bind to DNA, and generate chemicals such as IFN [42, 43].
TRIF can also stimulate MyD88-independent NF-κB activation in TLR-4 signalling pathways. Like the MyD88-dependent route, TRIF recruits TRAF6 and activates TAK1 via ubiquitination-dependent processes, activating the NF-κB and MAPK pathways [44]. In addition, TRIF induces MyD88-independent NF-κB activation via binding adaptor RIP1 [45]. In addition, RIP1 can bind to TRIF, resulting in Fas-associated death domain-dependent apoptosis and NF-κB activation [41]. Therefore, TRIF activates IRFs via interactions with TBK1 and IRAK1 and NF-κB via interactions with RIP1 molecules. Subsequently, when the TLR-4 is incorporated into endosomes, it stimulates Type-I IFN production via the adaptor proteins TRAM and TRIF [46].
Irrespective of the TLR-4 pathways, reports have been published that TLR-4 inhibitors can reduce the levels of inflammatory factors in LPS-stimulated cells [47]. They still cannot completely block the production of TNF-α. This observation was also shown in nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain containing 3 (NLRP3) and [apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD)] (ASC) downstream of TLR-4. TLR-4 receptor inhibitors only partially reduced LPS-induced NLRP3 and ASC expression. This observation explains the presence of a less explored TLR-4 independent LPS detection mechanism, which is discussed further.
The LPS from extracellular bacteria, which is either endocytosed or transfected into the cytosol of host cells or cytosolic LPS produced by intracellular bacteria, is recognized by cytosolic proteases (Figure 3). Recent reports have reported caspase 11 as a direct sensor of intracellular LPS [48]. The caspase 11 molecule was first discovered and characterized in mice. It was called caspase 11 due to its sequence homology to other caspases involved in apoptosis [49]. It is a protein enzyme that plays a pivotal role in the innate immune response, particularly in detecting intracellular pathogens and the induction of pyroptosis [50, 51], a highly inflammatory form of programmed cell death. Activation of caspase 11 triggers the release of crucial pro-inflammatory cytokines, including IL-1β and IL-18 [51]. In humans, caspase 11 is known as caspase 4. It serves a similar role in innate immunity and inflammatory responses [52].
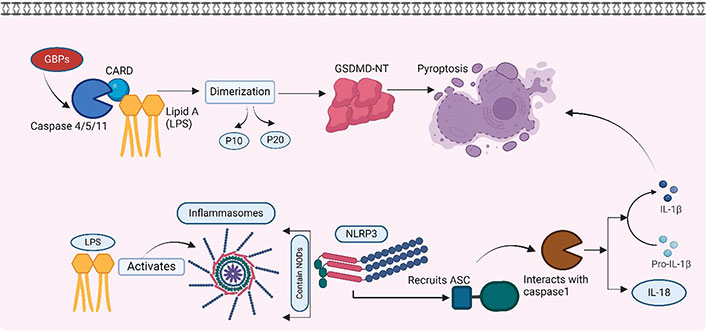
Cytosolic detection of endocytosed LPS. Detection of LPS by caspase 4/5/11 and LPS activating intracellular inflammasomes. GBPs: guanylate-binding proteins; NODs: nucleotide-binding oligomerization domains; GSDMD-NT: N-terminal fragment of gasdermin D. Created with BioRender.com
Intracellular proteases such as caspases have structural protein motifs. These are referred to as caspase-activated recruited domain (CARD). The CARD domain of caspase 11 binds to the lipid A component of LPS [53]. This binding is based on the electrostatic adsorption of LPS to the positively charged CARD domain, which is very similar to the binding of LPS to the TLR-4-MD2 complex [54]. Such binding of caspase 11 with LPS activates the GSDMD-NT, ultimately resulting in pyroptosis (discussed later).
Intracellular LPS also leads to the activation of inflammasomes. Cells have large intracellular multiprotein complexes called inflammasomes. These play a central role in innate immunity by detecting and responding to a variety of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). These inflammasomes contain a member of the NOD-like receptor (NLR) family, such as NLRP3 and NLR family CARD-domain containing protein 4 (NLRC4), by which they are defined. Upon interaction with LPS, the NLR protein recruits the ASC, which interacts with caspase 1, activating it. The activated caspase 1 promotes the maturation of the pro-inflammatory cytokines IL-1β and IL-18. Caspase 1 is also reported to induce lytic changes in the cells, resulting in a cell death process called pyroptosis [55].
Pyroptosis is a highly inflammatory type of cell death that differs from apoptosis. It is gasdermin-mediated programmed necrosis, characterized by cell swelling, membrane rupture, and the release of cytoplasmic contents, including pro-inflammatory cytokines and DAMPs [50]. GSDMD-NT forms pores on the cell membrane that further lead to cell lysis. These pores allow the release of pro-inflammatory cytokines, such as IL-1β and IL-18, which are crucial for immune responses [56].
Pyroptosis has been reported to occur in various inflammatory diseases, such as coronavirus disease 2019 (COVID-19), colitis, rheumatoid arthritis, Crohn’s disease, neuroinflammatory injury, and even periodontitis [57–62]. Though such a response protects against bacterial infections, an exaggerated or excessive activation under pathogenic circumstances leads to the progression of inflammatory conditions, leading to destruction. Apical periodontitis, a local inflammation of the periapical tissues due to pulpal infection [63], simulates localized periodontal destruction. The host defense response to pathogenic microbes triggers inflammation and the destruction of periradicular tissues [63]. A correlation between clinical apical periodontitis intensity and pyroptosis levels is now well-established [64]. Similarly, increased IL-1β and IL-18 levels are reported to be significantly elevated in tissues undergoing pyroptosis. This process further leads to the activation of osteoclastic bone resorption and further periodontal bone destruction [65].
In contrast to the canonical system, the noncanonical pathway involves the dual role of caspase 4/5 in humans and caspase 11 in mice as both sensor and effector molecules in the induction of pyroptosis by LPS. Following the identification of LPS, the process of caspase 4/5/11 dimerization and autoproteolysis takes place, resulting in the liberation of the p10 and p20 domains [66, 67]. The latest developments in this study area have revealed that several GBPs have a role in the signalling of caspase 4 triggered by LPS [68, 69]. Nevertheless, further research is needed to explore this phenomenon in the context of periodontitis. The role of pyroptosis in periodontitis is explored and reported in various research [65, 70].
Despite pyroptosis being identified as one of the mechanisms of cell death in periodontitis [62], there are conflicting reports about the production of IL-1b and related alveolar bone loss in NLRP3 deficient mice [71] and in circumstances where caspase 1 is deficient. A hypothesis of different aged mice used in various experiments is quoted for this observation. However, this needs further research to be certain.
Despite pyroptosis having been demonstrated to have distinct morphological features from apoptosis, numerous studies have demonstrated crosstalk between apoptosis and pyroptosis. Classically, apoptosis is described to be activated via the intrinsic or the extrinsic pathway by caspase 3 and 8, respectively. To begin with the intrinsic pathway, recent studies have demonstrated caspase 3/gasdermin E (GSDME) as a switch between apoptosis and pyroptosis [72], where the authors demonstrated the potential of caspase 3 to cleave GSDME, thereby inducing pyroptosis [72]. Similar observations were reported by Rogers et al. [73] in 2017 using human embryonic kidney cells in periodontitis. Butyrate-initiated pyroptosis is reported to be following the same pathway [74]. Also, cadmium, an apoptosis-triggering agent, is known to induce pyroptosis via the activation of caspase 1 [75]. Caspase 8 is also reported to induce pyroptosis by directly binding to gasdermin D (GSDMD) and gasdermin C (GSDMC) leading to their cleavage in Yersinia-infected macrophages and Hela cells respectively [76, 77].
On the contrary, pyroptosis is also reported to induce apoptosis. Oligomerization of pyroptosis-initiating proteins such as NLRP3 or melanoma 2 (AIM2) is reported to induce apoptosis. This is via the recruitment of caspase 8 through ASC [78]. Also, the activated caspase 1 is reported to activate caspases 3/8/9 in two ways, thereby inducing apoptosis [79]. Also, the GSDME protein of pyroptosis is known to damage the mitochondrial membrane, releasing cytochrome c and further inducing apoptosis [80]. However, whether these mechanisms occur in periodontitis is yet to be ascertained.
Despite apoptosis and pyroptosis being reported to occur bi-directionally and together [81], tissues will normally undergo one specific type of programmed cell death, primarily depending on the intensity of expression of related molecules. Apoptosis predominates in tissues where GSDMs are deficient, and in tissues with caspase deficient tissues, pyroptosis dominates [62]. They are also reported to depend on specific cell types [80]. Despite multiple published reports of increased levels of caspase 1/4/5/11/3/8, no studies have tried evaluating the bi-directional relationship between apoptosis and pyroptosis in understanding periodontitis. This explanation requires further evaluation.
Necroptosis is another programmed cell death that distinctly differs from pyroptosis in its pathway and morphological changes. Cells that undergo necroptosis show loose cellular detachment and exaggerated cellular swelling compared to pyroptosis [82]. To briefly describe necroptosis, the activation of various receptors, such as TNFR, death receptor, TLR, or type I IFN receptor (IFNAR), results in the assembly of complex I (on the cellular membrane), comprising many components such as TNFR-associated death domain (TRADD), Fas-associated death domain (FADD), TRAFs, receptor-interacting protein kinase (RIPK) 1, and cellular inhibitor of apoptosis protein 1 and 2. Subsequently, the recruitment of RIPK3 and the subsequent assembly of the RIP complex occurs, resulting in the phosphorylation and subsequent activation of mixed lineage kinase domain-like (MLKL). The process of pore formation is facilitated by the activation of MLKL, leading to the subsequent release of inflammatory agents and cellular components. This finally results in the collapse of the membrane [83].
Although necroptosis has a pathway, many factors involved in necroptosis are reported to be involved in pyroptosis pathways too. In addition, periodontal pathogens such as P. gingivalis LPS (LPS-G) are reported to cascade both pyroptosis and necroptosis pathways [84, 85]. In addition, the involvement of RIPK1, RIPK3, and MLKL in the mediation of NLRP3/caspase 1 activation has been found to potentially contribute to the activation of pyroptosis in periodontitis [86].
It is well-established that neutrophils play a significant role in periodontitis. Neutrophils exert their effects on cytotoxicity and phagocytosis when exposed to pathogens by releasing neutrophil extracellular traps (NETs). This process is called NETosis. NETosis is typically triggered by the recognition of pathogens or inflammatory signals. Upon activation, neutrophils undergo morphological changes and initiate the process of NETosis [87]. One of the hallmark features of NETosis is the decondensation of chromatin (DNA and associated proteins, such as histones) within the neutrophil’s nucleus [88]. The process of extruding decondensed chromatin from neutrophils involves the rupture of the cell membrane, leading to the release of NETs into the extracellular space [89]. These NETs trap and kill pathogens by acting as physical barriers that trap and immobilize pathogens. NETs contain enzymes, such as neutrophil elastase and myeloperoxidase, that can directly kill pathogens by degrading their components. NETs also facilitate the recognition of pathogens by other immune cells, such as macrophages and dendritic cells [90]. The impact of NETs on the pathogenesis of periodontitis and their role in inflammatory reactions and gingival tissue destruction was emphasized by Magán-Fernández et al. [91] in 2020, where the authors exclusively reviewed NETs in periodontitis.
Further, the same team investigated NETs in the gingival tissues of patients with periodontitis and compared them with controls, including patients with gingivitis. Electron microscopy and immunofluorescence revealed increased neutrophil expression in periodontal tissue compared to controls, indicating NET formation. The analysis of specific NET components showed higher citrullinated histone H3 expression in gingivitis samples than in periodontitis samples. The study concluded that NETs were present in periodontitis tissues, serving as extracellular components of chromatin and neutrophil enzymes, with a higher expression observed in gingivitis, suggesting their association with the acute phases of the periodontal inflammatory process [92].
NETosis is classified as a biological process that plays a significant role in the autoimmune response. Therefore, information about its role in periodontitis and periodontal pathogens is scarce [93, 94]. The induction of NETosis by P. gingivalis was found to rely on gingipains, which facilitated the proteolysis of several components of NETs [94]. This phenomenon can potentially enhance the buildup of NETs in the context of periodontitis, hence exacerbating the tissue damage inflicted by neutrophils.
Furthermore, it has been documented that these pathogens induced by NETosis can also elicit pyroptosis in several cell types inside periodontal tissues. The precise determinants that govern the decision of neutrophils to engage in NETosis, pyroptosis, or phagocytosis in response to the same stimuli and pathogens still need to be better understood. The controversy about the ability of neutrophils to undergo pyroptosis remains unresolved [95]. The large amount of neutrophil proteases and the low expression level of caspases and inflammasomes determine the priority for NETosis in neutrophils [62]. Furthermore, this phenomenon was further supported by the observation of neutrophil protease-mediated activation of GSDMD. Additionally, the activation of inflammasomes and GSDMD contributed to the release of chromatin in a feed-forward loop, ultimately promoting NETosis rather than pyroptosis [95, 96]. However, further research is required since these reports are not published exclusively for periodontitis.
Despite multiple distinct cell death mechanisms in inflammatory diseases, a significant amount of crosstalk occurs between these mechanisms and is complex and intriguing. All mechanisms are interconnected in various ways, often depending on the cellular context and specific signalling pathways. When LPS activates the TLR-4 mediated pathway, it activates caspase 8, RIPK1, RIPK3. This activates necroptosis. However, caspase 8 is considered to play an important role in regulating and modulating inflammatory responses concerning inflammasome activation and cell death, involving apoptosis, pyroptosis, necroptosis, and NETosis [97]. Further, inhibitor of apoptosis proteins (IAPs) can directly bind to caspase 8, 3, and 7, thereby inhibiting apoptosis. Any inhibitory activity of AIPs will lead to caspase 8-mediated increased apoptosis and further cleavage of IL-1b, and RIPK1-mediated necroptosis and activation of NLRP3 further leads to enhanced IL-1b production. Crosstalk can occur through caspase 8, which cleaves GSDMD to induce pyroptosis-like membrane permeabilization [98]. Similarly, caspase 8 can suppress necroptosis by cleaving key regulators of the necroptotic pathway, such as RIPK1 and RIPK3 [99]. Further, gasdermin family proteins can contribute to cell membrane disruption in necroptosis and pyroptosis [51]. Regarding crosstalk between NETosis, inflammatory conditions can lead to “apoptotic NETosis”, where neutrophils undergoing apoptosis exhibit features of NETosis [100]. Further, pyroptosis can release cytokines that attract neutrophils, which can then undergo NETosis [101].
Over the years, researchers have employed numerous strategies to overcome the adverse effects of LPS. The methods for mitigating the effects of LPS are illustrated in Figure 4. This section of the review will shed light on such strategies.
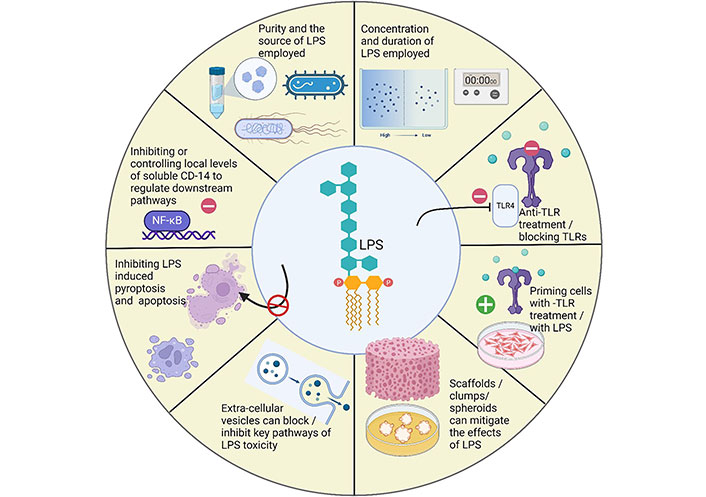
Strategies employed to mitigate the adverse effects of LPS in periodontitis. Created with BioRender.com
In 2016, Andrukhov et al. [102] compared the response of periodontal ligament stem cells (PDLSCs) to LPS-G and Escherichia Coli (E. Coli)-derived LPS. No significant differences between the two sources of LPS in the absence of CD14 were reported. The “purity” of LPS was more significant than the source of the LPS. The “ultrapure” variety of LPS lacks contaminants, thereby inducing a specific and defined LPS-mediated immune response. However, a few “standard” varieties of commercially available LPS contain additional components of gram-negative bacteria, such as peptidoglycans or lipopeptides, resulting in a non-standardized immune response in response to a stimulus [103]. 2017, Parusel and colleagues [103] studied differences between the standard LPS and ultrapure LPS. Their observations showed standard LPS to activate both TLR-2- and TLR-4-dependent signalling, whereas ultrapure LPS activated specifically TLR-4-dependent signalling alone. This confirms the additional contaminants in addition to a specific TLR-4 ligand that is present in standard LPS. Consequently, the standard LPS is unsuitable for investigating a specific TLR-4-dependent signalling pathway.
Many researchers, to date, have already demonstrated that LPS at low concentrations is cytoprotective [104, 105]. Numerous published studies demonstrate that LPS at lower concentrations are indeed cytoprotective and enhance PDLSC proliferation. The authors also report that the actual proliferative response of PDLSCs depends on the degree of inflammation [106–109]. Yu Sen Hou et al. [105] demonstrated that human umbilical cord-derived mesenchymal stem cells (MSCs) undergo apoptosis when treated with LPS, but pre-treatment with low-dose LPS prevents apoptotic cell death. This observation indicates that low-dose LPS can be cytoprotective. It is reported that pre-treating MSCs can benefit cells by conditioning them to produce specific extracellular vesicles that can polarize macrophages towards the anti-inflammatory M2 phenotype [110].
Activation of the classical Wnt signalling pathway induces the formation and regulation of dozens of lymphoid enhancer-binding factor 1-β-catenin complexes and the target gene that encodes cyclin D1 protein, thereby altering the cell cycle and promoting the proliferation of PDLSCs [111]. Other factors that are reported to enhance PDLSCs proliferation in lower concentrations of LPS include enhanced histone deacetylase in PDLSCs disrupts the actual balance of acetylation that affects the transcription of inflammatory factors [112]. C-X-C motif chemokine ligand 8 and chemokine (C-C motif) ligand 5 (CCL5) upregulation are also reported to play a role in the enhanced proliferation of PDLSCs to lower concentrations of LPS [113].
Schröder et al. [114] reported that low-dose LPS exposure may stimulate regulatory immune responses, such as the expansion of regulatory T cells (Tregs) and the release of anti-inflammatory cytokines like IL-10. These responses can contribute to immunomodulation and protection against excessive inflammation [114]. Similarly, Low-dose LPS exposure may induce a state of endotoxin tolerance in cells and the immune system. This tolerance can reduce the inflammatory response to subsequent, potentially harmful LPS exposure, thereby protecting tissues and organs from excessive inflammation [115]. Similarly, Sangaran et al. and Mizobuchi et al. [116, 117] suggested that low-dose LPS exposure may promote tissue repair and regeneration by stimulating the production of growth factors and other molecules that support tissue healing.
Li et al. [118] investigated the effects of LPS and its concentration on human dental pulp stem cells (hDPSCs). At 1 µg/mL and 10 µg/mL concentrations, LPS promoted cell adhesion, with 1 µg/mL showing a stronger effect. However, LPS had a dual effect on cell migration. It increased migration at 1 mg/mL but significantly decreased it at 10 µg/mL. Further, 1 µg/mL LPS significantly enhanced the gene expression of adhesion molecules and chemotactic factors. The research also explored the role of NF-κB MAPK signalling pathways in LPS-induced migration and adhesion of hDPSCs. Specific inhibitors for NF-κB and various MAPK pathways (ERK, JNK, P38) were used and found to counteract the effects of LPS significantly. The study specifically noted that inhibitors of the stromal cell-derived factor-1 (SDF-1)/C-X-C chemokine receptor type 4 (CXCR4) pathway, particularly AMD3100, significantly reduced LPS-induced migration of hDPSCs [118].
Given the role of TLR-4 in initiating the inflammatory response to bacterial LPS, blocking TLR-4 or using anti-TLR-4 approaches to counteract the adverse effects of LPS in periodontitis is widely researched.
A synthetic TLR-4 antagonist, FP7, in various inflammatory conditions, demonstrated that it effectively countered TLR-4 activation, reducing pro-inflammatory cytokines by human monocytes and dendritic cells and preventing dendritic cell maturation upon TLR-4 activation by LPS. FP7 selectively targeted TLR-4 stimulation without affecting other TLR pathways. Furthermore, FP7 protected mice from lethal influenza virus infection, reducing lung inflammation and acute lung injury, likely by mitigating TLR-4-dependent cytokine release [119].
This strategy is employed therapeutically in various conditions, including sepsis, neurodegenerative diseases, neuroinflammation, drug abuse, inflammatory disorders, and neuropathic pain [120]. Various TLR-4 modulators are identified in the literature. However, their use in periodontitis is still to be ascertained.
Similarly, secretory leukocyte protease inhibitor (SLPI) inhibits serine proteases [121]. Intracellular SLPI moves into the nucleus and binds to NF-κB binding regions in the promoter region of the TNF-α and IL-8 [C-X-C-motif chemokine ligand 8 (CXCL-8)] genes. This binding antagonizes NF-κB activation [122]. The secretory form of SLPI has been shown to interact with LPS and thus inhibit the binding of LPS to its cell surface receptors. Periodontal stem cells are reported to express SLPI, and cellular levels of SLPI inversely correlate with the stimulation of pro-inflammatory cytokines like IL-6 and monocyte chemoattractant protein-1 (MCP-1) by LPS [123].
Even though the PDLSCs express all stem cell surface markers, it is important to emphasize that they weakly express CD14, a well-recognized membrane protein that regulates the effects of LPS, among many others, in immune cells. Over the years, CD14 has been a great research interest and is one of the most discussed membrane proteins that is present on the cell membrane of most immune cells, such as macrophages. As discussed earlier, physiologically, LPS binds to the CD14/MD2 complex, further translating the effects of LPS via the TLR-4-induced NF-κB activation and subsequent cytokine production and tissue destruction. This weak expression of CD14 in PDLSCs mandates a significantly higher dosage of LPS (> 1,000 times) than those other immune cells require. This property of PDLSCs and their well-recognized immunomodulatory effects make them suitable candidates in cell-based therapies.
Since PDLSCs express minimal CD14, soluble CD14 (sCD14) plays a crucial role in augmenting the response of human PDLSCs (hPDLSCs) to bacterial LPS. In 2016, Andrukhov et al. [102] evaluated the response of PDLSCs to LPS-G and E. Coli LPS in their comparative experiment. The response of hPDLSCs to both LPS-G and E. Coli LPS was significantly enhanced by sCD14 compared to when sCD14 was absent. The responses to LPS were lower compared to the response to Pam3CSK4, a TLR-2 agonist. Therefore, local levels of sCD14 levels are reported to regulate the response to LPS [102].
Pre-conditioning cells before LPS exposure results in immunomodulation, proliferation, migration, osteogenic differentiation, and polarization towards anti-inflammatory phenotype in PDLSCs and various other cell types.
Ballerini et al. [124], in 2017, investigated the impact of conditioned medium (CM) from human PDLSCs from healthy donors and relapsing-remitting multiple sclerosis (RR-MS) patients on the inflammatory response induced by LPS-G in human monocytoid and oligodendrocytes. LPS-G increased pro-inflammatory cytokine expression and protein levels. However, treatment with PDLSCs-CM from healthy donors and RR-MS patients reduced the LPS-induced inflammation and TLR-4 levels [124]. Similarly, another study investigated the immunomodulatory and neuroprotective effects of CM derived from healthy hPDLSCs and those obtained from RR-MS patients [125]. Mouse NSC34 (motor neuron-like cell line model) motoneurons were stimulated with LPS in vitro. LPS stimulation led to increased levels of TLR-4 and NF-κB, along with reduced levels of IκBα in motoneurons. However, pre-conditioning with an hDPSC-CM modulated these markers. In addition, the CM influenced the expression of inflammatory cytokines (TNF-α, IL-10), neuroprotective markers [Nestin, NFL (neurofilament light chain) 70, nerve growth factor (NGF), growth-associated protein 43 (GAP43)], and apoptotic markers (Bax, Bcl-2, p21). Additionally, extracellular vesicles from the hDPSC-CM contained anti-inflammatory cytokines like IL-10 and TGF-β.
Yang et al. [126] recently assessed the effects of concentrated growth factors-conditioned media (CCM) on PDLSCs and stem cells from apical papilla (SCAPs) with or without LPS. CCM dose-dependently enhanced PDLSCs and SCAPs migration, proliferation, and differentiation, alleviating LPS-inhibited differentiation, and inflammatory cytokine up-regulation. Concentrated growth factor (CGF) induction increased newly regenerated tissue and micro-vessels. Similarly, Liu et al. [127] in 2019 showed that the CM pre-treated PDLSCs could polarize macrophages towards M2 phenotype, enhancing periodontal regeneration after stem cell transplantation and potentially altering the immune microenvironment by influencing the early stage of tissue repair.
The regulatory effects of the human antimicrobial peptide LL-37 on LPS-treated bone marrow MSCs were evaluated. LL-37 showed several positive effects, including promoting their proliferation, migration, and osteogenic differentiation. Further, LL-37 demonstrated anti-inflammatory properties by inhibiting the expression of IL-1β, TNF-α, and RANKL. It also attenuated the inhibition of osteogenesis induced by LPS. The study confirmed the expression of purinergic P2X receptor 7 (P2X7R) in bone marrow stem cells (BMSCs). Blue brilliant G (BBG), a P2X7R antagonist, significantly reduced LL-37-promoted osteogenesis, suggesting the involvement of P2X7R in LL-37’s effects. Also, LL-37 increased the phosphorylation of ERK1/2 and JNK without affecting p38 phosphorylation. Inhibitors specific to ERK1/2 and JNK attenuated the effects of LL-37 on the expression of genes related to osteogenesis, indicating the involvement of the MAPK signalling pathway [128].
Recently, the effects of low-intensity pulsed ultrasound (LIPUS) on PDLSCs were assessed [129]. LIPUS increased cell viability dose-dependently and elevated the levels of immunomodulatory factors. When irradiated cells were co-cultured with peripheral blood mononuclear cells (PBMCs) stimulated by LPS, the levels of inflammatory factors in PBMCs were reduced. LIPUS irradiation also promoted osteogenic gene expression in hPDLSCs. Further, LIPUS inhibited the NF-κB pathway as indicated by changes in the levels of IκBα, phosphorylated IκBα (p-IκBα), and the p65 subunit of NF-κB. However, when hPDLSCs were treated with both LPS and LIPUS, the inhibitory effect of LIPUS irradiation on the NF-κB pathway was partially reversed, resulting in a decrease in the immunoregulatory and osteogenic differentiation abilities of hPDLSCs.
Mitochondrial reactive oxygen species (mtROS) generation is reported to play a crucial role in the pro-inflammatory response of gingival fibroblasts to LPS. Inhibition of mtROS production using mitochondrial-targeted exogenous antioxidant called mito-TEMPO or by transfection with manganese superoxide dismutase (MnSOD) transfection prevents the upregulation of cytokines and the activation of key signalling pathways, such as p38, JNK, and inhibitor of NF-κB. It also prevents the nuclear localization of NF-κB [130].
MicroRNAs (miRNAs) are small RNA molecules that post-transcriptionally regulate gene expression by binding to target messenger RNAs (mRNAs). The miRNAs play critical roles in various biological processes, including development, cell differentiation, immune response, and the maintenance of tissue homeostasis. Dysregulation of miRNA expression is associated with many diseases.
MicroRNA-138 (miR-138) regulates PDLSCs and their impact on homeostasis in animal models of periodontitis, where it was significantly upregulated. Overexpression of miR-138 in PDLSCs inhibited the expression of osteocalcin (OC), Runt-related transcription factor 2 (Runx2), and collagen I. Further, IL-6 and LPS increased in miR-138 expression, but conversely, the expression of OC and Runx2 was significantly decreased in response. Knockdown of miR-138 partially reversed the LPS-induced downregulation of OC expression and promoted osteoblast differentiation [131]. Similarly, miR-302b is upregulated in periodontitis, and tetramethylpyrazine (TMP) is reported to alleviate the adverse effects of LPS on cell viability, inflammation, and apoptosis in PDLSCs. Importantly, TMP downregulates the miR-302b levels in LPS-stimulated cells [132]. Another miRNA, miR-142-3p, is reported to repress the osteogenic abilities of PDLSCs by reducing serum- and glucocorticoid-regulated kinase (SGK) 1 expression [133].
Cells are traditionally cultured in Petri dishes, where they adhere to the surface and then flatten to form monolayers. Once confluent, due to the absence of additional adhesive surface area and appropriate cell-cell connections, further growth and functional differentiation are constrained. Consequently, they cease proliferating and begin to die. These methods must provide adequate conditions for the cells’ growth and functional replication compared to their in vivo counterparts. The complex macromolecular network surrounding the cells serves an essential function in signalling the cells appropriately with precise information. Consequently, tissue health depends on the cells directly involved in generating its functionality; however, the surrounding cells and extracellular microenvironment consistently affect their optimal behavior. Frequently, the contribution of supporting cells and extracellular matrix (ECM) to maintaining the functional integrity of cells and tissues is remarkable [134]. As a result, when it comes to comprehending the actual behavior of specific cells or their response to a particular substance or stimulus, it is impossible to obtain an accurate image by growing cells as monolayers in isolation.
MSCs in 3D spheroid cultures, when exposed to LPS, exhibit self-activation, which is partially triggered by intracellular stress responses and results in PGE2 production. PGE2 can convert stimulated macrophages from a predominantly pro-inflammatory M1 to a predominantly anti-inflammatory M2 phenotype. This indicates that 3D spheroid cultures of human MSCs can modulate inflammation through PGE2 production and macrophage polarization [135]. Similarly, 3D clumps/ECM complexes of PDLSCs are clumps of mesenchymal stem cells (C-MSCs) and are reported to be more resistant to the effects of LPS and the cells retain osteogenic potential than cells grown in monolayer cultures [136]. Similarly, many studies conducted on 3D clumps/ECM complexes have reported significant beneficial effects in periodontal regeneration experiments, both in vitro and in vivo [137, 138].
In contrast, a study by Liu et al. [139] demonstrated a significant difference in the organization of F-actin, a structural protein, between spheroid and monolayer cultures. Spheroids were reported to exhibit junctional F-actin at cell-cell contact points. At the same time, monolayer cells had stress fibres of actin and more prominent F-actin localized at the cell base. Further, responses of 3D spheroids to LPS and the traditional monolayer cultures of A549 lung epithelial cells and HepG2 hepatocytes were compared. The A549 spheroids consistently secreted higher levels of IL-6 and IL-8 than monolayer cultures. HepG2 spheroids also responded to LPS by releasing a significant amount of IL-8, which was not observed in monolayer cultures [140]. However, the mechanisms of creation of 3D spheroids and the cells employed differ between the studies reviewed here.
TLR-3 recognizes viral pathogens and activates a distinct signalling pathway compared to TLR-4, which responds chiefly to LPS. Individual stimulation of hPDLSCs with Poly I:C (TLR-3 agonist) or Pam3CSK4 (TLR-2 agonist) has been shown to induce the production of IL-6, IL-8, MCP-1, and osteoprotegerin (OPG) in a published report. The magnitude of the responses to these stimuli was comparable. However, a synergistic effect was observed when hPDLSCs were simultaneously exposed to Poly I:C and Pam3CSK4. All inflammatory mediators’ levels were significantly higher than those induced by individual stimuli or the summed response of the two. Poly I:C and Pam3CSK4 increased the gene expression and protein levels of OPG, a protein involved in bone metabolism. However, OPG levels were substantially lower when stimulated simultaneously with Pam3CSK4 and Poly I:C than when stimulated with Pam3CSK4 alone. The study suggests that the simultaneous activation of TLR-2 and TLR-3 in hPDLSCs induces a synergistic increase in pro-inflammatory cytokine production [140]. This effect was not observed for the protein OPG, which regulates bone metabolism. The activation of TLR-3 by viral infections may contribute to the progression of periodontitis [140].
LPS and its adverse effects have been extensively studied over many years. Enormous progress is achieved in understanding the intricate molecular interactions in chronic inflammatory conditions. This review has provided an overview of bacterial LPS and the mechanisms involved in its detection. Numerous studies have extensively studied and reported TLR-4 mediated mechanisms in detecting LPS; however, a less explored intracytoplasmic mechanism of detection of LPS and its further consequences needs to be explored and understood more. This is especially true about periodontitis. Though well-established crosstalk between cytosolic LPS-activated apoptosis, necroptosis, pyroptosis, and NETosis is reported, conclusions about specific types of programmed cell death in periodontal disease are yet to be ascertained.
Strategies mitigating the adverse effects of LPS are reviewed. Though numerous methods are reported to be successful, most studies are conducted in vitro or animal models. It is pivotal that such mechanisms and strategies be further explored in clinical trials, specifically addressing periodontitis.
ASC: apoptosis-associated speck-like protein containing a caspase-activated recruited domain
CARD: caspase recruitment domain
CD14: cluster of differentiation 14
CM: conditioned medium
E. Coli: Escherichia Coli
ECM: extracellular matrix
ERK: extracellular signal-regulated kinase
GSDMD: gasdermin D
GSDMD-NT: N-terminal fragment of gasdermin D
GSDME: gasdermin E
hDPSCs: human dental pulp stem cells
hPDLSCs: human periodontal ligament stem cells
IFN: interferon
IKK: inhibitor κB kinase
IL: interleukin
IL-1R: interleukin-1 receptor
IRAK: interleukin-1 receptor-associated kinase
IRFs: interferon-regulatory factors
IκB: inhibitor κB
JNK: c-Jun N-terminal kinase
LBPs: lipid-binding proteins
LIPUS: low-intensity pulsed ultrasound
LPS: lipopolysaccharides
LPS-G: Porphyromonas gingivalis lipopolysaccharides
MALP: macrophage-activating lipopeptide
MAPK: mitogen-activated protein kinase
MD2: myeloid differentiation factor 2
miR-138: microRNA-138
miRNAs: microRNAs
MLKL: mixed lineage kinase domain-like
MMPs: matrix metalloproteinases
MSCs: mesenchymal stem cells
MyD88: myeloid differentiation factor 88
NETs: neutrophil extracellular traps
NF-κB: nuclear factor-kappa B
NLRP3: nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3
NOD: nucleotide-binding oligomerization domain
OC: osteocalcin
OPG: osteoprotegerin
P. gingivalis: Porphyromonas gingivalis
P2X7R: purinergic P2X receptor 7
PDLSCs: periodontal ligament stem cells
PGE2: prostaglandin E2
RANKL: receptor activator of NF-κB ligand
RIP: receptor-interacting protein
RIPK: receptor-interacting protein kinase
RR-MS: relapsing-remitting multiple sclerosis
sCD14: soluble cluster of differentiation 14
SLPI: secretory leukocyte protease inhibitor
TAB: transforming growth factor β-activated kinase-binding protein
TAK: transforming growth factor β-activated kinase
TBK: TANK binding kinase
TIR: Toll/interleukin-1 receptor
TLR: Toll-like receptor
TNF: tumour necrosis factor
TNFR: tumour necrosis factor receptor
TRAF: tumour necrosis factor receptor-associated factor
TRAM: Toll-interleukin-1 receptor domain-containing adaptor inducing interferon-β-related adaptor molecule
TRIF: Toll-interleukin-1 receptor domain-containing adaptor inducing interferon-β
The authors thank the Institute for Research Development and Innovation (IRDI) at the International Medical University (IMU) and the members of the Research Office.
SRB: Writing—original draft, Investigation, Data curation. ELT: Supervision, Writing—review & editing, Project administration, Validation. FD: Supervision, Writing—review & editing. SPK: Conceptualization, Supervision, Writing—review & editing.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This research was supported by the Ministry of Higher Education (MoHE) under the Fundamental Research Grant Scheme (FRGS), with a reference number of [FRGS/1/2016/SKK14/IMU/01/1] and Project ID-10093 and selected Grant FRGS 2016-1. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

