 Review
Review

Affiliation:
Faculté de Santé, Sorbonne Université, 75013 Paris, France
Email: valerie.lannoy@laposte.net
ORCID: https://orcid.org/0009-0005-6760-4233
Explor Endocr Metab Dis. 2026;3:101464 DOI: https://doi.org/10.37349/eemd.2026.101464
Received: January 17, 2026 Accepted: March 05, 2026 Published: March 18, 2026
Academic Editor: Amedeo Lonardo, Azienda Ospedaliero-Universitaria di Modena, Italy
The hypothesis that early-life antibiotic exposure predisposes to obesity has, over the past decade, gained substantial traction across both biomedical literature and public discourse. Its appeal lies in a seemingly coherent mechanistic framework: disruption of the developing intestinal microbiota is presumed to induce long-lasting alterations in metabolic homeostasis, thereby promoting increased adiposity. However, reported effect sizes are consistently modest, with odds ratios typically ranging from 1.1 to 1.3, values that approach the limits of residual confounding and statistical imprecision. Studies incorporating rigorous adjustment for familial environment, socioeconomic status, and dietary patterns observe a complete loss of the association, underscoring the dominant influence of these confounders. This review revisits the conceptual emergence and sustained prominence of the antibiotic-obesity paradigm, positioning it as a case study in the amplification of weak epidemiological signals through mechanistic plausibility. Notwithstanding its methodological limitations, the hypothesis has exerted a constructive influence by fostering more judicious antibiotic use and stimulating renewed investigation into host-microbiota interactions within the context of metabolic disease.
Obesity is now recognized as a chronic, relapsing, multifactorial disease characterized by excess adiposity associated with increased cardiometabolic risk, systemic inflammation, and endocrine dysfunction. Its global prevalence has risen dramatically over the past decades, now affecting hundreds of millions of individuals worldwide, including an increasing proportion of children and adolescents [1]. Rather than representing a simple imbalance between caloric intake and expenditure, obesity is increasingly conceptualized as a complex systems disease involving host genetics, immune maturation, neuroendocrine regulation, environmental exposures, and microbial ecosystem dynamics [2]. Among environmental factors, early-life antibiotic exposure has emerged as a potential contributor to long-term metabolic programming. While these associations remain subject to confounding, they have fueled intense interest in the role of the gut microbiome as a potential mechanistic intermediary between environmental exposures and metabolic disease risk.
A major conceptual turning point in the microbiome-obesity field came from transplantation experiments in germ-free animal models. In a landmark study, fecal microbiota from monozygotic human twin pairs discordant for obesity were transplanted into germ-free mice. Mice colonized with microbiota from the obese twin developed increased adiposity compared with those receiving microbiota from the lean twin, despite identical diet exposure [3]. This study provided compelling experimental evidence supporting a causal contribution of microbiota composition and function to host metabolic phenotype. However, human population studies have complicated the narrative. Early work suggested that obesity was associated with reduced gut microbial diversity. Large-scale metagenomic and population-level analyses indicate that diversity differences between obese and lean individuals are often modest and frequently fall within the range of normal inter-individual variation observed among healthy populations [4]. These findings suggest that diversity alone is unlikely to represent a robust biomarker or primary driver of metabolic disease.
Similarly, the previously dominant Firmicutes/Bacteroidetes—the two main phyla among gut microbiota—ratio hypothesis has progressively lost explanatory power. While early sequencing studies suggested directional shifts associated with obesity, subsequent meta-analyses failed to demonstrate reproducible patterns across cohorts, geographic regions, and sequencing platforms [5]. The field is therefore shifting toward function-centric frameworks, focusing on microbial metabolic output rather than taxonomic composition. In this context, microbial metabolites, particularly short-chain fatty acids (SCFAs), including acetate, propionate, and butyrate, have emerged as candidate mediators linking microbiome activity to host metabolic regulation. SCFAs influence energy harvest, gut barrier function, immune signaling, and enteroendocrine hormone secretion [6]. Yet their role remains mechanistically complex. While SCFAs can enhance insulin sensitivity and promote anti-inflammatory signaling, they also represent a quantifiable caloric substrate, and elevated SCFA levels have been reported in some obese populations [7, 8].
In the early 2010s, a series of observational studies that will be discussed in the current review ignited a striking claim: antibiotic exposure during infancy could increase the risk of obesity in later life [9–11]. The idea quickly gained traction, both because it resonated with the zeitgeist of microbiome science and because it carried a moral undertone, suggesting that the life-saving tools of modern medicine might, in excess, turn against us. The hypothesis rested on a chain of logic: antibiotics disrupt the gut microbiota; the gut microbiota regulates metabolism; therefore, antibiotics must disturb metabolic homeostasis. This syllogism was elegant, emotionally charged, and almost impossible to resist. But elegance, as history repeatedly shows, is no guarantee of truth [11]. Between antibiotic exposure and obesity lies a tangle of confounders—genetics, socioeconomic status, maternal weight, infant feeding practices, and healthcare behaviors. To conflate the correlation with causation in this context is to overlook the complexity of human development and the multiplicity of pathways that lead to adiposity.
Nonetheless, the concept became a powerful narrative. It echoed long-standing cultural anxieties about overmedication, the “unnaturalness” of modern life, and the loss of microbial balance. The idea that a brief course of antibiotics could permanently alter one’s metabolism offered a story both tragic and moralizing. It makes obesity not a product of diet or the environment, but of modern medicine’s microbial recklessness. The problem, as this review will argue, is that the data never justified the drama. The association, though statistically reproducible, is quantitatively negligible. Once familial confounding variables are properly controlled, the signal vanishes. Yet the hypothesis persists, fueled by mechanistic enthusiasm, media amplification, and the seductive power of the microbiome narrative.
The conceptual seed of the antibiotic-obesity hypothesis was planted decades before its epidemiological flowering. Since the 1950s, low-dose antibiotics have been used in agriculture as growth promoters. Subtherapeutic antibiotic feed supplements reliably increase weight gain and feed efficiency in livestock [12]. The extrapolation to humans seemed natural. To put it provocatively, if antibiotics make pigs and chickens fatter, why not children? The analogy was rhetorically compelling—but biologically naive. Livestock growth promotion involves chronic exposure to low-dose antibiotics over months or years, typically in feed, under carefully controlled nutritional and environmental conditions. By contrast, pediatric antibiotic use in immunocompetent patients is rather short and episodic, usually five days per course. To equate the two is to conflate chronic ecological conditioning with transient therapeutic intervention. The cumulative antibiotic exposure of a farm animal outweighs that of a human child by several orders of magnitude. Moreover, the diets, housing, genetics, and microbial ecology of livestock differ radically from those of human infants. The agricultural analogy, while rhetorically powerful, thus collapses upon quantitative scrutiny.
Antibiotics encompass diverse structural classes whose antibacterial activity derives from the selective inhibition of essential microbial processes. β-lactams block peptidoglycan transpeptidation by binding penicillin-binding proteins (PBPs), leading to cell lysis [13]. Glycopeptides such as vancomycin bind termini of cell-wall precursors, preventing cross-linking in Gram-positive bacteria [14]. Protein synthesis inhibitors include aminoglycosides, which induce misreading at the 30S ribosome [15]; tetracyclines, which prevent aminoacyl-tRNA binding at the 30S site [16]; macrolides and lincosamides, which bind the 50S subunit to inhibit peptide elongation [17]; and oxazolidinones, which disrupt initiation-complex formation [18]. Other agents target nucleic-acid metabolism: fluoroquinolones inhibit DNA gyrase and topoisomerase IV [19], while rifamycins block RNA polymerase [20]. Metabolic inhibitors such as trimethoprim–sulfamethoxazole interfere with folate synthesis [21], whereas nitroimidazoles generate reactive intermediates causing DNA strand breaks in anaerobes [22]. All these mechanisms correlate with characteristic toxicities. However, a Canadian clinical study has revealed that the most prevalent side effect of antibiotics is gastrointestinal disorders, including nausea, vomiting, diarrhea, abdominal pain, loss of appetite, and bloating, because they are known to perturb the intestinal microbiota [23].
The modern version of the hypothesis began with a handful of retrospective cohort studies using hospital health records. Trasande et al. [24], analyzing 11,532 children from the Avon Longitudinal Study of Parents and Children, reported that antibiotic exposure before six months of age was associated with an increased body mass index (BMI) at 38 months. The adjusted odds ratio (OR) is 1.22, 95% confidence interval (CI) 1.06–1.40. Bailey et al. [10], using a USA cohort of 64,580 infants, had the highest rate ratio (RR) = 1.16 (95% CI 1.06–1.29) for obesity by age five. The effect sizes were small, but consistent, and thus appeared persuasive. Yet beneath the surface of statistical consistency lay a striking fragility: none of these associations exceeded the conventional threshold of epidemiological credibility (OR or RR > 1.5).
In any other research domain, such small effects would be treated as hypothesis-generating at best, artefactual at worst. Actually, the theory fitted the cultural moment. The 2010s marked the rise of the microbiome as the grand unifying theory of chronic diseases. A mechanistic explanation linking antibiotic exposure, microbial disruption, and obesity fits seamlessly into that paradigm. The story was simple, plausible, and optimistic—it suggested that by preserving or “restoring” our microbes, we might reverse a global epidemic. But the very simplicity that made the idea compelling also made it misleading. When public fascination meets statistical ambiguity, the narrative tends to triumph over nuance.
An OR of 1.1 implies a 10% increase in relative risk. If 10% of unexposed children become obese, the prevalence rises to 11% among the exposed. Such a change is clinically trivial, easily overwhelmed by measurement errors or uncontrolled lifestyle variables. Nevertheless, these small differences generated major headlines. The Journal of the American Medical Association (JAMA) Pediatrics and the International Journal of Obesity published studies with enormous samples and minuscule ORs and RRs. In the hierarchy of epidemiological credibility, these values sit barely above random noise. As Grimes and Schulz [25] mentioned, an RR that does not exceed 2 in a cohort study should not be considered credible, tending to residual confounding and reporting bias. According to clinician epidemiologists, an OR greater than 4 in case-control studies is considered strong support for causation [26]. When effect sizes are small, any residual confounding—no matter how minor—can fully explain the observed association. The power of big data lies in its ability to detect small effects—but that is also its peril. With hundreds of thousands of participants, even trivial differences yield p-values far below 0.001. Statistical significance thus becomes a function of sample size, not effect magnitude. The result is what Ioannidis called “the tyranny of the p-value”: large datasets producing significant but meaningless results [27].
Meta-analyses soon followed the disclosure of the link between early antibiotic exposure and obesity. A systematic review reported the highest OR = 1.24 (95% CI 1.09–1.43) [28]. Miller et al. [29] in 2018 confirmed an almost identical estimate (pooled OR = 1.05; 95% CI 1.00–1.11) across over 500,000 children. Children prone to infections or with delayed immune maturation are more likely to receive antibiotics and to develop later metabolic irregularities [30]. Furthermore, infants who receive more antibiotics often do so because they suffer from recurrent respiratory or ear infections. It is these infections themselves—rather than antibiotic treatments—that may alter feeding patterns, appetite, or growth [30]. If studies do not carefully control for the frequency and severity of infections, it is not methodologically sound to attribute weight changes to antibiotics. For example, overweight and obese children exhibit low-grade inflammation [31], which makes them more susceptible to urinary tract infections [32]. In this context, antibiotic exposure is better interpreted as a marker of underlying vulnerability than as a causal factor. In other words, we are not observing causation, but confounding disguised as robust statistics.
Familial factors are potent predictors of both antibiotic exposure and obesity. Parents with a higher BMI are more likely to have obese children and to seek medical care frequently, leading to more antibiotic prescriptions. Sibling-comparison studies offer a rare window into causality because they automatically control for shared genetics, upbringing, and socioeconomic context. In a nationwide New Zealand cross-sectional study of 132,852 mothers and their children, the crude association between early-life antibiotic exposure and obesity at 4 years (including an apparent dose-response) vanished in sibling and twin fixed-effect models, indicating that shared familial factors, rather than antibiotics per se, explained the association [33]. In a large pediatric network cohort, early antibiotic exposure within the first 6 months of life was not associated with excess weight gain up to age 7 years; in twin pairs discordant for exposure, there was no measurable difference in body weight between the exposed and unexposed twin [34]. Such null results are not trivial: they suggest that the entire effect seen in conventional analyses arises from familial clustering rather than biological causality. In other words, antibiotics “predict” obesity because families that use more antibiotics also share other obesity risk factors—genetic, dietary, or behavioral.
One invoked limitation in the nationwide New Zealand cohort analysis of sibling-controlled studies is an ecological mechanism of intra-household microbial exchange [35]. According to this view, the gut microbiota of cohabiting siblings is so intertwined that any antibiotic-induced disruption in one child would be rapidly normalized through microbial transfer from the other. This concept is supported by evidence from animal studies. Ridaura et al. [3] demonstrated that when mice colonized with twin human “obese” or “lean” microbiota were cohoused, the transfer of microbes between cages prevented the increase in adiposity normally seen in recipients of the obese microbiota. The juxtaposition, however, is misplaced. Co-housed mice engage in coprophagy—a direct, continuous, and bidirectional transfer of intestinal contents that rapidly homogenizes their gut communities within days. Human siblings, by contrast, share only intermittent and environmentally mediated exposure. The microbial overlap observed in families reflects shared ecology rather than fecal exchange. The critical role of the gut microbiota in obesity and its metabolic consequences is further supported by a substantial body of literature [36].
In a comparison of 94 spouse and 83 sibling pairs from the Wisconsin Longitudinal Study, spouses displayed higher microbiota similarities and shared more bacterial taxa than siblings. No difference was detected between siblings and unrelated pairs, and these findings remained after controlling for dietary intake. Interestingly, the effect was specific to couples reporting very close relationships; those with less intimacy showed microbiota profiles indistinguishable from unrelated individuals [37]. It was confirmed in a cohort from the Fiji Islands, where common barriers to bacterial transmission are absent [38]. In this study, the household members who showed the highest levels of strain similarities in their gut microbiomes were the mother-child pairs and the spouses [38]. If early-life antibiotics truly exerted a profound and lasting metabolic disruption, such an insult would not be erased by the mere sharing of toys, towels, and intestinal bacteria between siblings. The claim that a perturbation vanishes through casual microbial exchange tacitly concedes that the effect was never devastating in the first place. The microbiota may be shared, but so is the diet, the parental genome, and the socioeconomic context—each infinitely more influential on weight gain than a bout of amoxicillin.
Ajslev et al. [39], analyzing 28,354 Danish mother-child dyads, found the highest association between early antibiotics and later BMI in children of obese mothers (adjusted OR = 2.59; 95% CI 1.28–5.23), even slightly lower than children of obese mothers who did not receive antibiotics (adjusted OR = 2.85; 95% CI 2.25–3.62). Therefore, maternal weight before pregnancy is among the strongest predictors of offspring BMI. Mothers with higher BMI are more likely to develop gestational diabetes, undergo caesarean delivery, and request antibiotic treatment during infancy—all pathways that inflate the apparent link between antibiotics and obesity. Furthermore, perinatal factors—such as caesarean birth, formula feeding, and early weaning—alter the microbiota and metabolic development independently of antibiotics. Any model ignoring these factors is bound to overestimate antibiotic effects.
Antibiotic exposure is also a social phenomenon. Families with higher healthcare engagement, insurance coverage, or anxiety about infection are more inclined to obtain antibiotic prescriptions [40]. These same families may exhibit different feeding habits, screen time, and dietary environments—all powerful confounders; families who use more antibiotics tend to have other, more relevant risk factors for obesity. Crucially, studies that adjust only crudely for demographic variables tend to report a small association between early-life antibiotic exposure and later obesity (relative risks in the ~1.2 range) [10]. Childhood obesity is a profoundly familial condition. Parental BMI, household diet, and social environment shape a child’s metabolic trajectory far more powerfully than transient antibiotic exposures.
Mechanistic plausibility is the hypothesis’s strongest and weakest pillar. The idea that antibiotics disrupt the gut microbiota, reducing diversity and altering energy harvest, seems consistent with our understanding of host-microbe symbiosis. But evidence supporting a sustained metabolic impact in humans is scarce. Early mouse studies, such as Cho et al. [12], showed that chronic antibiotic exposure in neonatal mice increased adiposity. More precisely, the authors developed a murine experimental model of sub-therapeutic antibiotic treatment (STAT), where mice are exposed at weaning in their drinking water for 7 weeks. STAT did not alter the microbiota richness [12]. Nevertheless, STAT exposure substantially increased the gut SCFAs, supporting mechanisms for the STAT-induced adiposity phenomenon (Figure 1). This observation is surprising, knowing that antibiotic treatment decreases the level of fecal SCFAs in a healthy rat model compared to nontreated rats, confirming the vanishing of the gut bacteria [41].
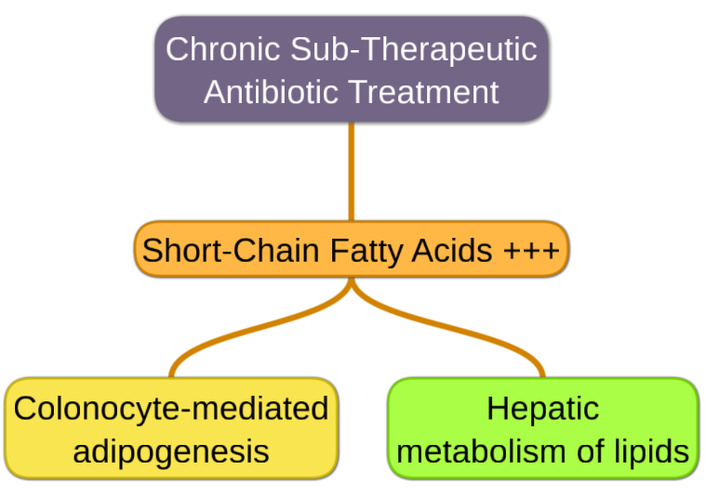
Metabolic mechanisms by which sub-therapeutic antibiotic treatment favors adiposity through intestinal microbiota reshaping.
However, we have to keep in mind that the STAT experimental model implies sublethal doses of antibiotics. Sublethal exposure is increasingly recognized as a potent ecological and metabolic modulator of the gut microbiota, capable of enhancing SCFA. Subinhibitory antibiotic concentrations induce bacterial stress responses, including oxidative stress and translational perturbations, which can favor fermentative metabolism as a means of maintaining redox balance under constrained conditions [42]. Disruption of trophic networks also plays a critical role: the loss of key microbial populations involved in lactate, succinate, or hydrogen utilization can lead to the accumulation and redirection of intermediates toward terminal SCFAs [43]. Importantly, these changes may reflect a shift toward a high-flux but metabolically inefficient ecosystem, in which increased SCFA levels represent an overflow phenotype rather than a marker of optimal symbiotic function. Such observations challenge the prevailing view of SCFAs as uniformly beneficial metabolites and suggest that, in the context of chronic sublethal antibiotic exposure, elevated SCFA production may instead signify stress-induced metabolic rewiring of the gut microbiome.
The team that developed the STAT model then elaborated a murine model of low-dose penicillin (LDP), delivered from birth [44]. Penicillin is a β-lactam antibiotic that exerts its bactericidal effect by binding to PBPs, thereby inhibiting peptidoglycan cross-linking, weakening the bacterial cell wall, and ultimately causing osmotic lysis of actively dividing cells. These mice significantly displayed higher adiposity than those exposed to LDP at weaning. LDP also aggravated the effect of high-fat diet-induced obesity. Growth promotion is transferable to germ-free mice by LDP-selected microbiota, showing its causal role [44]. These findings provided the mechanistic scaffolding for human extrapolation.
These models use antibiotic exposures incomparable to clinical practice—weeks to months of continuous treatment, in genetically uniform animals on fixed diets. A child’s five-day amoxicillin course bears little resemblance to a mouse’s chronic exposure to antibiotic cocktails. Some links in the evidential chain suffer from methodological fragility—from exposure definition to outcome measurement. Most studies rely on prescription records, not verified intake. Parents may not fill prescriptions or may discontinue treatment early. Over-the-counter antibiotic use, common even in regulated countries, is rarely recorded [40]. Such misclassification biases affect estimates toward null or create spurious associations when correlated with family variables. One of the most relevant criteria for causality is the presence of a dose-response gradient. Leong et al. [33] conducted a meta-analysis stratifying by the number of antibiotic courses. Children receiving 1–2, 3–5, or > 6 courses showed nearly identical pooled ORs for obesity in sibling and twin fixed-effects models. No gradient emerged, and without dose dependency, the hypothesis lacks a cornerstone of biological credibility.
Early exposure may refer to antibiotics before 6, 12, or 24 months, depending on the study. The resulting heterogeneity inflates statistical noise. Trasande’s [24] 2013 analysis defined obesity at 38 months, and Bailey’s [10] 2014 study used obesity at age five. Such variability undermines comparability across cohorts and exaggerates apparent reproducibility. Most studies treat antibiotic exposure in the first two years as a uniform variable. Nonetheless, the developing gut microbiome progresses through three distinct stages: a developmental phase (3–14 months), a transitional phase (15–30 months), and a stable phase (31–46 months) [45]. Among all variables, the intake of breast milk—whether exclusively or in combination with other foods—emerged as the strongest determinant of microbiome composition [45]. Lumping these intervals erases temporal resolution, while antibiotic use often reflects prior infection burden, blurring causality altogether.
Longitudinal metagenomic analyses reveal that the infant microbiota, though sensitive to perturbation, is remarkably resilient. Yassour et al. [46] followed 39 children aged 2–36 months across two years and found that antibiotic-induced gut disruptions in microbial diversity normalized within weeks. Their results shed light on the resilient infant microbiota, related to the ability of an ecosystem to equilibrate following perturbations [47]. Also, Yassour et al. [46] did not observe any increase in BMI at the age of three in children repeatedly exposed to antibiotics. Bokulich et al. [48] confirmed similar findings: even after repeated exposures, microbiota composition converged toward a stable adult-like configuration, a diet-dominated profile by age three, parallelizing the dietary transition to solid foods, an important driver for childhood microflora maturation [49]. Alfa-diversity was not decreased in individual children immediately following antibiotic administration. Rather, exposures altered the α-diversity trajectory during the first year of life, but transient effects are indistinguishable between exposed and non-exposed infants at 12 months of age at the operational taxonomic unit (OTU) level [48].
The rhetoric of “microbial loss” exaggerates the causal power of diversity metrics. The microbiota is not a fragile relic of premodern life but a dynamic ecosystem that reorganizes in response to diet and environment. Diet-induced changes in microbiota composition far exceed those induced by short antibiotic courses [50]. A meta-analysis has shown that the difference between the Shannon diversity of obese and nonobese individuals, while significant, is at best around 2% [4]. To elucidate the link between obesity and early antibiotic exposure, Nobel et al. [51] designed an appropriate murine model of early life pulsed antibiotic treatment (PAT), shortly after weaning, to mimic early life use in human children. The amoxicillin PAT showed no effect on body composition [51], which is substantial, knowing that this is the most pediatrically prescribed antibiotic [52]. Similar to penicillin mentioned above, amoxicillin is a broad-spectrum β-lactam that inhibits bacterial cell wall synthesis by binding to PBPs, thereby blocking peptidoglycan cross-linking and leading to bacterial cell lysis.
In a prospective birth cohort study, Chelimo et al. [53] enrolled 6,853 children before birth between 2009 and 2010. Anthropometric measurements, including height and weight, were longitudinally collected until 4.5 years of age. Data on systemic antibiotic dispensations were retrieved from the New Zealand Pharmaceutical Collection. Of the 5,128 children retained for analysis, 95% received at least one antibiotic prescription before the age of 4, and 9% met the criteria for obesity at 4.5 years of age. Exposure to more than nine antibiotic treatments was associated with a 2.4-fold increase in the OR of obesity (95% CI 1.07–5.41), compared with non-exposed children. However, the near-universal exposure rate (95%) in this cohort highlights the need for cautious interpretation when extrapolating microbiota-mediated mechanisms from observational associations.
In 2014, provincial healthcare records were linked to clinical data from a Canadian longitudinal birth cohort study [9]. Infants exposed to systemic antibiotics during the first year of life exhibited a higher prevalence of subsequent overweight compared with non-exposed peers. After multivariate adjustment for birth weight, breastfeeding status, maternal overweight, and additional covariates, this association remained statistically robust in boys (adjusted OR = 5.35; 95% CI 1.94–14.72), but not in girls (adjusted OR = 1.13; 95% CI 0.46–2.81). Sex-stratified analyses revealed a consistent dimorphism across adiposity-related outcomes. At 9 years of age, early antibiotic exposure was associated with increased odds of overweight in boys (adjusted OR = 2.19; 95% CI 1.06–4.54), whereas no significant association was detected in girls (adjusted OR = 1.20; 95% CI 0.53–2.70). Importantly, the unusually broad CI surrounding the strongest association in boys (95% CI 1.94–14.72) signals substantial statistical imprecision, likely reflecting model instability. In the cohort, boys were significantly more exposed to antibiotics during the first year of life (p = 0.03) than girls [9], which was not taken into consideration by the authors.
Preterm neonates are subjected to repeated, often prolonged prophylactic antibiotic courses in neonatal intensive care units [54]. If antibiotics were obesogenic, this group would display clear long-term weight gain. Yet follow-ups show the opposite: preterm children often have lower BMI in later childhood [55]. The most heavily antibiotic-exposed population on Earth thus directly contradicts the hypothesis. Very recently, some stool gut microbiotas were collected from 700 children in the Copenhagen Prospective Studies on Asthma in Childhood 2010 (COPSAC 2010) cohort [56]. In this deeply characterized longitudinal birth cohort, no association between early-life microbiota and BMI in later childhood was observed. It suggests that if such associations exist, they may be influenced by later factors, including lifestyle changes. Finally, from the same COPSAC 2010 cohort, children exposed to antibiotics had similar BMI and body composition between 1 and 6 years of life, in comparison with the unexposed group [57].
Early mouse studies, such as the STAT model, used since 1946 in livestock [58], showed that chronic antibiotic exposure in neonatal mice increased adiposity [12]. Seminal experiments conducted over half a century ago, demonstrating that the growth-promoting effect of STAT vanishes in germ-free chicks, established an early principle: the ability of antibiotics to modulate host growth is contingent upon the presence of a gut microbiota [59]. Recently, a Chinese study has reported that recruited children presented associations between overweight or obese status and veterinary antibiotics in their morning urine samples [60]. It advocates that exposure to certain types of antibiotics, mainly through food and drinking water, is correlated with an increased risk of pediatric obesity.
In line with these observations, a recent cross-sectional case-control study, including 3,730 primary school students, in which 10 antibiotics in urine samples were measured by liquid chromatography-tandem mass spectrometry [61]. Elevated OR of obesity was observed in children exposed to low sulfa-monomethoxine (OR = 1.91; 95% CI 1.05–3.47) or middle levels of tetracycline (OR = 1.96; 95% CI 1.05–3.66), with no association found in any high-level antibiotic exposure groups [61]. While the microbiota was not investigated, these new data suggest that low- to middle-level contacts with certain environmental antibiotics may contribute to the risk of childhood obesity. This outline appears closer to the situation in animal husbandry, namely chronic exposure to low doses, and may represent the missing link in the antibiotic-obesity hypothesis, deserving further exploration.
The rate of increase in overweight and obesity continues to rise [62]. If consumption of antibiotics promoted obesity, global epidemiology should reflect it, but it does not. Antibiotic overuse is rampant in low- and middle-income countries (LMICs), where over-the-counter sales are common, and regulation is minimal. A systematic review has divulged that in many LMICs, 60–80% of antibiotics are obtained without prescription [40]. Yet obesity remains low in these same regions, with childhood obesity rates often below 5% (Figure 2B). If antibiotics were obesogenic, one would expect obesity epidemics where antibiotic access is unregulated. This contradiction suggests that the antibiotic-obesity hypothesis captures not a biological mechanism but a socioeconomic pattern. The theory thus fails the ecological test: its predicted patterns contradict global data.
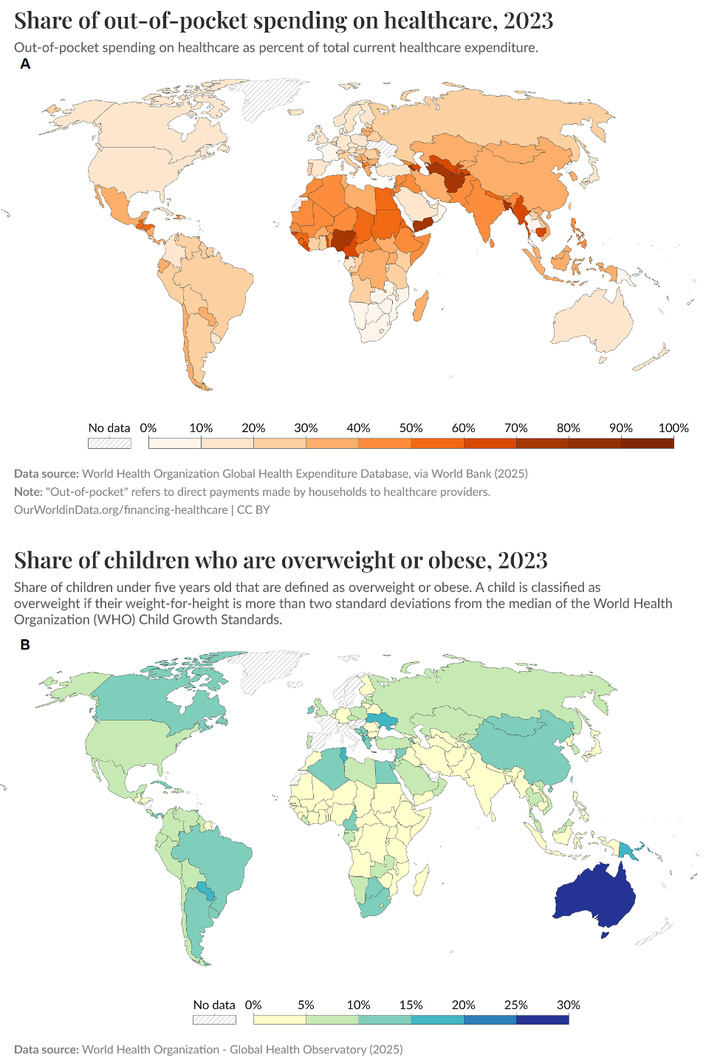
Comparison between the world’s repartitions of poor healthcare systems and childhood overweight or obesity. (A) Global Health Expenditure Database. Adapted from [68]. Accessed December 30, 2025. © 2025 Our World in Data. CC BY. (B) Global Health Observatory. Adapted from [69]. Accessed December 30, 2025. © 2025 Our World in Data. CC BY.
This incoherence arises from “epidemiological parochialism”—the tendency to generalize findings from high-income nations to the world at large [63]. Epidemiology, when disconnected from geography, becomes tautological: it explains local patterns to itself. Several analyses show that non-prescription antibiotic use is common where primary care access, regulation, and surveillance are frail [64] (Figure 2A); therefore, weak health systems are associated with higher antibiotic self-medication (over-the-counter, leftovers) [65]. In many LMICs, antibiotics are not only easily obtainable without prescription, but they are also actively administered by caregivers to infants and young children for common, mostly self-limiting respiratory and febrile illnesses [66]. Recent meta-analyses estimate that roughly one-third to two-fifths of children under five in such settings receive antibiotics without medical oversight [67].
From Figure 2 [68, 69], in countries with massive antibiotic consumption, the true public-health crisis is antimicrobial resistance, not obesity. In sub-Saharan Africa and South Asia, widespread antibiotic access has fueled resistant pathogens [70].
The 2010s witnessed a transformation in biomedical discourse: the gut microbiota became the new genetic code—a master key promising to explain every chronic condition. Within this intellectual climate, the antibiotic-obesity hypothesis required little evidence to be embraced. It satisfied an emotional need to locate modern pathology in modern disruption. The narrative resonated with cultural archetypes: a fall from microbial grace brought about by technological excess. Antibiotics, once symbols of medical triumph, were reimagined as ecological villains. This reversal appealed to both public skepticism and scientific curiosity. Once a few statistically significant studies appeared, the feedback loop between science and media took over. Headlines such as “Antibiotics can make young children heavier” [71] or “Giving antibiotics to babies may lead to obesity” [72] transformed subtle associations into deterministic proclamations. Popular reporting rarely mentions OR values or CIs. A weak increase in risk, framed as “a link to obesity”, becomes irresistible clickbait. Scientists, wary of sounding uncertain, rarely correct them. Thus was born a moralized narrative: the overuse of antibiotics as a symbol of modern hubris. The microbiome boom brought new funding streams, new journals, and new career incentives—projects linking microbiota to noncommunicable diseases multiplied exponentially after 2010 [73].
While this review discusses the link between obesity and early antibiotic exposure, some insights from studies on adults may complete the understanding. We already mentioned that antibiotic exposure is better interpreted as a marker of underlying vulnerability than as a causal factor in children. In adults, multinational epidemiological studies have disclosed that the risk of overweight is significantly elevated in the presence of IgG antibodies to Helicobacter pylori (H. pylori; OR = 1.86; 95% CI 1.34–2.60) [74]. However, no experimental works have demonstrated a direct effect of H. pylori on body fat accumulation [75]. The role of these infections, thus, might remain speculative. Previous studies have reported inconsistent findings regarding the association between H. pylori infection and obesity. For instance, an analysis of 235,107 individuals identified an increase in BMI among those infected with H. pylori [76]. In contrast, a retrospective study conducted in China found no significant relationship between H. pylori infection and obesity [77]. Notably, neither study accounted for age as a potential confounding factor, which may partly explain these divergent results. Very recently, a research group addressed this question [78]. A cohort of individuals who underwent routine physical examinations at Taizhou Hospital between 2017 and 2022 was analyzed. Univariate analysis identified age, blood pressure, glucose levels, and lipid profiles as potential risk factors for excess body fat. H. pylori infection (OR = 0.881; 95% CI 0.798–0.972) was significantly inversely associated with excess body fat [78].
After adjustment for confounding variables—including age, sex, blood lipids, blood pressure, and glucose—multivariable logistic regression confirmed that H. pylori infection remained significantly negatively associated with excess body fat [78]. Among the total cohort, 571 middle-aged and elderly participants underwent at least two physical examinations. Based on H. pylori infection status at the first and last visits, individuals were categorized into four groups: persistently negative, newly infected, persistently infected, and eradicated infection. A significant reduction in the prevalence of excess body fat was observed in both the persistently infected group and the newly infected group, compared with the persistently negative group [78]. No statistically significant difference was found between the eradicated group and the persistently infected group. In conclusion, H. pylori infection is inversely associated with excess body fat in middle-aged and elderly populations; such an association remains prolonged following infection eradication [78]. Converging epidemiological and interventional observations point toward a previously underappreciated relationship between H. pylori colonization and host energy homeostasis. Although post-eradication weight gain has been intermittently reported, such analyses extend these findings by divulging a robust inverse association between national H. pylori prevalence and obesity burden [79]. From a mechanistic standpoint, the bacterium may act as a surrogate marker of broader environmental exposures—particularly hygiene-related factors observed in the Western world—that shape the composition and function of the gastrointestinal microbiome, thereby influencing metabolic phenotypes. Within this framework, high H. pylori prevalence could reflect ecological conditions that preserve a metabolically protective microbial milieu [79].
Accumulating epidemiological evidence indicates that obesity is associated with an increased incidence of bacterial infections. Large population-based cohorts provide the strongest support for this association. In a prospective analysis of 39,163 Swedish adults, obesity was associated with a significantly increased risk of incident infections across multiple anatomical sites, including those predominantly bacterial in origin, such as skin (hazard ratio [HR] = 1.76; 95% CI 1.47–2.12 for obese females, and HR = 1.74; 95% CI 1.33–2.28 for obese males) [80]. Among bacterial infections, skin and soft tissue infections (SSTIs) represent the most robustly associated category. Multiple cohort and case-control studies summarized in a comprehensive review by Dobner and Kaser [81] report a consistent increase in SSTI incidence in obese individuals, with effect sizes often exceeding an OR of 2.0. This strong association likely reflects a convergence of mechanical (skin folds, microtrauma), vascular (impaired perfusion), and immunological (dysregulated innate immunity) factors. Obesity has been consistently identified as a risk factor for healthcare-associated infections, including surgical site infections and catheter-related infections, where raised incidence is well documented [82]. Taken together, these data suggest that obesity appears to function as a broad susceptibility state, increasing not only the probability of adverse outcomes once bacterial infection occurs, but also the likelihood of infection onset itself.
Despite its empirical frailty, the antibiotic-obesity episode left behind constructive legacies—scientifically valuable, and perhaps morally instructive. The hypothesis, though exaggerated, helped renew public and professional attention to the overuse of antibiotics in early childhood. The message resonated even if the mechanism did not. Physicians became more judicious in prescribing, parents more cautious in demanding, and public-health agencies more explicit in advocating narrow-spectrum stewardship. In this sense, a flawed hypothesis served a noble function: it contributed to a cultural shift toward restraint. By associating antibiotics with unintended harm—even mistakenly—it reinforced the imperative to preserve their efficacy. Antibiotic stewardship, not antibiotic guilt, should be the lasting legacy of this debate.
More constructively, the controversy catalyzed a wave of legitimate research into the metabolic functions of the microbiota. Studies exploring microbial metabolites, bile acid signaling, and SCFA pathways have since deepened our understanding of host-microbe metabolic dialogue [83]. Even if antibiotics are not causal agents of obesity, the wider question—how microbial ecosystems influence host energy balance—remains scientifically fertile. The field that emerged from this flawed premise has matured into one of the most interdisciplinary areas of modern biomedicine. Major experimental demonstrations, like neonatal low-dose antibiotic models producing lasting metabolic shifts [44], together with syntheses by leading groups [84], helped to focus attention on the microbiome as a mechanistic axis for noncommunicable diseases. Bibliometric analyses confirm a steep rise in microbiome-obesity publications after 2010, consistent with these papers acting as important catalysts for the field [85].
The relationship between SCFAs and obesity remains one of the most debated and conceptually unstable areas in metabolic microbiome research. While SCFAs have historically been framed as beneficial mediators of metabolic health, accumulating evidence suggests a more nuanced, context-dependent role. A major unresolved paradox lies in the dual role of SCFAs as both energy substrates and metabolic signaling molecules. SCFAs can contribute approximately 5–10% of host caloric intake, suggesting that increased fermentation efficiency following microbiome remodeling could theoretically enhance energy harvest [86]. Another priority is improving the spatial resolution of SCFA exposure assessment. Luminal SCFA concentrations differ substantially from portal and systemic levels, and most SCFAs are consumed locally by colonocytes or extracted by the liver before reaching systemic circulation [36]. Plasma SCFA measurements, therefore, likely underestimate biologically relevant exposure at the tissue level. Finally, future conceptual models should move beyond metabolite-centric frameworks. SCFAs likely function within a wider ecological signaling network integrating bile acid metabolism, host nutrient sensing, microbial density signaling, and inflammatory tone. Systems biology approaches integrating microbial ecology and environmental exposures will be required to determine whether SCFAs are causal mediators, context-dependent amplifiers, or simply biomarkers of deeper host-microbiome metabolic reprogramming.
The antibiotic-obesity hypothesis thus embodies science’s paradoxical virtue: even wrong ideas can be productive. It reminds us that research progress depends not on the infallibility of hypotheses, but on our willingness to let evidence, not fashion, decide their fate. In the end, antibiotics do not make babies fat. But the idea that they might has made the scientific community leaner in its reasoning, and healthier in its habits of skepticism. New findings raise the possibility that sustained, low-to-moderate exposure to environmental antibiotics—rather than short therapeutic courses—could contribute to childhood obesity risk. This exposure profile more closely mirrors the growth-promoting antibiotic regimens historically used in animal husbandry, characterized by chronic subtherapeutic dosing. Such parallelism offers a biologically plausible, yet underexplored, framework that could help reconcile inconsistencies in the antibiotic–obesity literature and warrants systematic investigation.
BMI: body mass index
CI: confidence interval
COPSAC 2010: Copenhagen Prospective Studies on Asthma in Childhood 2010
H. pylori: Helicobacter pylori
HR: hazard ratio
LDP: low-dose penicillin
LMICs: low- and middle-income countries
OR: odds ratio
PAT: pulsed antibiotic treatment
PBPs: penicillin-binding proteins
RR: rate ratio
SCFAs: short-chain fatty acids
SSTIs: skin and soft tissue infections
STAT: sub-therapeutic antibiotic treatment
I warmly thank Dr. Daria Augustyniak (Department of Pathogen Biology and Immunology, University of Wroclaw, Poland) for her thoughtful reading of the manuscript and her friendly suggestions that improved its clarity.
VL: Writing—original draft, Writing—review & editing, Visualization. The author read and approved the submitted version.
The author declares that she has no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 2031
Download: 75
Times Cited: 0
