 Case Report
Case Report

Affiliation:
1Pediatrics and Adolescent Medicine, Faculty of Medicine, University of Augsburg, 86156 Augsburg, Germany
ORCID: https://orcid.org/0000-0001-7488-6885
Affiliation:
2Biomedical Informatics, Data Mining and Data Analytics, Faculty of Applied Computer Science and Medical Faculty, University of Augsburg, 86159 Augsburg, Germany
Affiliation:
1Pediatrics and Adolescent Medicine, Faculty of Medicine, University of Augsburg, 86156 Augsburg, Germany
3Pediatric Hematology/Oncology, Department of Pediatrics, Otto-von-Guericke University, 39120 Magdeburg, Germany
ORCID: https://orcid.org/0000-0002-2672-4045
Affiliation:
4Human Genetics, Faculty of Medicine, University of Augsburg, 86156 Augsburg, Germany
ORCID: https://orcid.org/0000-0003-2477-9277
Affiliation:
5Institute of Toxicology, University Medical Center of the Johannes Gutenberg University Mainz, 55131 Mainz, Germany
Affiliation:
2Biomedical Informatics, Data Mining and Data Analytics, Faculty of Applied Computer Science and Medical Faculty, University of Augsburg, 86159 Augsburg, Germany
ORCID: https://orcid.org/0000-0002-5896-4086
Affiliation:
6Pathology, Faculty of Medicine, University of Augsburg, 86156 Augsburg, Germany
ORCID: https://orcid.org/0000-0003-2617-8766
Affiliation:
3Pediatric Hematology/Oncology, Department of Pediatrics, Otto-von-Guericke University, 39120 Magdeburg, Germany
ORCID: https://orcid.org/0000-0002-1732-1869
Affiliation:
1Pediatrics and Adolescent Medicine, Faculty of Medicine, University of Augsburg, 86156 Augsburg, Germany
ORCID: https://orcid.org/0000-0002-8857-6148
Affiliation:
1Pediatrics and Adolescent Medicine, Faculty of Medicine, University of Augsburg, 86156 Augsburg, Germany
Email: Michaela.Kuhlen@uk-augsburg.de
ORCID: https://orcid.org/0000-0003-4577-0503
Explor Endocr Metab Dis. 2025;2:101429 DOI: https://doi.org/10.37349/eemd.2025.101429
Received: January 17, 2025 Accepted: April 09, 2025 Published: April 17, 2025
Academic Editor: David Torpy, Royal Adelaide Hospital, The University of Adelaide, Australia
Pediatric adrenocortical tumors (pACTs) are rare endocrine neoplasms with variable prognosis, commonly associated with germline pathogenic variants (PVs) in the tumor suppressor gene TP53. Here, we report the case of a 3.1-year-old female presenting with virilization and Cushing syndrome due to a left-sided adrenal mass. The tumor was completely resected and confirmed as stage II adrenocortical carcinoma (ACC) based on the Wieneke index. Comprehensive molecular profiling revealed heterozygous germline PVs in BRCA2 [c.9382C>T p.(Arg3128*)] and CHEK2 [c.1232G>A p.(Trp411*)]. These findings suggest a potential role of impaired DNA damage repair in ACC pathogenesis, as both PVs are associated with hereditary breast and ovarian cancer (HBOC) syndromes and genomic instability. This case expands the genetic spectrum of pACT and underscores the importance of advanced molecular analyses in identifying rare germline alterations that may inform personalized treatment strategies and cancer prevention programs. Although no additional treatment was required in this case, BRCA2 status highlights the potential for tailored therapeutic approaches, including poly(ADP-ribose) polymerase (PARP) inhibitors, in selected patients. Further research is warranted to explore the specific contributions of BRCA2 and CHEK2 PVs to ACC tumorigenesis and their implic ations for targeted therapies.
Pediatric adrenocortical tumors (pACTs) are very rare endocrine neoplasms that arise from the cortex of the adrenal gland and have a variable prognosis. The mainstay of therapy is radical surgical resection, while the benefit of aggressive systemic therapy in advanced disease, including mitotane and platinum-based chemotherapy, remains to be discerned [1, 2].
pACTs are recognized as childhood cancer with strong association to syndromes. Germline pathogenic variants (PVs) in the tumor suppressor gene TP53 as cause of the Li-Fraumeni syndrome are reported in 50−65% of pediatric patients overall and up to 95% in patients from Southern Brazil due to the endemic TP53 p.Arg337His founder variant [3]. Less frequently, pACT is associated with other genetic conditions such as Beckwith-Wiedemann syndrome or multiple endocrine neoplasia. Recently, pACT has been linked to Lynch syndrome [4].
Growing evidence suggests that PVs in genes related to DNA damage response and repair may play a role in childhood cancer; their exact contribution is not fully understood yet [5]. Comprehensive molecular tumor analysis is essential for determining their significance for cancer development and personalized cancer care [6].
We present the case of a 3.1-year-old female with virilization and Cushing syndrome along with a left-sided adrenal mass. Urinary steroid hormone profiling raised the suspicion of adrenocortical carcinoma (ACC). Complete tumor resection was performed, revealing a tumor volume of 420 mL and classified as a stage II (T2 N0 M0) according to the American Joint Committee on Cancer (AJCC) 7th staging system, with tumor-free resection margins. Malignancy was confirmed based on the Wieneke index. Germline testing of TP53 was unremarkable. No additional treatment was administered, and eight years later the patient remained in first complete remission.
To further characterize the tumor, comprehensive molecular profiling was performed within the EpiGenPAT project, an in domo screening platform for pediatric ACC patients. Blood and fresh-frozen tumor tissue samples were collected with written informed consent from the patient’s legal guardians. Tumor DNA was isolated using the Qiagen AllPrep DNA/RNA Mini Kit, while blood DNA was extracted using the Maxwell RSC Blood DNA Kit. DNA methylation analysis was performed on tumor DNA using Illumina’s Infinium MethylationEPIC (850K) array, with raw data processed in R utilizing the ‘IlluminaHumanMethylationEPICmanifest’ and ‘conumee’ packages. Examination of copy number variations (CNVs) derived from DNA methylation revealed numerous alterations, such as a gain of chromosome 13 (Figure 1A), which encodes BRCA2, amongst others. The majority of the observed CNVs correspond to whole chromosome or chromosomal arm aneuploidies.
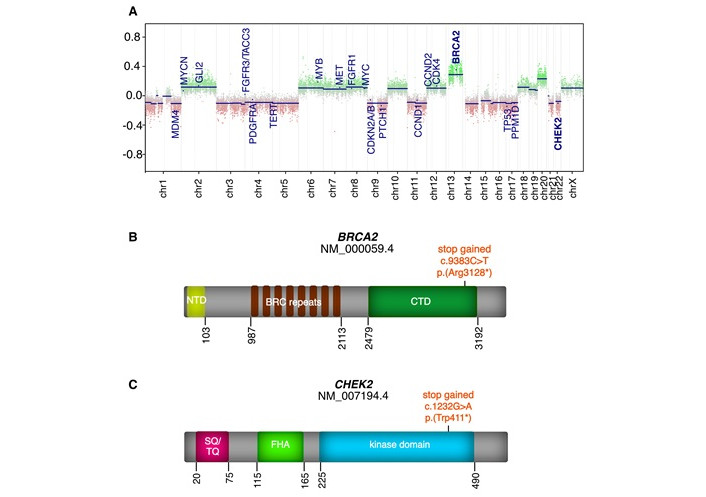
CNV profile and gene-specific mutations in the tumor sample. (A) Copy number variation (CNV) profile of the tumor, derived from the Illumina EPIC methylation array. Selected oncogenic and tumor suppressor genes, including BRCA2 and CHEK2, are highlighted; (B) Observed mutation in the BRCA2 gene; (C) Observed mutation in the CHEK2 gene. NTD: N-terminal domain; BRC: BRCA2 repeat clusters; CTD: C-terminal domain; SQ/TQ: serine-glutamine/threonine-glutamine cluster domain; FHA: forkhead-associated
Whole-exome sequencing (WES) of tumor and blood DNA was conducted by Eurofins Genomics to diagnostic standards (ISO 17025) with a coverage of 50×. Raw sequencing data were adapter-trimmed using TrimGalore, mapped to the hg19 reference genome with Burrows-Wheeler Aligner Maximum Exact Matches (BWA-MEM) and duplicates were marked using Picard. Variants were called using six different algorithms (Freebayes, HaplotypeCaller, Bcftools mpileup, Strelka, Platypus, and VarScan2) and filtered for quality and coverage parameters. Variants were annotated with visual evoked potential (VEP) and filtered against gnomAD and in-house control datasets. Detailed parameters and protocols are available from the corresponding author upon request. Using this approach on tumor DNA, a heterozygous PV in BRCA2 (NM_000059.4): c.9382C>T p.(Arg3128*) in exon 25 of 27 (Figure 1B) was found with a variant allele frequency (VAF) of 58%, alongside a heterozygous PV in CHEK2 (NM_007194.4): c.1232G>A p.(Trp411*) in exon 11 of 15 (Figure 1C) with a VAF of 34%. Both variants were confirmed to be of heterozygous germline origin through genetic testing using the patient’s blood sample. According to the American College of Medical Genetics and Genomics (ACMG) criteria 2015 both variants are classified as pathogenic (class 5). Degradation of the variant BRCA2 and CHEK2 mRNA through nonsense-mediated decay (NMD) likely leads to little or no expression of the variants.
The patient’s family history was unremarkable, with no reported cases of breast cancer, ovarian cancer, pancreatic ductal adenocarcinoma (PDAC), ACC, or other malignancies in first- or second-degree relatives. Information on cascade testing was not available.
WES of germline and tumor DNA identified an additional pathogenic (class 5) variant in IGSF3 (NM_001007237.3): c.1724G>A p.(Trp575Ter). IGSF3 encodes an immunoglobulin superfamily protein involved in cell adhesion and signaling; however, a role in ACC tumorigenesis has not been reported yet. No further high-confidence somatic single nucleotide variants (SNVs) in known cancer-related genes were detected.
Here, we report the case of a child with ACC, harboring double heterozygous germline PVs in BRCA2 and CHEK2. The c.9382C>T variant in BRCA2, resulting in a stop gain in exon 25, is listed in the population database gnomAD with a very low frequency. PVs in BRCA2 are related to hereditary breast and ovarian cancer (HBOC). CHEK2 is considered to be a moderate risk gene for HBOC. The c.1232G>A variant in CHEK2 results in a stop gain in exon 11 and is not found in gnomAD.
Given that ACC is a prototypic tumor entity that displays substantial genomic instability, we assume that the stop codons introduced into BRCA2 and CHEK2 contribute to tumorigenesis by impairing DNA-damage repair. In the largest pan-genomic characterization of 91 adult [4] and 37 pediatric ACTs [3] to date, no PVs were reported in BRCA2 and CHEK2.
El Ghorayeb et al. [7] found a c.8765delAG variant of germline origin in BRCA2 in a 50-year-old male with ACC, supported by somatic tumor analyses showing loss-of-heterozygosity in the tumor, which suggested a causal link to ACC tumorigenesis. In another case involving a 48-year-old female with ACC, the c.1100delC variant in CHEK2 was reported without molecular tumor analysis, leaving uncertainty regarding its association with tumorigenesis [8]. However, to our knowledge, no previous reports have described the co-occurrence of germline BRCA2 and CHEK2 variants in ACC. The co-occurrence of PVs in CHEK2 and other predominantly high penetrance cancer predisposing genes has been previously documented in other tumor entities such as HBOC [9]. This includes double heterozygosity with PVs in BRCA1 or BRCA2 and less frequently in ATM, CHEK2, and other moderate risk genes. Double heterozygosity may potentially cause synergistic or additional effects leading to a more severe phenotype [10–12].
Given the involvement of BRCA2 and CHEK2 in repairing DNA damage, it is plausible that a compromised ability to effectively repair DNA results in an accumulation of genetic alterations and consequently increases the risk of tumor formation. Germline PVs in both genes may increase the cancer risk even further compared to having either PV alone. Thus, the PVs could contribute to genomic instability, which is a well-known characteristic associated with cancer development, particularly for ACCs. The observed CNVs are likely to disrupt normal gene function and regulatory elements further, thus serving as an additional mechanism in cancer initiation and/or progression.
In contrast to the typical biallelic inactivation observed in breast and ovarian cancers, BRCA2 remained heterozygous in this case, with a VAF of 57% in blood and 53% in tumor, and no evidence of loss of heterozygosity (LOH) or a second-hit mutation. However, CHEK2 exhibited LOH, with its VAF increasing from 34% in blood to 85% in tumor, suggesting biallelic inactivation. While CHEK2 plays a key role in DNA damage response via checkpoint regulation, it is not classified as a core DNA repair deficiency gene, as it primarily functions in damage sensing rather than direct repair. Nevertheless, its inactivation in this case may have contributed to genomic instability through defective checkpoint control, which could indirectly impair DNA repair processes. WES analysis of BRCA1, PALB2, and CHEK1 revealed no PVs, suggesting that genomic instability in this case is primarily linked to CHEK2 inactivation, with a potential contributory role of BRCA2.
Trials assessing BRCA status have demonstrated an improved response to platinum agents; recently resulting in United States Food and Drug Administration (FDA)- and European Medicines Agency (EMA)-approval of poly(ADP-ribose) polymerase (PARP) inhibitors for adults with selected BRCA-positive cancers [13]. Extraordinary response to platinum-based chemotherapy was reported in a patient with metastatic pancreatic carcinoma associated with double heterozygous PVs in BRCA2 and CHEK2 [14]. In our patient’s case of completely resected stage II ACC, no additional treatment was necessary as she remained disease-free after eight years. However, BRCA2 status may open new avenues for personalized cancer care in certain patients with ACC, warranting further preclinical investigations of PARP-inhibition in ACC. A case report described an ACC patient with a germline BRCA2 variant who received the PARP inhibitor rucaparib following chemotherapy, but the treatment showed only limited efficacy [15].
Patients with germline pathogenic BRCA2 variants have a significantly increased lifetime risk of developing breast and ovarian cancer. CHEK2 PVs are associated with a moderately increased breast cancer risk but have an unclear role in ovarian cancer predisposition. Given this patient’s BRCA2 variant, she may be at elevated risk for these malignancies later in life. Long-term surveillance, including breast magnetic resonance imaging screening and consideration of risk-reducing strategies such as mastectomy or salpingo-oophorectomy in adulthood, is recommended per established clinical guidelines, while no risk-reducing surgical strategies exist for ACC. Our findings connect pediatric ACC with hereditary genetic variations in BRCA2 and CHEK2. Comprehensive analyses of ACCs are crucial for understanding the role of PVs in tumor development and tailoring personalized treatment, while additional research will be necessary to explore the specific roles of PVs in BRCA2 and CHEK2 in ACC development.
ACC: adrenocortical carcinoma
CNVs: copy number variations
HBOC: hereditary breast and ovarian cancer
LOH: loss of heterozygosity
pACTs: pediatric adrenocortical tumors
PARP: poly(ADP-ribose) polymerase
PVs: pathogenic variants
VAF: variant allele frequency
WES: whole-exome sequencing
VEF: Investigation, Data curation, Writing—original draft, Visualization. IS: Data curation. M Kunstreich: Investigation. MMG: Data curation, Writing—original draft. TGH and MS: Writing—review & editing. RC: Writing—original draft. AR: Investigation, Writing—review & editing. PDJ: Data curation, Writing—original draft, Supervision. M Kuhlen: Conceptualization, Investigation, Data curation, Writing—original draft, Supervision. All authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
The MET studies were approved by the ethics committees of the University of Luebeck (IRB 97125) and Otto-von-Guericke-University Magdeburg (IRB 174/12 and 52/22), Germany. The EpiGenPAT project was approved by the Ethics Committee of the Ludwig Maximilians University Munich, Germany (IRB 21-0997).
The informed consent to participate in the study was obtained from legal guardians.
The informed consent to publication in the study was obtained from legal guardians.
The data presented in this study are available upon reasonable request from the corresponding author. The data are not publicly available due to restrictions.
The research on pediatric cancer predisposition/double heterozygosity is supported by the Deutsche Forschungsgemeinschaft ([KU3764/3-1] and [HO2438/7-1]) and the EpiGenPAT project by intramurale Forschungsförderung, University of Augsburg (M Kuhlen). M Kuhlen is supported by Deutsche Krebshilfe [DKH 70115888]. PDJ is supported by the Max Eder Program of the Deutsche Krebshilfe. VEF receives a scholarship from the Konrad Adenauer Stiftung. The German MET studies were supported by Deutsche Kinderkrebsstiftung (grant numbers [DKS 2014.06], [DKS 2017.16], [DKS 2021.11], and [DKS 2024.16]) and Mitteldeutsche Kinderkrebsforschung. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

