 Review
Review

Affiliation:
College of Life Sciences, Shandong Agricultural University, Tai’an 271018, Shandong, China
†These authors contributed equally to this work.
Affiliation:
College of Life Sciences, Shandong Agricultural University, Tai’an 271018, Shandong, China
†These authors contributed equally to this work.
Affiliation:
College of Life Sciences, Shandong Agricultural University, Tai’an 271018, Shandong, China
†These authors contributed equally to this work.
Affiliation:
College of Life Sciences, Shandong Agricultural University, Tai’an 271018, Shandong, China
†These authors contributed equally to this work.
Affiliation:
College of Life Sciences, Shandong Agricultural University, Tai’an 271018, Shandong, China
†These authors contributed equally to this work.
Affiliation:
College of Life Sciences, Shandong Agricultural University, Tai’an 271018, Shandong, China
Email: haifangli@sdau.edu.cn
ORCID: https://orcid.org/0000-0002-2827-5925
Explor Endocr Metab Dis. 2026;3:101470 DOI: https://doi.org/10.37349/eemd.2026.101470
Received: February 28, 2026 Accepted: April 07, 2026 Published: May 05, 2026
Academic Editor: Stavros Dimopoulos, Onassis Cardiac Surgery Center, Greece
Obesity, diabetes mellitus (DM), metabolic dysfunction-associated steatotic liver disease (MASLD), and cardiovascular diseases (CVDs) share pathogenic mechanisms like oxidative stress and inflammation. Resveratrol (RSV) offers therapeutic potential by activating the sirtuin (SIRT) signaling network. This review synthesizes RSV’s pharmacological impacts on adipose, pancreatic, hepatic, and cardiovascular tissues, focusing on the AMPK/SIRT1/PGC-1α axis, PI3K/AKT pathways, and epigenetic modulations. Despite robust preclinical data, a significant translational gap exists. Clinical evidence is heterogeneous, often contradicting animal studies due to varying dosages, durations, and population characteristics. RSV acts as a pan-SIRT activator, though its precise SIRT1 activation mechanism, direct or via NAD+ modulation, remains debated. In obesity, RSV promotes adipose beiging and thermogenesis, yet clinical weight loss is modest. For DM, it preserves β-cell function and improves insulin sensitivity, primarily benefiting diabetic populations with variable glycemic outcomes. In MASLD, RSV ameliorates steatosis and fibrosis in models, but large-scale human trials confirming histological benefits are lacking. Regarding CVDs, RSV protects against endothelial dysfunction and inflammation, showing minor improvements in biomarkers like blood pressure, though hard endpoints need validation. Major limitations hinder clinical efficacy, such as poor oral bioavailability, rapid metabolism, and significant interindividual pharmacokinetic variability. The lack of standardized formulations further complicates systemic exposure. Nevertheless, the RSV-SIRT axis remains a unified metabolic and epigenetic modulator, stabilizing cellular microenvironments across organ systems. It represents a promising target for complex metabolic syndromes. Future research should prioritize overcoming bioavailability challenges through novel delivery systems and investigating synergistic combinatorial therapies to bridge the gap between preclinical promise and clinical reality.
Metabolism-related diseases precipitate a spectrum of pathologies, including obesity, diabetes mellitus (DM), metabolic dysfunction-associated steatotic liver disease (MASLD), and cardiovascular disorders (CVDs), which are driven by systemic energy imbalance and disrupted cellular metabolic networks [1–4]. These disorders are accompanied by a well-documented state of cellular stress characterized by oxidative stress, chronic low-grade inflammation, and mitochondrial dysfunction [2, 4]. The molecular basis of this condition is an impaired adaptation of cellular energy-sensing networks to nutrient overload. A central node in this network is the NAD+-dependent deacetylase sirtuins (SIRTs), whose activity is compromised by the reduced NAD+/NADH ratio inherent to this metabolic state [5, 6]. The functional decline of SIRTs impairs the cell’s capacity for adaptive transcriptional responses, thereby perpetuating a self-reinforcing cycle of metabolic decay [6, 7]. Consequently, the pharmacological restoration of SIRTs activity represents a primary therapeutic objective to counteract metabolism-related diseases.
The mammalian SIRT family comprises seven NAD+-dependent deacylases (SIRT1–7) with distinct subcellular localizations, catalytic activities, and tissue distributions [8]. SIRT1, SIRT6, and SIRT7 are predominantly nuclear, where they regulate chromatin architecture, DNA repair, and ribosomal RNA transcription [9]. SIRT2 is primarily cytoplasmic, deacetylating α-tubulin and modulating cell cycle progression, though it translocates to the nucleus during mitosis [9, 10]. The mitochondria harbor the largest SIRT subgroup, SIRT3, SIRT4, and SIRT5, reflecting the critical importance of mitochondrial metabolic regulation [8]. SIRT3, the primary mitochondrial deacetylase, governs oxidative phosphorylation, fatty acid oxidation, and the tricarboxylic acid cycle [11]. SIRT4 exhibits ADP-ribosyltransferase activity and regulates insulin secretion and glutamine metabolism [8, 12]. Notably, SIRT4 serves as an early sentinel of metabolic health, with its circulating levels declining precipitously in obesity and fatty liver disease, thereby signaling compromised oxidative balance and cardiovascular integrity [12–14]. SIRT5 displays weak deacetylase activity but robustly removes negatively charged acyl modifications (malonylation, succinylation, glutarylation) from metabolic enzymes. SIRT6 maintains genomic stability through histone H3K9 deacetylation and modulates glucose homeostasis via HIF‑1α regulation [15]. SIRT7, localized to the nucleolus, controls ribosomal RNA transcription and couples nutrient sensing to protein synthesis [8, 16]. This compartmentalized distribution enables SIRTs to integrate metabolic cues across nuclear, cytoplasmic, and mitochondrial domains, forming a coordinated network that maintains cellular energy homeostasis [17].
In this context, the natural polyphenol resveratrol (RSV; 3, 5, 4’-trihydroxy-trans-stilbene) has become a focus of significant scientific investigation. Found abundantly in grapes, peanuts, and berries, RSV exhibits antioxidant, anti-inflammatory, and anti-apoptotic effects [18]. It phenocopies several metabolic benefits of caloric restriction, which is largely attributed to its capacity to engage and activate the SIRT1 pathway [19]. However, the precise molecular mechanism by which RSV mediates this activation has been a subject of major scientific debate for over a decade and represents a key unresolved question in cellular pharmacology [20]. Current evidence indicates that RSV functions not through direct binding to SIRT1, but by remodeling the cellular energetic state [21]. The proposed key mechanism involves enhancing the function of the rate-limiting enzyme in the NAD+ salvage pathway, nicotinamide phosphoribosyl transferase (NAMPT), to increase intracellular NAD+ bioavailability [22]. The theoretical advantage of this strategy is that it does not target an individual member of the SIRT family but instead systematically enhances the global catalytic activity of the entire family, including the nuclear SIRT1/SIRT6 and mitochondrial SIRT3, by increasing the availability of the requisite co-substrate, NAD+ [23]. This “upstream substrate supply” model provides a unified theoretical basis for explaining how RSV exhibits broad biological functions across different tissues and pathological conditions.
The downstream effects of this core mechanism for RSV at the physiological level reconstruct the pathogenic processes driven by nutrient overload. Its impact is first manifested in adipose tissue, the center of energy storage, where it directly intervenes in the pathogenesis of obesity by promoting fatty acid oxidation and driving the browning of white adipose tissue (WAT) [24]. This control over the systemic source of lipotoxicity extends to the amelioration of type 2 diabetes, reflected in restored peripheral insulin sensitivity and the preservation of pancreatic β-cell function [25]. The protective effects of this axis are also concentrated in the liver, the central organ for processing systemic metabolic flux. It exerts a molecular inhibitory effect at multiple key stages in the progression of MASLD, from steatosis to fibrosis and carcinogenesis [26]. Ultimately, these effects in key metabolic organs converge to confer protection on the cardiovascular system, evidence for which includes anti-atherosclerotic, cardioprotective, and endothelial-preserving outcomes [27]. This review will, therefore, critically examine the molecular evidence underpinning the role of the RSV-SIRT axis across this disease spectrum, with the aim of constructing a unified regulatory framework and providing a theoretical basis for future therapeutic strategies targeting this axis.
Obesity, recognized as a relapsing chronic disease, is pathophysiologically defined by aberrant expansion and ectopic deposition of WAT [28]. The development of obesity is driven by the interaction of multiple etiological factors, including energy metabolism imbalance, dysregulated gut-brain axis signaling, adipose tissue pathology, hormonal secretion disorders, and sleep insufficiency [29]. Individuals with a body mass index (BMI) of 30.0 kg/m2 or higher are defined as obese, and there is a continuing rise in obesity worldwide. In addition to its negative impact on quality-of-life indices, obesity constitutes a dominant nexus for cardiometabolic multimorbidity, conferring a markedly increased risk for dysglycaemia, dyslipidaemia, hypertension, MASLD, reproductive dysfunction, selected cancers, and cardiovascular diseases (CVDs) [2, 3, 30, 31].
The primary goal of obesity treatment is to reduce excess fat stores and lower the risk of obesity-related comorbidities. Several animal studies indicate that RSV induces anti-adiposity efficacy and promotes systemic metabolic homeostasis via SIRT activation [32, 33]. Sleep deprivation induces obesity accompanied by decreased SIRT1 and forkhead box O1(FOXO1) levels, whereas RSV alleviates this effect by restoring the SIRT1/FOXO1/ATGL pathway and enhancing lipolysis [34]. RSV reduces body weight in mice and inhibits inflammation by elevating SIRT1 and reducing Akt2 activity [35]. Notably, maternal RSV treatment even exhibits therapeutic potential for managing obesity and preventing its transmission to the offspring [36]. Maternal RSV treatment restores SIRT1 in retroperitoneal tissue, down-regulates factor-related apoptosis (FAS), and up-regulates lipoprotein lipase (LPL), thereby mitigating maternal obesity, preventing high-fat diet-aggravated offspring obesity, and alleviating adverse effects on embryonic development [37].
Additionally, due to the specific energy expenditure capability of brown and beige adipocytes, inducing the development and thermogenic activity of these cells could also effectively combat obesity [38, 39]. The RSV-SIRT1-PGC-1α axis increases energy expenditure and thermogenesis, ultimately reducing lipid accumulation in WAT [40]. It has been reported that RSV enhances thermogenic capacity in brown adipose tissue (BAT) through activation of SIRT1 and PGC-1α, leading to upregulation of uncoupling protein 1 (UCP1) and amelioration of metabolic disturbances and increased adiposity induced by hormonal changes following ovariectomy [41]. In interscapular BAT and skeletal muscle, RSV administration up-regulates mitochondrial transcription factor A (TFAM) and cytochrome c oxidase subunit 2 (COX2), selectively elevates UCP1 protein abundance, and promotes mitochondrial biogenesis [40]. Moreover, the interaction between RSV and the gut microbiota plays a key role in host health and can suppress obesity through microbiota-derived metabolic pathways [42]. For example, RSV is metabolized by the gut microbiota into 4-hydroxyphenylacetic acid (4-HPA), and related metabolites activate the SIRT1 signaling pathway, thereby up-regulating the expression of browning genes [43].
Despite compelling mechanistic evidence from rodent studies demonstrating the anti-obesity function of RSV [35–37, 40–43]. However, findings from human trials remain controversial [44]. Have reported that RSV supplementation exerts no significant effects on body weight, body composition, or metabolic parameters in the treatment of obesity [45]. These discrepancies can be attributed to several factors. Differences in administration routes can affect absorption. The oral bioavailability of RSV is extremely low, with studies indicating that its absorption is rate-limited by human intestinal Caco-2 cells [46]. Nevertheless, the combination of physical activity and oral RSV supplementation (250 mg/day) for three months has been shown to improve total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), conferring beneficial effects in the management of metabolic syndrome [47]. The effects of RSV also vary depending on dosage and population characteristics. The optimal effective dose for human supplementation remains to be established. Short-term (4-week) high-dose RSV supplementation (500 mg/day) in obese men demonstrated no significant differences in total body weight, lean body mass, total fat mass, or visceral and abdominal subcutaneous adipose tissue volumes [48]. Conversely, another study found that short-term (30-day) RSV supplementation (150 mg/day) enhanced the caloric restriction effects on energy metabolism and metabolic profiles in obese individuals, increasing intramyocellular lipid levels while reducing intrahepatic lipid content [49]. Furthermore, a 12-week RSV supplementation (75 mg/day) yielded no beneficial metabolic effects in non-obese, postmenopausal women with normal glucose tolerance [50].
In summary, the RSV-SIRT axis acts directly on host tissues and indirectly via microbiota-derived metabolites to counteract obesity in rodents (Table 1), providing multiple theoretical bases for potential obesity therapies in humans. However, in practical applications, the dosage of RSV in humans is lower than in rodents, and high doses may induce adverse effects [51]. Therefore, identifying appropriate dosages and delivery strategies represents a critical breakthrough required for the successful clinical application of RSV in human obesity management.
Mechanisms of RSV-targeted SIRTs in the regulation of obesity.
| RSV-targeted SIRTS | Mechanisms of action | Regulation of substrates and pathways | Roles in diseases | Research objects | References |
|---|---|---|---|---|---|
| SIRT1 | ↑SIRT1↑FOXO1↑ATGL↑Lipolysis↓Lipid accumulation | RSV activates SIRT1, restores the expression of FOXO1 and ATGL, and thereby reverses fat accumulation. | Alleviates obesity by increasing lipid accumulation. | Male Sprague-Dawley rats and 3T3-L1 preadipocytes | [34] |
| SIRT1 | ↑SIRT1↑PGC-1α↑NFR-1/PPAR/PXR/PAR↑TFAM/UCP1 | RSV activates SIRT1, increases the level of UCP protein expression and TFAM, thereby enhancing energy expenditure. | Alleviates obesity by enhancing energy expenditure and thermogenesis. | Rats | [40] |
| SIRT1 | ↑SIRT1↓Akt2↓mTORC1↓p-mTOR↓p-S6K1↓Adipose inflammation | RSV inhibited adipose inflammation by activating the SIRT1/Akt2/mTOR/S6K1 pathway. | Alleviates obesity by inhibiting lipid accumulation. | Mice | [35] |
| SIRT1 | ↑4-HPA↑SIRT1↑PGC-1α/PPARγ/UCP1↑Mitochondrial biogenesis↑Browning of white adipocytes↓Lipogenesis | RSV is metabolized by the gut microbiota to produce 4-HPA and related metabolites, which activate the expression of SIRT1 pathway genes. | Inhibits obesity through gut microbiota-derived metabolic pathways. | Male C57BL/6J mice | [43] |
4-HPA: 4-hydroxyphenylacetic acid; Akt2: protein kinase B 2; ATGL: adipose triglyceride lipase; FOXO1: forkhead box O1; mTORC1: mechanistic target of rapamycin complex 1; NFR-1: nuclear factor related-1; PAR: poly (ADP-ribose); PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; p-mTOR: phosphorylated mTOR; PPAR: peroxisome proliferator-activated receptor; p-S6K1: phosphorylated ribosomal protein S6 kinase beta-1; PXR: pregnane X receptor; RSV: resveratrol; SIRT: sirtuin; TFAM: mitochondrial transcription factor A; UCP: uncoupling protein.
DM represents a significant global health challenge characterized by the destruction of blood glucose homeostasis, which is finally manifested as hyperglycemia. DM can be classified into type 1 and type 2 diabetes [52]. Type 1 diabetes is resulted by deficiency of insulin secretion. Type 2 diabetes is caused by a reduction of insulin action (insulin resistance) with progressive loss of β-cell function, often involving relative insulin deficiency and typically disruption of glucose-dependent insulin secretion [53]. The treatment of DM focuses on decreasing insulin resistance, improving insulin sensitivity, enhancing insulin secretion, and maintaining glucose homeostasis [54].
As an antioxidant and anti-inflammatory polyphenol, RSV shows glucose metabolism regulation function [26]. Existing studies have proved that its effect on DM involves the activation of SIRT proteins, particularly SIRT1 and SIRT3 [55–58]. For example, RSV could reverse ethanol-induced β-cell dysfunction through the SIRT1-UCP2 axis [55]. Recent research reveals that exercise can reduce the aging of β-cells in type 2 DM through the SIRT1/mTOR pathway [56], indicating the application possibility of RSV in simulating exercise effects. It has been established that the RSV-SIRT axis repressed oxidative stress-induced hyperglycemia and cytokine toxicity via deacetylating FOXO1 and the nuclear factor kappa-B (NF-κB) subunit p65 on β-cells, respectively [59]. In high glucose-induced human umbilical vein endothelial cells, RSV restores the anti-glycation defense system through SIRT1 to prevent cell damage [60]. It is notable that SIRT3 indirectly affects glucose metabolism by maintaining mitochondrial functional stability, and RSV could enhance mitochondrial respiration rates and ATP production in the diabetic rat heart via activation of SIRT3, leading to the deacetylation of TFAM [58].
Surprisingly, the RSV-SIRT axis also plays an important role in managing diabetic complications. For instance, RSV has been shown to reduce the symptoms of peripheral neuropathy in diabetic mouse models [61, 62]. Another research indicates that RSV can prevent renal cell apoptosis and oxidative stress, which are the main causes of renal damage in diabetic nephropathy, via activation of AMPK-SIRT1-PGC-1α and the consequent effects on its target molecules, peroxisome proliferator-activated receptor (PPAR)-1α and the phosphatidylinositol 3-kinase (PI3K)-Akt-pFOXO3a pathway [57]. The combination of RSV with hypoglycemic drugs has demonstrated synergistic effects in preclinical studies, improving glucose dysregulation, insulin sensitivity, and metabolic syndrome-related indicators [63, 64]. This combination strategy not only enhances the efficacy of a single drug, but also may reduce the side effects of long-term medication through a multi-target mechanism.
Although RSV/SIRT-targeted strategies have become an active research area and show promise as adjunctive therapies, they are not yet incorporated into routine clinical practice. A randomized controlled trial revealed that 24 weeks of supplementation of RSV resulted in a significant reduction in glycosylated hemoglobin A1c (HbA1c) (6.31%), fasting insulin (9.96%), homeostatic model assessment of insulin resistance (HOMA-IR) (17.96%), high-sensitivity C-reactive protein (hs-CRP) (13.12%), IL-6 (13.27%), and microalbuminuria (13.48%) (p < 0.05) compared to the control. The comparison of TC (+1.31%), triglyceride (−3.59%), low-density lipoprotein cholesterol (LDL-C) (2.07%), and HDL-C (1.10%) levels between RSV and the control group was non-significant [65]. The tolerance of RSV as a clinical drug in diabetes treatment trials is temporarily lacking in long-term trials, but no obvious adverse traits have been observed for whether this will endanger human health.
Collectively, the RSV-SIRT axis, primarily through SIRT1 and SIRT3 activation, represents a promising therapeutic target for DM by targeting multiple pathological mechanisms (Table 2). Furthermore, the axis also shows significant potential in managing diabetic complications and synergistic therapies with existing hypoglycemic agents [61–64]. However, there are also some unstable results and problems, such as the effects of RSV in some studies are contradictory or vague [65], and the utilization rate of RSV is low. In order to solve these problems, long-term trials of the therapeutic effect of RSV in diabetes will also become the focus of future research. But this does not affect the important role of the RSV-SIRT axis in diabetes. We believe that RSV will be more widely used as a diabetes drug after further trials.
Mechanisms of RSV-targeted SIRTs in the regulation of type 2 diabetes mellitus.
| RSV-targeted SIRTS | Mechanisms of action | Regulation of substrates and pathways | Roles in diseases | Research objects | References |
|---|---|---|---|---|---|
| SIRT1 | ↑NAD+/NADH↓UCP2 | RSV, increasing NAD/NADH ratio and further regulating the SIRT1/UCP2 axis, protects β-cells against ethanol-induced dysfunction. | Protects β-cell function under ethanol exposure. | Rat insulinoma INS-1 cells | [55] |
| SIRT1 | ↓FOXO1↓NF-κB | SIRT1 via deacetylating FOXO1 and the NF-κB subunit p65 represses oxidative stress-induced hyperglycemia. | Overexpression of SIRT1 prevents cytokine toxicity and maintains normal insulin-secreting responses to glucose. | RINm5F cells and rat islets | [59] |
| SIRT1 | ↑AMPK↑PGC-1α↑SOD1↑SOD2 | RSV treatment increased the AMPK and SIRT1 and decreased PI3K-Akt signaling to improve diabetes-induced renal damage. | Reduce renal cell death and oxidative stress induced by type 2 diabetes mellitus. | C57BLKS/J db/m and db/db mice | [57] |
| SIRT3 | ↑SIRT3↑TFAM↑ATP↓TGF-β/Smad | RSV activates SIRT3, SIRT3 indirectly affects glucose metabolism by decreasing the acetylation status of TFAM, maintaining the stability of mitochondrial function. | Regulates the acetylation status of TFAM and preserves the mitochondrial function along with cellular size in the diabetic rat heart. | Rat-cardiomyoblast cells (H9C2) | [58] |
Akt: protein kinase B; AMPK: AMP-activated protein kinase; FOXO1: forkhead box O1; NADH: nicotinamide adenine dinucleotide; NF-κB: nuclear factor kappa-B; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PI3K: phosphatidylinositol 3-kinase; RSV: resveratrol; SIRT: sirtuin; Smad: Sma- and mad-related protein; SOD: superoxide dismutase; TFAM: mitochondrial transcription factor A; TGF-β: transforming growth factor-β; UCP: uncoupling protein.
MASLD constitutes a spectrum of pathologically interconnected and progressively worsening conditions, including hepatic steatosis, fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) [66, 67]. The nomenclature of MASLD has evolved from non-alcoholic fatty liver disease (NAFLD), reflecting a paradigm shift from an exclusionary diagnosis to an affirmative, etiology-based framework. This change emphasizes the central role of metabolic dysfunction in disease pathogenesis while reducing stigma associated with the term “fatty” [66]. The MASLD commences with excessive accumulation of triglycerides within hepatocytes, termed simple hepatic steatosis. When steatosis is accompanied by hepatocyte injury and inflammation, it progresses to steatohepatitis, marking a critical transition into an active inflammatory and necrotic phase that is pivotal for the development of fibrosis [68]. Persistent liver injury activates hepatic stellate cells (HSCs), prompting their transformation into myofibroblasts, which synthesize and deposit excessive extracellular matrix, forming fibrous scars [69, 70]. The end-stage result of hepatic fibrosis is cirrhosis, characterized by diffuse fibrous septa formation and regenerative nodules of parenchyma, leading to severe architectural distortion and significant functional decline [71]. The cirrhotic microenvironment promotes hepatocyte dedifferentiation, genomic instability, and creates a fertile ground for tumorigenesis, resulting in the most severe outcomes of chronic liver disease, HCC [72].
RSV multi-targetedly intervenes in various stages of the aforementioned MASLD by modulating SIRTs, particularly SIRT1, SIRT3, and SIRT6, thereby significantly reducing serum triglyceride levels [73, 74]. It has been reported that RSV activates SIRT1, which downregulates the activity of lipogenic transcription factors such as sterol regulatory element-binding proteins (SREBPs), while promoting the deacetylation and activation of PGC-1α [75]. This dual action reduces hepatic de novo lipogenesis and enhances mitochondrial fatty acid β-oxidation, effectively alleviating hepatic steatosis [76]. Through SIRT1-mediated mechanisms, RSV improves insulin signaling and upregulates the expression of the long-form leptin receptor, enhancing leptin sensitivity and helping to correct systemic and hepatic metabolic disturbances [77].
It has been established that RSV could inhibit hepatic fibrosis and cirrhosis progression via modulating HSC fate, regulating core fibrogenic signaling pathways, or combating cellular senescence and inflammation [78]. By activating SIRT1, RSV modulates the PPARγ/SIRT1 balance, helping to maintain HSCs in a quiescent state and inhibiting their activation. Concurrently, it suppresses the excessive proliferation of activated HSCs via crosstalk such as the SIRT1-signal transducer and activator of transcription 3 (STAT3) pathway [79]. RSV directly inhibits the key pro-fibrogenic transforming growth factor-β (TGF-β)/Sma- and mad-related protein (Smad) signaling pathway and attenuates chronic inflammation driving fibrosis by modulating the SIRT1/NF-κB axis [80]. Through SIRT1-mediated deacetylation of heterogeneous nuclear ribonucleoprotein U (HnRNP U), RSV suppresses p53-related cellular senescence and NOD-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome activation, thereby ameliorating the senescence-associated secretory phenotype and inflammatory environment linked to fibrosis [81]. Through the aforementioned anti-fibrotic and metabolic-improving actions, RSV may help delay the progression of cirrhotic architectural distortion.
In addition, RSV is able to suppress HCC progression through inhibiting oncogenic signaling, maintaining genomic and mitochondrial stability, and remodeling the tumor immune microenvironment [82]. It is indicated that RSV, via SIRT1-mediated deacetylation of β-catenin, inhibits its nuclear translocation and transcriptional activity, thereby attenuating the oncogenic effects of the Wnt/β-catenin pathway and limiting cancer stem cell-like properties in HCC. By activating SIRT6, RSV helps suppress long interspersed elements-1 (LINE-1) retrotransposition activity and maintain chromatin stability [83]. Through activation of SIRT3, it enhances mitochondrial oxidative phosphorylation, maintains mitochondrial DNA stability (via deacetylating TFAM), and reduces reactive oxygen species (ROS) production, thereby counteracting metabolic reprogramming and apoptosis resistance in tumor cells [84, 85]. RSV can also influence the polarization of tumor-associated macrophages and regulate immunometabolic networks within the tumor, potentially improving anti-tumor immune responses, although direct evidence in MASLD-related HCC is currently limited [86].
According to current clinical practice guidelines for the management of MASLD, primary interventions remain centered on lifestyle modification, encompassing dietary optimization and regular physical activity [87]. Among dietary patterns, the Mediterranean diet has been demonstrated to significantly reduce the risk of MASLD [88]. For individuals with obesity or advanced disease, bariatric surgery and emerging pharmacological agents, including PPAR, farnesoid X receptor (FXR), and thyroid hormone receptor-beta (THR-β) agonists, have entered clinical practice or late-stage trials, showing considerable therapeutic potential [89]. In this context, RSV has exhibited hepatoprotective effects in preclinical models of MASLD [90–92]. However, despite robust evidence from animal studies, the clinical translation of RSV remains constrained, and further investigation is required to establish its efficacy and safety profile in humans [74]. Collectively, MASLD progression is driven by lipotoxicity, oxidative stress, mitochondrial dysfunction, and fibrogenesis. By activating SIRTs, RSV confers multi-stage protection and represents a promising holistic therapy (Table 3).
The role of the RSV-SIRT axis in the treatment of MASLD.
| RSV-targeted SIRTS | Mechanisms of action | Regulation of substrates and pathways | Roles in diseases | Research objects | References |
|---|---|---|---|---|---|
| Not applicable | ↑Thrombospondin-2 as a predictive biomarker | Thrombospondin-2 levels correlate with HCC risk after HCV cure. | Predicts HCC occurrence post-DAA treatment. | HCV-infected patients treated with DAAs | [72] |
| SIRT1 | ↑SIRT1↓Lipogenesis↓Inflammation | RSV activates SIRT1, which decreases lipogenesis and inflammation. | Alleviates hepatic steatosis in obese mice. | High-fat diet-fed mice | [73] |
| SIRT1 | ↑SIRT1↓TGF-β/Smad signaling | SIRT1 activation inhibits TGF-β/Smad pathway. | Ameliorates liver fibrosis. | Rhesus monkeys with liver fibrosis; aged rats with liver fibrosis | [71] |
| SIRT1 | ↑SIRT1↓p53-related senescence↓NLRP3 inflammation | Deacetylation of HnRNP U by SIRT1 inhibits cellular senescence and suppresses NLRP3 inflammasome activation in liver fibrosis. | Contributes to the amelioration of liver fibrosis. | Aged rats with liver fibrosis | [81] |
| SIRT1-independent | ↑OBRb expression, ↑Leptin sensitivity | RSV increases long-form OBRb content, enhancing leptin-induced STAT3 phosphorylation and reducing triglyceride accumulation. | Improves hepatic steatosis by restoring leptin signaling. | Palmitate-induced steatotic HepG2 cells | [77] |
| SIRT1 | ↑SIRT1 activity↓STAT3 phosphorylation↓HSC proliferation | Pterostilbene (RSV analog) inhibits excessive proliferation of activated HSCs through crosstalk between SIRT1 and STAT3 pathways. | Alleviates liver fibrosis by reducing HSC activation. | Activated HSCs | [79] |
| SIRT1 | ↑SIRT1, regulation of SREBPs↓Lipid synthesis | SIRT1 regulates SREBPs to control lipid metabolism. | Contributes to the improvement of NAFLD and metabolic syndrome. | Palmitate-induced steatotic HepG2 cells | [77] |
| SIRT1 | ↑SIRT1, deacetylation of PGC-1α↑Mitochondrial fatty acid oxidation | SIRT1-mediated deacetylation of PGC-1α improves mitochondrial fatty acid oxidation. | Alleviates hepatic steatosis in type 2 diabetes. | Animal model of type 2 diabetes mellitus with hepatic steatosis | [75] |
DAAs: direct-acting antivirals; HCC: hepatocellular carcinoma; HCV: hepatitis C virus; HnRNP U: heterogeneous nuclear ribonucleoprotein U; HSCs: hepatic stellate cells; MASLD: metabolic dysfunction-associated steatotic liver disease; NAFLD: non-alcoholic fatty liver disease; NLRP3: NOD-like receptor family, pyrin domain-containing 3; OBRb: leptin receptor isoform b; p53: tumor protein p53; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; RSV: resveratrol; SIRT: sirtuin; SREBPs: sterol regulatory element-binding proteins; STAT3: signal transducer and activator of transcription 3; TGF-β: transforming growth factor-β.
CVDs are the leading cause of global mortality and a major contributor to disability, encompassing various pathological conditions such as metabolic cardiomyopathies, heart failure, ischemic heart disease, hypertension, and atherosclerosis [93]. Current studies suggest that oxidative stress plays a pivotal role in CVD pathogenesis. A decline in endogenous antioxidant enzyme activity elevates ROS levels, promoting the oxidation of LDL-C. The oxidized LDL accumulates within vascular walls, initiating plaque formation, thrombosis, and ultimately vascular occlusion [94, 95]. Additionally, the pathogenesis of CVDs may also be driven by abnormal metabolism of vascular smooth muscle cells, leading to inflammatory responses that facilitate vascular constriction, occlusion, or even rupture [96].
RSV has increasingly attracted attention for its cardioprotective potential [97–99]. Extensive in vivo and in vitro studies have demonstrated that RSV markedly suppresses the expression of inflammatory mediators and their receptors in vascular tissues [100] and alleviates myocardial injury by reducing oxidative stress and iron overload [101]. Notably, the SIRT family may represent an important target underlying the biological effects of RSV. For instance, SIRT1 regulates apoptosis via acetylation of p53 and FOXO1, while RSV enhances SIRT1 deacetylase activity, thereby suppressing pro-apoptotic signaling and improving cardiac function [102, 103]. Moreover, RSV restores SIRT1 expression suppressed under transverse aortic constriction and inhibits the TGF-β1/p-Smad3 pathway, thereby reducing myocardial hypertrophy and fibrosis [104]. Additional studies indicated that in SIRT1-knockout models, RSV failed to maintain mitochondrial bioenergetics in diabetic cardiomyopathy mice [105] and could not preserve antioxidant defense mechanisms [106]. These findings collectively underscore that SIRT1 activity plays a crucial role in mediating the antioxidant and cardioprotective effects of RSV (Table 4).
Mechanisms of RSV-targeted SIRT1 in the treatment of CVDs.
| RSV-targeted SIRTS | Mechanisms of action | Regulation of substrates and pathways | Roles in diseases | Research objects | References |
|---|---|---|---|---|---|
| SIRT1 | ↑SIRT1↓FOXO1 acetylation↓Bim | RSV enhances SIRT1 deacetylase activity, leading to decreased acetylation of FOXO1 and subsequent downregulation of the pro-apoptotic protein Bim. | Inhibits the apoptosis of myocardial cells and protects against cardiac aging and age-related cardiac dysfunction. | Male senescence-accelerated mice prone mice | [102] |
| SIRT1 | ↑SIRT1↓p53 acetylation↓Ferroptosis-related genes↓Lipid peroxidation | RSV activates SIRT1 deacetylase activity, thereby inhibiting cardiomyocyte ferroptosis and reducing lipid peroxidation stress. | Attenuates cardiomyocyte ferroptosis and decelerates heart failure progression. | C57BL/6 mice;Human-induced pluripotent stem cell-derived cardiomyocytes | [103] |
| SIRT1 | ↑SIRT1↓TGF-β1↓p-Smad3 | RSV restores SIRT1 function suppressed by pressure overload, thereby suppressing TGF-β1/Smad3-mediated programs of cardiac hypertrophy and fibrosis. | Attenuates pressure overload-induced cardiac hypertrophy and fibrosis. | Rat;NCMs | [104] |
| SIRT1 | ↑SIRT1↓PGC-1α/NRF-1/NRF-2 /TFAM↑Mitochondrial↑ATP/↓ROS | RSV activates SIRT1 to deacetylate PGC-1α and improve mitochondrial function in diabetic cardiomyopathy. | Alleviates cardiac dysfunction in diabetic cardiomyopathy by preserving mitochondrial function and energy metabolism. | SIRT1flox5-6/flox5-6;Myh6-Cre+ transgenic mice;H9c2 cardiomyoblast cell;H9c2 embryonic rat heart-derived cell | [105] |
| SIRT1 | ↑SIRT1↑NRF2↑Antioxidant gene↓ROS | RSV synergizes with FGF1 to activate SIRT1-NRF2 signaling and enhance antioxidant defense. | Attenuates doxorubicin-induced cardiotoxicity by reducing oxidative stress and improving cardiac function. | C57BL/6J male mice;H9c2 cardiomyoblast cell | [106] |
Bim: Bcl-2-interacting mediator of cell death; CVDs: cardiovascular diseases; FGF1: fibroblast growth factor 1; FOXO1: forkhead box O1; NCMs: neonatal rat cardiomyocytes; NRF2: nuclear factor erythroid 2-related factor 2; p53: tumor protein p53; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; p-Smad3: phosphorylated Sma- and mad-related protein 3; ROS: reactive oxygen species; RSV: resveratrol; SIRT: sirtuin; TFAM: mitochondrial transcription factor A; TGF-β: transforming growth factor-β.
Although current studies have provided new insights into the treatment of CVDs, it should be noted that the majority of available evidence is derived from preclinical research, and corresponding clinical data remain limited and, in some cases, inconsistent [97]. For instance, several human interventional studies have suggested that RSV supplementation may improve endothelial function, reduce blood pressure, and modulate lipid profiles and inflammatory markers in individuals with cardiovascular risk factors [107, 108]. However, other studies have failed to demonstrate significant benefits in major cardiovascular outcomes [109]. These discrepancies indicate that the clinical efficacy of RSV may depend on factors such as dosage, treatment duration, and patient characteristics. Furthermore, recent discussions have reported the results of the role of polyphenol/antioxidant intervention measures in large-scale trials, but they have failed to provide strong evidence for reducing the risk of CVDs [110, 111]. Taken together, these findings highlight the ongoing challenges in translating promising preclinical results into meaningful clinical benefits. Safety considerations are also critical for clinical translation. RSV may be generally regarded as safe, but its potential effects on bleeding risk, interactions with antiplatelet or anticoagulant therapies, and its influence on cardiac electrophysiology have not been fully elucidated [112–114]. Therefore, the safety and efficacy of RSV in high-risk CVD populations require more systematic evaluation. Future studies should focus on well-designed, large-scale clinical trials targeting diverse patient populations to better clarify the therapeutic potential and safety profile of RSV in CVDs.
In conclusion, mounting studies have established that RSV functions as a pivotal regulator of the SIRT family, particularly SIRT1, SIRT3, and SIRT6, orchestrating the restoration of metabolic homeostasis across multiple organ systems. This polyphenol significantly ameliorates hepatic steatosis and fibrosis by upregulating mitochondrial biogenesis via PGC-1α deacetylation and dampening pro-inflammatory cascades through the inhibition of NF-κB and NLRP3 pathways. Furthermore, RSV augments insulin sensitivity and safeguards pancreatic β-cell integrity by counteracting oxidative stress, while concurrently stimulating adipose tissue thermogenesis and mitigating cardiovascular risk factors. Therefore, its therapeutic application constitutes a promising strategy for managing intricate metabolic syndromes (Figure 1).
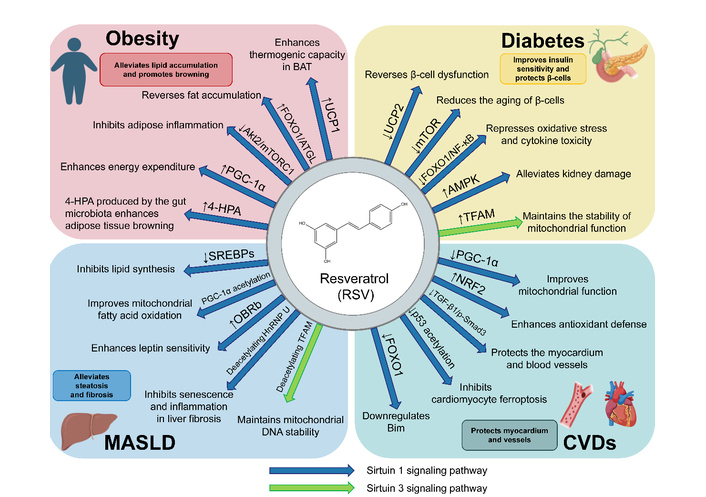
Resveratrol (RSV) improves obesity, diabetes, MASLD, and CVDs through activating the SIRT1/3 Pathways. RSV regulates energy metabolism, lipid catabolism, antioxidant defense, and mitochondrial function to improve tissue health and mitigate pathology in obesity, diabetes, MASLD, and CVDs, acting on key factors such as FOXO1, PGC-1α, AMPK, and TFAM. 4-HPA: 4-hydroxyphenylacetic acid; Akt2: protein kinase B 2; AMPK: AMP-activated protein kinase; ATGL: adipose triglyceride lipase; BAT: brown adipose tissue; Bim: Bcl-2-interacting mediator of cell death; CVDs: cardiovascular diseases; FOXO1: forkhead box O1; HnRNP U: heterogeneous nuclear ribonucleoprotein U; MASLD: metabolic dysfunction-associated steatotic liver disease; mTORC1: mechanistic target of rapamycin complex 1; NF-κB: nuclear factor kappa-B; NRF2: nuclear factor erythroid 2-related factor 2; OBRb: leptin receptor isoform b; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; SREBPs: sterol regulatory element-binding proteins; TFAM: mitochondrial transcription factor A; TGF-β1/p-Smad3: transforming growth factor-β1/phosphorylated Sma- and mad-related protein 3; UCP: uncoupling protein.
Nevertheless, clinical implementation is currently impeded by constraints regarding bioavailability and dose-dependent variability in human subjects. Future investigations should prioritize the engineering of novel delivery systems and the evaluation of synergistic combinations with other bioactive compounds to optimize their pharmacokinetic profile and clinical efficacy. Strategic advances should also encompass systematic head-to-head comparisons of natural polyphenols, NAD+ precursors, and next-generation synthetic modulators to identify the most promising strategies for metabolic disease intervention. Despite existing challenges, strategic advances in formulation science, combination regimens, and mechanistic understanding position RSV and SIRT-targeted interventions as compelling candidates for the future prevention and treatment of metabolic dysfunction-associated diseases.
BAT: brown adipose tissue
COX2: cytochrome c oxidase subunit 2
CVDs: cardiovascular diseases
DM: diabetes mellitus
FOXO1: forkhead box O1
FXR: farnesoid X receptor
HbA1c: hemoglobin A1c
HCC: hepatocellular carcinoma
HDL-C: high-density lipoprotein cholesterol
HOMA-IR: homeostatic model assessment of insulin resistance
HSCs: hepatic stellate cells
LDL-C: low-density lipoprotein cholesterol
MASLD: metabolic dysfunction-associated steatotic liver disease
NF-κB: nuclear factor kappa-B
NRF2: nuclear factor erythroid 2-related factor 2
PPAR: peroxisome proliferator-activated receptor
ROS: reactive oxygen species
RSV: resveratrol
SIRT: sirtuin
Smad: Sma- and Mad-related protein
TC: total cholesterol
TFAM: mitochondrial transcription factor A
TGF-β: transforming growth factor-β
UCP1: uncoupling protein 1
WAT: white adipose tissue
JY: Data curation, Formal analysis, Investigation, Methodology, Writing—original draft, Writing—review & editing. SC: Data curation, Formal analysis, Investigation, Methodology, Writing—original draft, Writing—review & editing. TZ: Data curation, Formal analysis, Investigation, Methodology, Writing—original draft, Writing—review & editing. YZ: Data curation, Formal analysis, Investigation, Methodology, Writing—original draft, Writing—review & editing. ZL: Data curation, Formal analysis, Investigation, Methodology, Writing—original draft, Writing—review & editing. HL: Conceptualization, Investigation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by a grant from the Natural Science Foundation of Shandong Province, China [No. ZR2025MS387]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 3904
Download: 44
Times Cited: 0
