Review
Review

Affiliation:
1Departament de Nutrició, Ciències de l’Alimentació i Gastronomia, Facultat de Farmàcia i Ciències de l’Alimentació, Institut de Biomedicina (IBUB), Universitat de Barcelona, 08021 Santa Coloma de Gramenet, Spain
ORCID: https://orcid.org/0000-0002-8476-2728
Affiliation:
2Departament de Nutrició, Ciències de l’Alimentació i Gastronomia, Facultat de Farmàcia i Ciències de l’Alimentació, Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, 08021 Santa Coloma de Gramenet, Spain
ORCID: https://orcid.org/0000-0003-4862-0850
Affiliation:
3Instituto de Química Médica (IQM, CSIC), 28006 Madrid, Spain
Email: mj.camarasa@iqm.csic.es
ORCID: https://orcid.org/0000-0002-4978-6468
Affiliation:
4Pharmacelera, Parc Científic de Barcelona (PCB), 08028 Barcelona, Spain
Email: tiziana.ginex@pharmacelera.com
ORCID: https://orcid.org/0000-0002-5739-8713
Affiliation:
1Departament de Nutrició, Ciències de l’Alimentació i Gastronomia, Facultat de Farmàcia i Ciències de l’Alimentació, Institut de Biomedicina (IBUB), Universitat de Barcelona, 08021 Santa Coloma de Gramenet, Spain
2Departament de Nutrició, Ciències de l’Alimentació i Gastronomia, Facultat de Farmàcia i Ciències de l’Alimentació, Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, 08021 Santa Coloma de Gramenet, Spain
Email: fjluque@ub.edu
ORCID: https://orcid.org/0000-0002-8049-3567
Explor Drug Sci. 2024;2:85–116 DOI: https://doi.org/10.37349/eds.2024.00037
Received: September 05, 2023 Accepted: November 04, 2023 Published: February 29, 2024
Academic Editor: Prasat Kittakoop, Chulabhorn Graduate Institute, Thailand
The influenza virus glycoprotein hemagglutinin (HA) participates in critical steps of the attachment of viral particles to the host cell membrane receptor and membrane fusion. Due to its crucial involvement in the initial phases of influenza A infections, HA emerges as a promising target in the search of novel drug-like candidates. Given its pivotal role in the early stages of influenza A infections, intense drug discovery efforts have been undertaken to target HA in the past decades. Drug discovery studies mainly rely on preventing the recognition of sialic acid units by the receptor binding site in the globular head (GH) domain, or the conformational rearrangement required for the fusion of viral and cell membranes. In this work, the aim is to summarize the progress made in HA-targeted development of small molecule fusion inhibitors. To this end, attention will primarily be focused on the analysis of the X-ray crystallographic structures of HA bound to fusion inhibitors. Furthermore, this study also aims to highlight the efforts made in exploiting the structural information in conjunction with molecular modeling techniques to discern the mechanism of action of the fusion inhibitors and to assist the design and interpretation of structure-activity relationships of novel lead compounds will be highlighted. The final section will be dedicated to elucidating novel and promising antiviral strategies proceeding from the transformation of known small molecule antivirals in proteolysis targeting chimera (PROTAC)-based targeted protein degradation. This knowledge will be valuable to assist the exploitation of classical and novel antiviral structure-based strategies, together with a deeper understanding of the mechanism of action and minimization of the impact of drug resistance.
Seasonal flu is an acute respiratory infection caused by influenza viruses. Although most people recover from the viral infection within a week without requiring medical attention, influenza can cause severe illness or even death among high-risk populations, as noted by the estimated 3–5 million severe cases and 294,000–518,000 respiratory deaths across the globe [1, 2]. The impact of seasonal flu may be aggravated by the emergence of pandemics, as exemplified by the outbreaks in 1918, also known as Spanish flu, the Asian influenza in 1957, the Hong Kong pandemic in 1968, and the swine flu in 2009 [3]. This underscores the latent risk of facing new pandemics, especially when one keeps in mind the occurrence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in 2019 following the previous SARS-CoV-1 infection in 2002 and the Middle East respiratory syndrome (MERS) in 2012 [4, 5]. This threatening scenario urges continued efforts for promoting influenza prevention, understanding the molecular mechanisms implicated in the viral infection, and developing novel effective influenza treatments, especially regarding the emergence of strains resistant to current treatments [6, 7].
Although flu vaccination is the main strategy to prevent influenza virus infections, the limited ability to generate new vaccines and the fluctuating efficacy against emerging strains make it necessary to complement vaccination with small molecule antivirals [8, 9]. In this context, the adamantane-based amantadine and rimantadine were the first drugs for the treatment of influenza [10]. They inhibit viral replication by blocking the matrix protein 2 (M2) proton channel, which is an integral transmembrane protein in the viral envelope that regulates the acidification of the interior of the virion through a pH-dependent proton transport mechanism in the influenza A virus. Amantadine and rimantadine are no longer recommended due to limited effectiveness against adamantane-resistant influenza A viral strains and influenza B virus, as well as to the adverse side effects in the gastrointestinal tract and the central nervous system. These limitations stimulated the design of novel M2 channel inhibitors [11, 12], and simultaneously the development of antiviral agents against novel targets, including (i) neuraminidase (NA), also called sialidase, which catalyzes the hydrolysis of sialic acid (SA) residues from the host cell receptor and the newly formed virions, (ii) hemagglutinin (HA), which is involved in the binding of the virion to the host receptors and in the fusion of viral and host membranes, and (iii) the RNA polymerase, which is a heteromeric complex that participates in the viral gene transcription and replication [13–18].
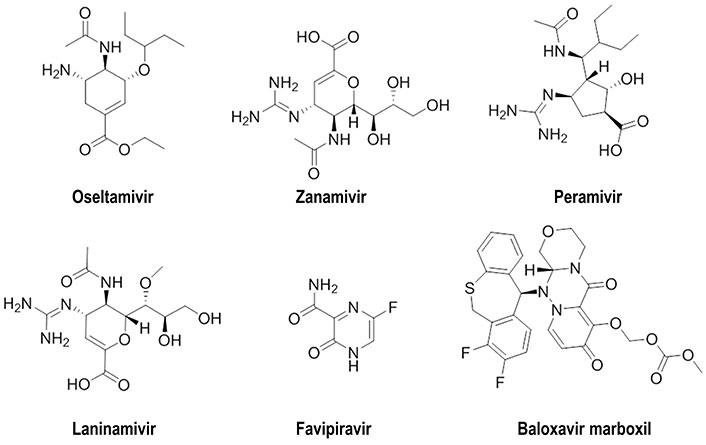
Currently, two classes of antiviral drugs that target NA (oseltamivir, zanamivir, peramivir, and laninamivir) and RNA polymerase (favipiravir, baloxavir marboxil) have been approved for influenza treatment and post-exposure prophylaxis (Figure 1) [17, 18]. Nevertheless, the emergence of resistant strains highlights the need to develop novel strategies [19, 20]. Regarding HA, the most successful therapeutic approach led to the discovery of arbidol, which has been approved in Russia and China, although multiple mechanisms of action have been proposed to explain its antiviral activity [21]. However, HA has proven to be more elusive than other viral proteins in finding small molecules able to interfere with the role played in mediating receptor binding and entry/fusion [19]. The reader is addressed to previous works, which have already provided a compilation of the advances made in the development of small molecules against different influenza A viral proteins [19, 22–28].
The aim of this contribution is to provide an overview of the progress made in the last years in the discovery of small molecules targeting HA. For the sake of brevity, the studies related to the interaction of HA with antibodies will not be examined here, limiting the discussion to the discovery of small molecule compounds. Accordingly, attention will primarily be focused on the analysis of the X-ray crystallographic structures solved for HA complexes with antiviral agents, paying emphasis to those compounds acting as inhibitors of the fusion of the viral and host membranes. Furthermore, attention will be given to efforts paid to exploit the available structural information along with molecular modeling techniques to understand the mechanism of action of fusion inhibitors. The concluding section will be devoted to unraveling innovative and promising antiviral approaches derived from the conversion of established small molecule antivirals into targeted protein degradation (TPD) compounds, commonly called as PROTACs. Overall, this review will provide structural information valuable to assist the design and interpretation of novel lead compounds.
The influenza A virus is formed by a viral envelope mainly composed by lipids and embedded proteins, such as HA, NA, and the M2 proton channel. The viral envelope encloses the viral genome, which consists of 8 negative RNA segments in complex with ribonucleoproteins in a 7 + 1 disposition (one central segment surrounded by other seven), holding the genetic information coding for the viral proteins: HA, NA, M2 proton channel, M1, non-structural proteins 1 and 2, and nuclear export protein, and RNA-dependent RNA polymerase (RdRp) and nucleoproteins (NPs) [29].
The generation of new virions in the host cell is mediated by a complex combination of viral and cellular mechanisms that are required to coordinate cell entry, replication, intracellular trafficking, and virion assembly and release (Figure 2) [30–32].
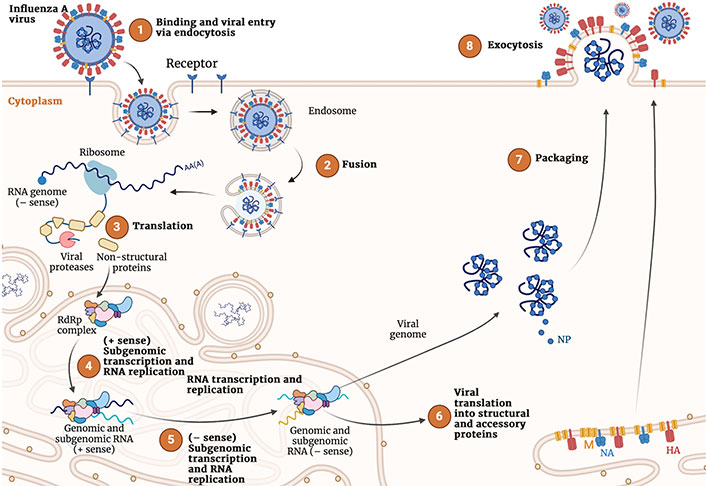
Representation of the complete viral cycle of influenza A virus in human cells. Created with Biorender.com
The first event of the viral infection is the recognition of the SA residues present in the host cell membrane glycoconjugated receptors by HA, which binds through the receptor binding site (RBS). This allows the virus to enter the cell either by clathrin-dependent receptor-mediated endocytosis or micropinocytosis. In the interior of the endosome, the low pH causes a large-scale conformational rearrangement in HA that involves multiple steps, including cleavage of HA by host cell proteases into two subunits, HA1 and HA2, and subsequent release of the fusion peptide (FP), which ultimately promotes the fusion of viral and host membranes. On the other hand, the M2 channel transports protons to the interior of the virus, which facilitates the disassembly of the packaged viral ribonucleoproteins (vRNPs) from M1 proteins.
After the fusion of the viral and cell membranes, the genetic material is released to the cytoplasm, where the nuclear import pathway participates in the transport of the viral RNA (vRNA) to the nucleus. This enables the transcription and replication of the vRNA, as well as messenger RNA (mRNA) transcription from the vRNA templates in a primed process named cap-snatching performed upon association of the polymerase acidic (PA) domain of the vRNA polymerase complex with the cellular RNA polymerase Ⅱ. The translation machinery of the host cell is recruited for the synthesis of the viral proteins. In this process, HA, NA, and M2 are expressed by using the endoplasmic reticulum (ER)-associated ribosomes, whereas cytosolic ribosomes are mediated in the expression of the other protein components. Finally, non-structural protein 2, nuclear export protein and M1 are assumed to assist the export of the vRNP from the nucleus to the cytoplasm, where the assembly and maturation of the novel virions occurs.
The whole replication cycle of the influenza virus requires 8–10 h. Relative to virus entry at time 0, attachment and entry may last up to 2 h, the endosomal release, genome replication, and viral protein translation between 2 h and 8 h, and the late stage of virion assembly and release of progeny viruses between 8 h to 10 h [33].
Overall, the preceding comments highlight the complexity of the multiple processes implicated in the generation of new virions. This has enabled the identification of druggable viral targets for drug discovery, which has benefited from the advances in solving the molecular structures of the target proteins and the characterization of the mode of action of small molecule inhibitors.
HA is a homotrimeric glycoprotein present on the viral envelope of influenza viruses, which are phylogenetically grouped into four genera (A, B, C, and D). Influenza A viruses are additionally classified based on the sequence and antigenic properties of HA and NA. There are 18 distinct HA subtypes and 11 NA subtypes. Phylogenetic analyses have categorized the HA subtypes into two groups, named 1 and 2. The former includes HA subtypes H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, H17, and H18, and the latter H3, H4, H7, H10, H14, and H15 [34, 35].
The assembly of three identical monomeric units leads to the formation of the functional HA structure. The monomeric protein is expressed as an inactive precursor, known as HA0, and maturation to the active HA species, which is composed of two chains known as HA1 and HA2 that remain cross-linked by a disulfide bridge, requires the proteolytic cleavage by host cell proteases in conjunction with post-translational modifications (e.g., glycosylation) [26].
Two structural domains can be distinguished in the structure of HA (Figure 3): the globular head (GH), which contains the receptor binding domain that mediates the recognition and binding to the host cell membrane, and the stem loop (SL), also known as stalk, which maintains the whole structure bound to the viral membrane. Both GH and SL define the ectodomain of the protein, which is attached through flexible linkers to the transmembrane domain, which anchors HA to the membrane.
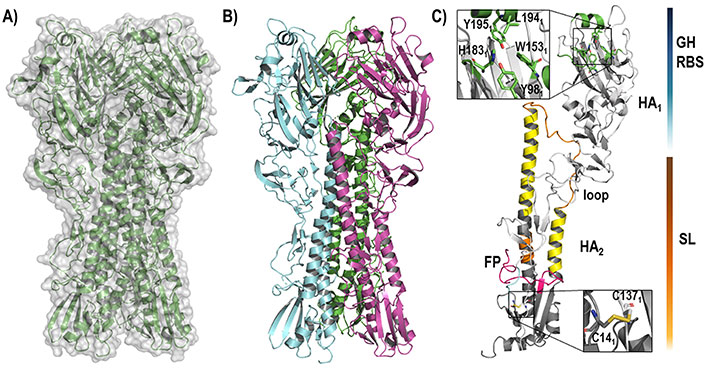
Representation of the HA structure. (A) The backbone of the whole homotrimeric structure [protein data bank (PDB) ID 6ONA] is shown enclosed in the molecular (gray) surface; (B) the three assembled monomers that define the functional trimeric HA are highlighted in green, blue, and pink; (C) each monomer is composed of two chains, called HA1 and HA2 (PDB ID 2VIU), that contain the GH (upper site) and the SL (lower site)
The RBS in the GH domain is a well conserved region through all HA isoforms. It has a negative (for group 1, H1, and H2), positive (for group 2 and especially for H3), or neutral (for H5) net charge [36]. The binding site is mainly shaped by residues Trp1531, His1831, Leu1941, and Tyr1951, all surrounded by structural elements like the 130-loop, 150-loop, 190-helix, and 220-loop [37, 38]. During the infectious process, the RBS recognizes the SA units present in the host receptor glycoproteins through the formation of a dense network of hydrogen bond interactions, which has enabled to rationalize the differences between distinct HA subtypes, particularly the molecular basis of the receptor binding specificity for SA receptors in α2,3 and α2,6 linkages of avian and human viruses, respectively [38].
The homotrimeric composition of HA, which implies a C3 symmetry axis, has enabled the analysis of multivalency effects [39] arising from the simultaneous binding of glycans to more than one of the three binding sites per HA trimer. In turn, although not the main subject of this review, it is worth noting that the concept of multivalency has been exploited for the design of divalent and trivalent HA inhibitors as an interesting therapeutic option against the influenza A virus [40–43], and also other viruses, such as SARS-CoV-2 [44].
On the other hand, the SL domain encodes the FP that participates in the fusion of viral and cell membranes following a pH-dependent conformational rearrangement [45–49]. This process is believed to involve the change in the net charge of protonatable residues, mainly histidine residues, which are present as histidine clusters, although residues Asp109 and Asp112 in the HA2 subunit also seem to influence the fusion process [50–52].
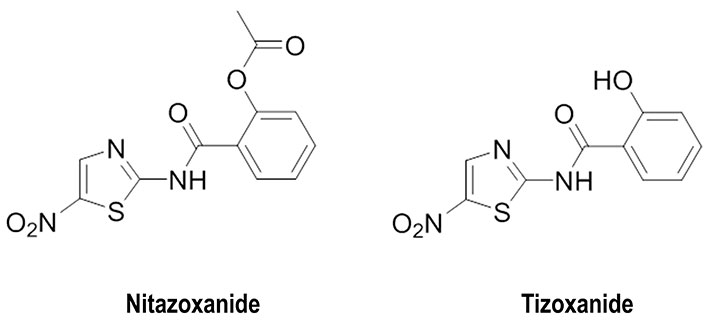
In this context, three main strategies focused on the usage of small molecules have been explored. Since the cleavage of HA0 is necessary for HA maturation and infectivity of influenza A viruses, efforts have been focused on the design of inhibitors to impede the action of host proteases in the airways of animals and humans (Figure 4) [53]. Another strategy has relied on preventing the recognition of the host receptor by HA resorting to the usage of SA-mimic peptides [28], and alternatively targeting the maturation of HA, as noted for the blockage of HA terminal glycosylation by nitazoxanide and its active circulating metabolite tizoxanide (Figure 5) [54]. Finally, the third approach, which will be examined below, has targeted the fusion between viral and host cell membranes by preventing the dynamical changes that trigger the release of the FP.
HA is a highly dynamical structure. The relevance of the structural plasticity of HA into the crowded environment of the influenza A virus envelope has been recently highlighted through mesoscale molecular simulations [55, 56], which have provided detailed information into the main molecular motions of HA, such as the ectodomain tilting and the GH breathing, and their implication in antigenically relevant conformational states. Furthermore, these studies have examined how structural properties, such as stalk height and secondary-site binding, affect the interaction with SA, and the interplay between SA binding sites in HA and NA in the attachment to the host cell receptors. Similarly, beyond the static spring-loaded fusion model of HA-mediated membrane fusion, both structural, spectroscopic, and computational studies are providing insights into the large-scale conformational rearrangements implicated in the release of the FP and remodeling of HA2, and the molecular mechanisms that underlie the acidification-promoted fusion of viral and cell membranes [57–62].
The dynamical flexibility of HA can be anticipated to be a challenge for the successful design of fusion inhibitors, which implies an additional level of complexity to the differences in the sequence of residues observed between the distinct HA subtypes, the potential effect of glycosylation sites, and the emergence of resistance-associated mutations. In this context, fusion inhibitors should stabilize the pre-fusion conformational state of HA, which in turn should prevent the occurrence of the conformational transitions necessary for membrane fusion. Nevertheless, since the low pH-induced refolding of HA involves the entire HA stem structure, HA fusion inhibitors may exert their inhibitory activity by targeting different HA binding pockets.
At this point, besides the RBS located at the GH domain, which will not be considered in this review, the analysis of the available X-ray structures deposited in the PDB [63] has disclosed the occurrence of two major binding sites, which are in the HA1-HA2 stem region (Figure 6). Site A corresponds to the binding pocket that accommodates tert-butylhydroquinone (TBHQ; PDB IDs 3EYK and 3EYM) and arbidol (PDB IDs 5T6N and 5T6S), and site B denotes the binding groove filled by fusion inhibitors JNJ4796 (PDB IDs 6CFG and 6CF7), (S)-F0045 (PDB ID 6WCR), and CBS1117 (PDB ID 6VMZ). This structural information, which will be discussed below, is highly valuable not only to explore the structure-activity relationships (SAR) of these inhibitors, but also to assess the reliability of putative binding modes for other fusion inhibitors identified from the screening of chemical libraries, and to extract guidelines (i.e., hotspots) that may assist the structure-aided search of novel chemical scaffolds.
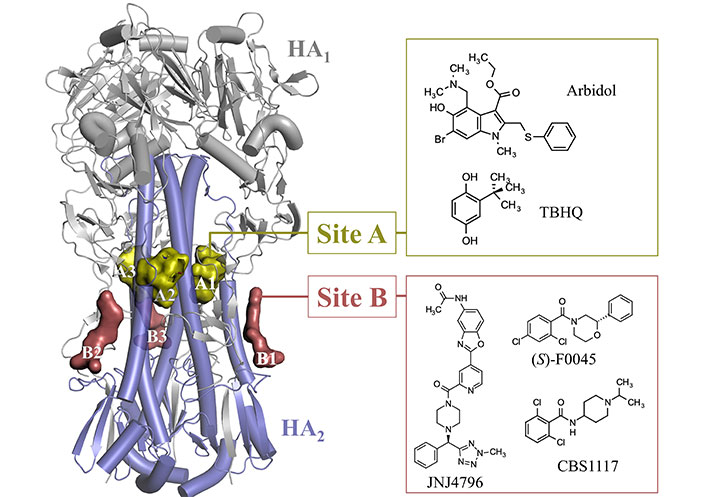
Representation of the binding sites observed in the stem of HA upon inspection of the X-ray crystallographic structures available in the PDB (PDB IDs 3EYK, 3EYM, 5T6N, 5T6S, 6CFG, 6CF7, 6WCR, and 6VMZ)
Gratifyingly, it is worth noting that these binding pockets have been identified as targets for small molecules and vaccine development in a computational study using the web-based protein binding site predictor (ProBiS) [64], which is a bioinformatics tool that enables the detection of structurally similar ProBiS through local pairwise alignment of crystallographic or nuclear magnetic resonance (NMR)-based protein structures [65]. Remarkably, the identification of the putative binding pockets has been made from the comparison of three-dimensional (3D) regions without reference to known binding sites or co-crystallized ligands and takes into account the entire protein surface.
TBHQ (Figure 7A) is a weak antiviral compound [50% inhibitory concentration (IC50) > 20 μmol/L] [66] and its antiviral activity was ascribed to inhibition of the conformational rearrangements required for membrane fusion by stabilizing the neutral pH structure (Figure 7). The stabilization exerted upon the binding of TBHQ was supported by thermal shift assays, which showed an increase of 3℃ in the melting temperature of H3 HA in the presence of 1 mmol/L TBHQ [67]. Moreover, TBHQ was found to inhibit H7 HA-mediated entry with an IC50 around 6 μmol/L [68].
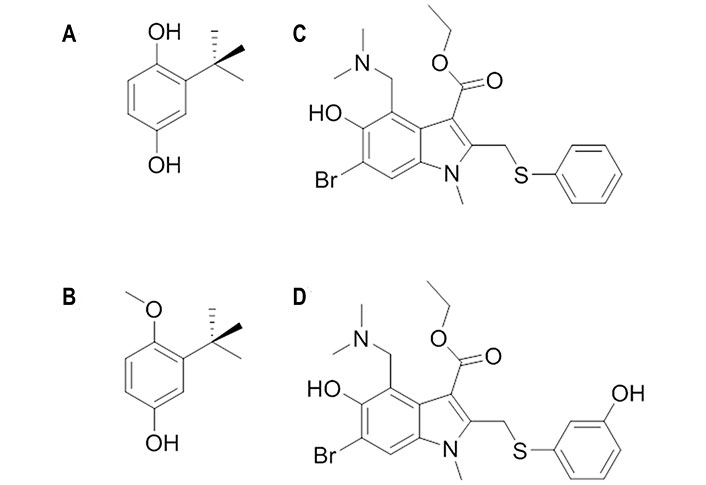
Chemical structures of TBHQ, arbidol and their derivatives. (A) TBHQ; (B) 3-tert-butyl-4-methoxyphenol; (C) arbidol; (D) der-arbidol
Knowledge of the structural details that mediate the recognition of THBQ by group 2 HA was gained from the X-ray structures of the complexes with H14 (A/Mallard/Astrakhan/263/1982) and H3 (A/Aichi/2/1968) HA (PDB IDs 3EYK and 3EYM, respectively) [67]. The binding site is found at the interface between two protomers in a region located further up the stem region, which is placed at around 16 Å from the FP (Figure 8A). This enables the formation of a TBHQ-HA complex with a stoichiometric ratio of 3:1. The binding mode is mainly characterized by hydrophobic interactions with Leu291’, Leu982’, and Ala1012’ of monomer 1, and with Leu552 and Leu992 of monomer 2. Furthermore, the aliphatic parts of the side chains of Arg542 and Glu972’ face the binding site, whereas the ionizable groups are oriented away from it. In addition, the carboxyl group of Glu572 forms a hydrogen bond with one of the oxygen atoms of TBHQ, and the other oxygen is hydrogen bonded with the main chain amide of Leu982’.
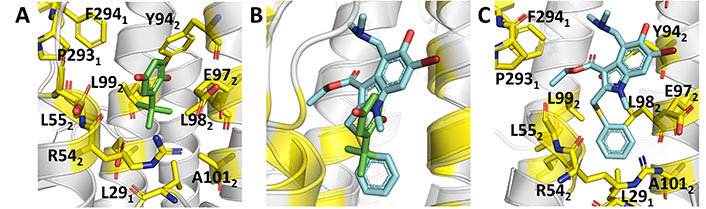
Binding mode of TBHQ and arbidol. (A) Representation of the binding mode of TBHQ (PDB ID 3EYK); (B) relative arrangement of TBHQ and arbidol after superposition of the protein backbone of their HA complexes; (C) representation of the binding mode of arbidol (PDB ID 5T6S)
The SAR analysis of TBHQ derivatives disclosed 3-tert-butyl-4-methoxyphenol (Figure 7B) as a promising candidate, since its IC50 value was close to 0.6 μmol/L [69]. NMR studies confirmed that this compound binds to the same site as TBHQ [69]. Molecular dynamics (MD) simulations for the complex with H7 HA pointed out that the aromatic ring and tert-butyl group of this compound adopt an arrangement that is almost identical to those of TBHQ, and that the presence of the methoxy group enables the formation of supplementary hydrophobic interactions with the side chains of Pro2841, Phe2851, Leu552, and Leu992 [69].
Arbidol (Figure 7C) is a broad spectrum inhibitor, as it is not only active against group 1 and 2 influenza viruses, but also against other viruses [21]. It binds with higher affinity to group 2 HAs than to group 1 HAs, as the dissociation constant ranges from 18 μmol/L to 45 μmol/L and from 5 μmol/L to 8 μmol/L in groups 1 and 2, respectively [70]. Furthermore, experimental assays have shown that arbidol increases influenza virus HA stability and prevents the low pH-induced HA transition to its fusogenic state [71]. Thus, the introduction of destabilizing or stabilizing mutations in or near the FP affects the viral sensitivity to arbidol and the degree of sensitivity was found to be proportional to the extent of FP stability on the pre-fusion HA [72].
X-ray crystallographic studies [73] disclosed the binding mode of arbidol to H3 (A/Hong Kong/1/1968; PDB ID 5T6N) and H7 (A/Shanghai/2/2013; PDB ID 5T6S) HAs. Inspection of the two X-ray structures reveals that arbidol adopts a similar orientation in the binding pocket, which partially overlaps with TBHQ (Figure 8B and C). Compared to TBHQ, the increased size of arbidol enables the formation of a larger number of contacts at the interface between adjacent protomers of the HA trimer, involving residues in the surrounding α-helices from HA2, and the C-terminal loops and an N-terminal β-hairpin from HA1.
In this binding mode the thiophenyl and indole rings are buried inside the cavity, whereas the polar substituents, hydroxyl, and bromine groups on the indole ring, are partially exposed to the bulk solvent (Figure 8C). The thiophenyl group forms hydrophobic interactions with Leu552, Leu992, Leu291’, Leu982’, and Ala1012’, which are supplemented with weak CH-π interactions between Tyr942’ and the indole ring as well as between Phe2941 and the methyl group of the ethyl acetate moiety of arbidol. Further, the carbonyl oxygen of the ethyl acetate moiety forms an intramolecular hydrogen bond with the arbidol dimethylamine group. This constrained conformation directs the ethyl acetate moiety towards the hydrophobic patch that contains Pro2931 and Phe2941.
Comparison with the apo structure of H7 HA (PDB ID 4LN6) reveals that adoption of this binding mode requires conformational rearrangements in the side chains of Arg3071 and Arg542. Thus, Arg3071 rotates by around 90°, and Glu902’ reorients to maintain the Glu902’-Arg3071 interaction, whereas the Glu902’-Lys3101’ interaction is broken. On the other hand, the Glu1032-Arg542 interaction is disrupted as the side chain of this latter residue rearranges to become more solvent-exposed, opening the cavity, while Glu572 and Glu972’ reorient to form new salt bridges with Arg542.
The need for higher dosages to attain a good therapeutic effect for arbidol stimulated the search for novel derivatives, such as der-arbidol (Figure 7D), which possesses a hydroxyl group in the meta position of the thiophene ring introduced to replace a water molecule in the binding pocket [74]. This small chemical change enhanced the antiviral activity against H1 HA, as the dissociation constant (Kd) changed from 47 μmol/L 1 μmol/L to 0.48 μmol/L 0.02 μmol/L, and H3 HA, with Kd values of 92 μmol/L 13 μmol/L and 0.0800 μmol/L 0.0008 μmol/L for arbidol and der-arbidol, respectively. The effective displacement of the water molecule by the hydroxyl group introduced in der-arbidol was observed in MD simulations (200 ns) performed for the complex with H3 HA (PDB ID 5T6N). The results also suggested a closer packing in the HA complex with der-arbidol compared to the parent compound, and a stronger stabilization due to non-polar interactions for der-arbidol, which agrees with the enhanced antiviral activity [75].
Based on the arbidol structure, influenza virus fusion inhibitors incorporating a variety of chemical scaffolds were explored, such as the series of N-(1-thia-4-azaspiro[4.5]decan-4-yl)carboxamide (spirothiazolidinone) compounds (Figure 9A–D), which showed strong activity against H3 HA [76]. Thus, the lead compound (Figure 9A) showed a median effective concentration (EC50) of around 3 μmol/L and around 6 μmol/L in activity assays using H3N2 A/X-31 and A/Hong Kong/2/68 virus-infected Madin-Darby canine kidney (MDCK) cells, and was completely inactive against H1N1, H5N1, and H7N2 strains. Docking calculations were performed to explore the binding to H3 HA in the TBHQ/arbidol pocket. The results showed multiple hydrophobic interactions with the surrounding residues, and the position of the aliphatic cyclic part was similar to that of TBHQ. Furthermore, the nitrogen atom of its carboxamide bridge formed hydrogen bonds with the side chain carboxyl of Glu572 and the main chain carbonyl of Arg542. The SAR analyses examined the effect of incorporating a variety of chemical units to the spirothiazolidinone nucleus [76]. The results showed that the imidazo[2,1-b][1,3]thiazole moiety could be replaced by 2-hydroxyphenyl (Figure 9B), 1-adamantyl (Figure 9C), or 5-chloro-2-hydroxyphenyl (Figure 9D), as the antiviral efficacy and selectivity were not drastically diminished [76–78]. Recent studies also showed the feasibility of incorporating an indole moiety, leading to a compound (Figure 9E) that showed an EC50 of 1 nmol/L in H3N2 virus-infected MDCK cells [79]. Docking calculations performed using the THBQ- and arbidol-bound complexes with HA showed that this latter compound adopts similar binding poses, with the azaspiro group being inserted into the hydrophobic pocket and the indole group oriented towards the pocket mouth. Recent studies have also examined the attachment of nicotinohydrazide moiety to the azaspiro nucleus, and the lead compound retained the activity against the H3N2 virus (EC50 of 5.2 μmol/L), but was inactive against the H1N1 strain [80].
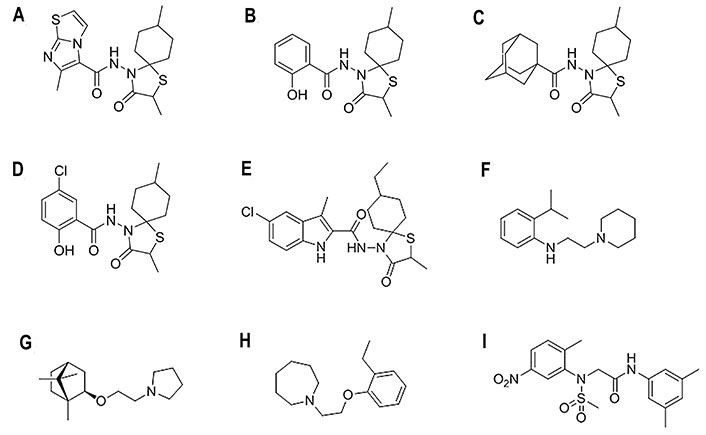
Chemical structures of selected fusion inhibitors binding at the TBHQ/arbidol binding site [27, 79, 81]. (A) 6-Methyl-N-(2,8-dimethyl-3-oxo-1-thia-4-azaspiro[4.5]dec-4-yl)imidazo[2,1-b][1,3]thiazole-5-carboxamide; (B) 2-hydroxy-N-(2,8-dimethyl-3-oxo-1-thia-4-azaspiro[4.5]dec-4-yl)benzamide; (C) N-(2,8-dimethyl-3-oxo-1-thia-4-azaspiro[4.5]decan-4-yl)adamantane-1-carboxamide; (D) 5-chloro-N-(2,8-dimethyl-3-oxo-1-thia-4-azaspiro[4.5]decan-4-yl)-2-hydroxybenzamide; (E) 5-chloro-N-(8-ethyl-2-methyl-3-oxo-1-thia-4-azaspiro[4.5]decan-4-yl)-3-methyl-1H-indole-2-carboxamide; (F) 2-isopropyl-N-[2-(piperidin-1-yl)ethyl]aniline; (G) (±)-1-(2-(1,7,7-trimethylbicyclo(2.2.1)heptan-2-yloxy)ethyl)pyrrolidine; (H) MBX-2329; (I) MBX-2546
The aniline scaffold was also exploited to develop a novel class of influenza fusion inhibitors targeting the TBHQ/arbidol binding site [82]. The anti-influenza A activity was evaluated in cell-based assays with H1N1 and H3N2 strains, showing group 1 specificity, and the fusion inhibiting effect was demonstrated using the polykaryon assay [81]. The inhibitory activity of the most potent compound (Figure 9F) was characterized by EC50 values of around 5 μmol/L and around 1.6 μmol/L against the A/PR/8/34 and A/Virginia/ATCC3/2009 strains, respectively. Remarkably, binding to H5 HA was confirmed by NMR data, which indicated that the aromatic moiety is in closest relative contact to the H5 HA surface and that the piperidine moiety is more distant [82].
Binding of the aniline-based compound (Figure 9F) to the TBHQ/arbidol binding site was examined by combining docking and subsequent refinement with MD (50 ns) simulations. The arrangement of this compound showed a significant overlap with the X-ray pose of both TBHQ and arbidol. In particular, the 2-isopropylaniline moiety was found in a subpocket shaped by Leu982, Leu992, and Leu1012, matching most of the region filled by TBHQ, and the piperidine ring occupies the inner part of the cavity, in agreement with the NMR data [82]. Moreover, this binding mode is assisted by hydrogen bonds between the piperidine and aniline nitrogen atoms with the backbone carbonyl groups of Glu572 and Thr542 in A/PR/8/34, and the carbonyl groups of Val552 and Ser542 in A/Virginia/ATCC3/2009. Likewise, attempts to dock this compound within the binding site of H3 HA (A/HK/7/87) models were unsuccessful, likely due to the reduced accessibility caused by the Arg542 side chain and the electrostatic repulsion with the positive charge of the protonated piperidine ring. Indeed, the electrostatic potential in the binding pocket showed a more pronounced negative potential in H1 HA compared to the H3 protein, which can stabilize positively charged ligands, whereas the binding site in H3 HA is less polar and hence better suited to accommodate hydrophobic ligands [82].
A structurally similar compound is the camphene derivative (Figure 9G), which showed broad in vitro antiviral activities against a panel of enveloped pathogenic viruses [81]. In particular, the EC50 determined against H1N1 (A/PR/8/34) was 45.3 μmol/L. Molecular modeling studies supported the potential binding of this compound to the TBHQ/arbidol pocket, although the results also suggested the possibility of binding to an alternative binding site proposed to accommodate M090 (see M090 section below), which is located slightly higher than the TBQH/arbidol site along the HA stem, close to the loop that connects the two α-helices [81]. While present results cannot permit to discriminate between the two putative binding sites, the hypothetical involvement of multiple binding sites cannot be ruled out.
Screening of a chemical library composed of over 106,400 compounds led to the discovery of MBX2329 (Figure 9H), an aminoalkyl phenol ether, and MBX2546 (Figure 9I), a sulfonamide derivative, which displayed concentration-dependent inhibitory activities with IC90 values of 8.6 μmol/L and 5.7 μmol/L, respectively, in pseudotype virus (H5) assays [83]. They inhibited H1N1 virus strains with IC50 values ranging from 0.3 μmol/L to 5.8 μmol/L, while being less active against H3N2 virus strain. Binding to H5 HA was confirmed by NMR studies, which revealed that binding takes place at the site filled by the antibody C179, although the two compounds seem to be recognized at different sites in the HA stem region [83]. By combining docking and MD (10 ns) simulations, MBX2329 was proposed to occupy the TBHQ site within the SL [83]. The binding mode involved interactions with Thr311 and Gln401, Val482, Thr492, Val522, and Asn532, which was consistent with the mutational effects caused by substitutions in Ile452, Val522, Asn532, and Ser542 on entry inhibition [84].
The feasibility of this binding mode was reinforced from the results of MD (100–200 ns) simulations performed for the complexes formed by MBX2329 with H1N1 (A/Washington/10/2008 and A/Florida/21/2008) HAs [85]. In agreement with the previous findings reported in [84], the MD simulations confirmed the stability of the binding mode, and the energetic analysis pointed out the relevant contribution played by van der Waals interactions to the binding of MBX2329.
Subsequent studies suggested that MBX2546 might bind to a location similar to that of the MBX2329 within the SL [86]. However, the precise nature of the binding mode is intriguing, as MBX2546 shows comparable levels of binding to H5 and H3 HAs, although this compound does not inhibit influenza A virus with H3 HA.
Broadly neutralizing antibodies targeting either the GH domain or the stem have been discussed elsewhere [87]. The stem-targeted antibodies recognize the highly conserved stem region and block the viral fusion machinery. This has promoted the design of small molecule mimetics of the key interactions that guide the recognition between HA and antibodies, although this strategy is challenged by the flat, large, and undulating (1,000–2,000 Å2) features of protein-ProBiS [88, 89]. As an example of this antibody-guided strategy, recent studies have disclosed small molecules as fusion inhibitors of human immunodeficiency virus type 1 (HIV-1) replication acting at the membrane proximal external region of the HIV-1 envelope spike [90, 91]. In the case of HA, a similar approach was adopted in conjunction with deep sequencing and protein design techniques to guide the search for small proteins and peptides with nanomolar binding affinity against HA [92, 93].
Following this strategy, a library of approximately 500,000 small molecules was screened with the aim of displacing HB80.4, which is a small protein with a very similar binding mode and fusion inhibition profile as CR6261, an antibody that broadly neutralizes most group 1 influenza A viruses [94]. This approach led to the discovery of JNJ7918 (Figure 10), which showed an EC50 of 1.1 μmol/L and 12.9 μmol/L against H1N1 A/PR/8/1934 and A/California/07/2009 HAs, respectively. The inhibitory potency was further increased upon the addition of an ester group at the benzylic group and the replacement of the terminal propargyl moiety by a heterocyclic ring, leading to compound JNJ6715 (Figure 10), which showed EC50 values of 0.14 μmol/L and 0.22 μmol/L against the same viral strains. Due to poor kinetic aqueous solubility and metabolic stability, this compound was evolved to JNJ4796 (Figure 10), where the methyl ester group at the benzylic position is replaced by 2-methyl tetrazole, the central benzene is substituted by pyridine, and the 6-methoxy benzothiazole is transformed into 6-methylamide benzoxazole. Overall, JNJ4796 retained the virus neutralization with an EC50 of 0.066 μmol/L against H1N1 (A/California/07/2009) HA and improved the pharmacological profile.
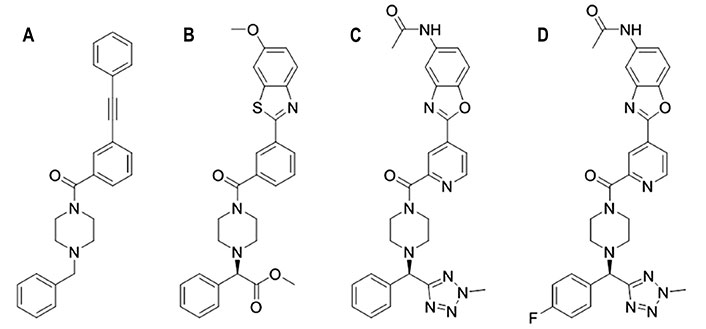
Chemical structures of JNJ4796 and related compounds. (A) JNJ7918; (B) JNJ6715; (C) JNJ4796; (D) (R)-N-(2-(2-(4-((4-fluorophenyl)(2-methyl-2H-tetrazol-5-yl)methyl)piperazine-1-carbonyl)pyridin-4-yl)benzo[d]oxazol-5-yl)acetamide
The X-ray structures of the complexes with H1N1 (A/Solomon Islands/3/2006; PDB ID 6CF7) and H5N1 (A/Vietnam/1203/2004; PDB ID 6CFG) HAs provided a structural basis to justify the antiviral activity of JNJ4796, which stabilizes the pre-fusion conformation of HA and prevents the conformational rearrangements leading to the post-fusion structure. JNJ4796 occupies the same position in the two structures and binds to a highly conserved hydrophobic groove at the HA1/HA2 interface in the HA stem, which comprises His181, Thr3181, His381-Leu421, Gly202, Trp212, and Thr412-lle562, primarily through hydrophobic and CH-π interactions (Figure 11). Overall, JNJ4796 buries around 453 (H1) Å2 and around 472 (H5) Å2 upon binding to the hydrophobic groove. Interestingly, the replacement of the hydrophobic residue Val40 in H1 HA by the polar, more flexible Gln40 in H5 may contribute to the decrease (approximately 25-fold) in binding affinity of JNJ4796 to H5 HA compared to H1 HA.
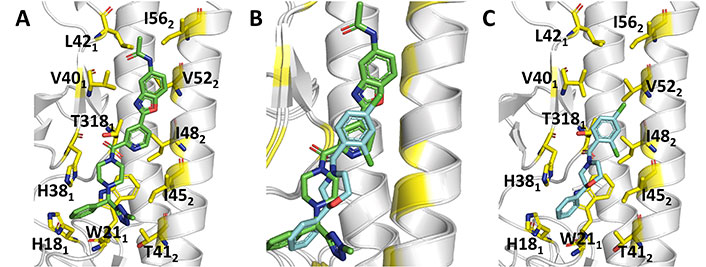
Comparison of the binding mode of JNJ4796 and (S)-F0045. (A) Representation of the binding mode of JNJ4796 (PDB ID 6CF7); (B) relative arrangement of JNJ4796 and (S)-F0045 after superposition of the protein backbone of their HA complexes; (C) representation of the binding mode of (S)-F0045 (PDB ID 6WCR)
Subsequent studies have examined the SAR of JNJ4796 by exploring the effect of introducing multiple chemical modifications in the rings (piperazine, benzene, benzoxazole, tetrazole) of the parent compound [95, 96]. The most promising compound, (R)-N-(2-(2-(4-((4-fluorophenyl)(2-methyl-2H-tetrazol-5-yl)methyl)piperazine-1-carbonyl)pyridin-4-yl)benzo[d]oxazol-5-yl)acetamide (Figure 10), showed an EC50 value of 0.03 μmol/L against H1N1 (A/PR/8/1934) HA, (EC50 > 100 μmol/L against H3N2 A/HanFang/359/95 HA), while it exhibited potent in vivo antiviral activity, and good pharmacokinetics, especially low potential cardiotoxicity.
By using a P7 peptide-based fluorescence polarization probe [93] selective for the stem epitope of group 1 HAs, a high-throughput screening of 72,000 compounds disclosed (S)-F0045 (Figure 12) [97]. This compound showed an EC50 of 1.9 μmol/L against H1N1 (A/PR/8/1934) HA, being around 20-fold more potent than its enantiomeric species. Similar trends were found for other H1 viral strains, as noted in dissociation constants (Kd) of 0.3 μmol/L and 4.7 μmol/L determined for (S)- and (R)-F0045 against H1N1 (A/PR/8/1934) HA. On the other hand, EC50 values of 5.4 μmol/L, 16.2 μmol/L, and 7.5 μmol/L were determined for H2 (A/Adachi/2/1957), H5 (A/Vietnam/1203/2004), and H6 (A/Taiwan/2/2013) HAs. The compound prevented H1 (A/PR/8/1934) HA from trypsin digestion, suggesting that its binding impedes the transition to the pH-induced post-fusion state.
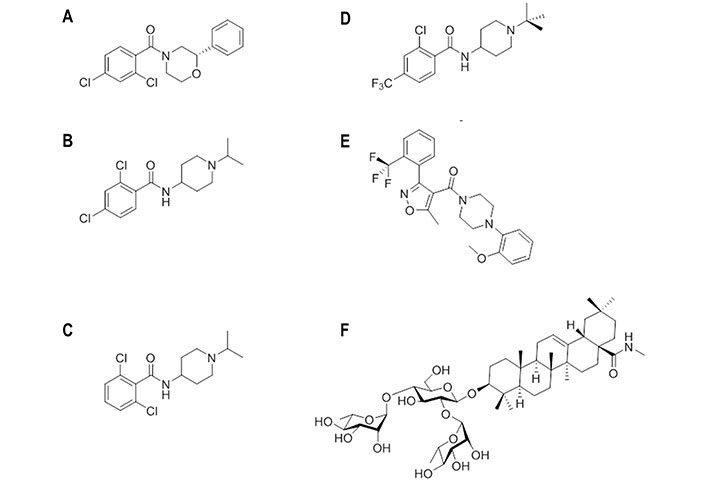
Chemical structures of small molecules experimentally bound or computationally predicted to bind to site B. (A) (S)-F0045; (B) CBS1116; (C) CBS1117; (D) N-(1-(tert-butyl)piperidin-4-yl)-2-chloro-4-(trifluoromethyl)-benzamide; (E) IY640; (F) oleanolic acid (OA)-10
The X-ray structure of the complex (PDB ID 6WCR) revealed that (S)-F0045 recognizes the hydrophobic cavity at the HA1-HA2 interface of the HA stem region. It is worth noting the close overlap of (S)-F0045 and JNJ4796 in Figure 11B. Binding of this compound involves the formation of both polar and non-polar interactions (Figure 11C). Possibly the most relevant features are the direct hydrogen bond of the amide carbonyl with the hydroxyl group of Thr3181, which is reinforced by a CH-π interaction between this residue and the dichlorobenzene ring, and between His181 and Trp212 with the phenyl ring. Furthermore, non-polar interactions involve residues Thr3181, Val401, Trp212, Thr412, IIe452, IIe482, and Val522. Finally, the group 1 HA binding specificity of (S)-F0045 may arise from the presence of a glycosylation site at Asn381 in group 2 HA, and the presence of Asn492 and Leu522 in helix A of group 2 H3 (A/Hong Kong/1/1968), which could enhance the steric hindrance.
A structurally related series of 4-aminopiperidines with an effective mechanism of action targeting HA was discovered through the screening of a chemical library of 19,200 compounds [98]. The screening disclosed CBS1116 (Figure 12B) and CBS1117 (Figure 12C), which showed IC50 values of 0.4 μmol/L and 0.07 μmol/L, respectively, against H1N1 (A/PR/8/34) HA. The SAR analysis pointed out that alkyl substituents might be introduced at the piperidine ring, as well as halogens at the phenyl ring, generally with a disubstituted pattern, which were better tolerated than alkyl or alkyloxy groups.
Indeed, the superposition of the protein backbone of the HA complexes formed with JNJ4796 (Figure 13A) and CBS1117 reveals that there is a large overlap between the chemical scaffolds of the two compounds (Figure 13B) [99]. The X-ray structure of the complex between CBS1117 and H5N1 (A/Vietnam/1203/04) HA (PDB ID 6VMZ) revealed that this compound binds at the hydrophobic gorge between the HA1 and HA2 subunits near the HA FP (Figure 13C). The binding mode was supported by hydrophobic interactions of CBS1117 with residues His381, Gln401, Trp212, and Ile452, which are supplemented with polar interactions established between the amide oxygen and Thr3251 and contacts between the chlorine atoms and the side chains of residues Thr3251 and Thr492.
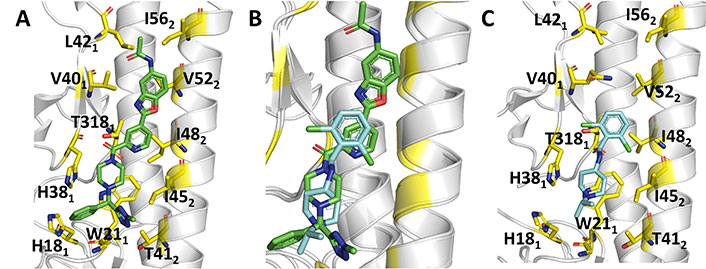
Comparison of the binding mode of JNJ4796 and CBS1117. (A) Representation of the binding mode of JNJ4796 (PDB ID 6CF7); (B) relative arrangement of JNJ4796 and CBS1117 after superposition of the protein backbone of their HA complexes; (C) representation of the binding mode of CBS1117 (PDB ID 6VMZ)
Starting from CBS1117, which showed an EC50 of 3.0 μmol/L against the H5N1 (A/Goose/Qinghai/59/05) HA, SAR studies led to a tert-butyl-N-substituted piperidine linked through an amide moiety to a 2-chloro-4-trifluoromethylbenzene (Figure 12D) [100]. The EC50 was reduced to 0.24 μmol/L. Replacement of the trifluoromethyl group by chlorine or tert-butyl by cyclohexyl had little effect on the inhibitory potency (EC50 values of 0.5 μmol/L and 0.9 μmol/L, respectively), but the bioisosteric substitution of the amide moiety by sulfonamide abolished the inhibitory potency. This exemplifies the sensitivity of the antiviral activity to seemingly mild changes in the chemical scaffold.
Docking computations suggested that the tert-butyl-N-substituted piperidinyl derivative might fill the hydrophobic groove located between HA1 and HA2, mimicking some of the interactions found for JNJ4796 [100]. In particular, the carbonyl oxygen was engaged in the hydrogen bond with Thr3181, the tert-butyl group formed hydrophobic contacts with the side chains of Val182, Asp192, and Gly202, the piperidine ring established cation-π interactions with His181 and Trp212, and a halogen-π bonding interaction was formed between Trp212 and the chlorine atom in ortho-position of the disubstituted aryl group.
Compound IY7640 (Figure 12E) was also identified from a screening of 9,687 chemicals [101]. This compound was found to be primarily effective against H1N1 viruses, as noted in EC50 values close to 0.6 μmol/L in viral cytopathic assays against H1N1 (A/Solomon Island/03/2006 and A/Brisbane/59/2007) strains, with higher EC50 values against other group 1 viruses, such as H5N1 (A/chicken/IS/06/2006; EC50 of 59.6 μmol/L) and H9N2 (A/chicken/Korea/01310/2001; EC50 of 33.4 μmol/L), and group 2 subtypes, such as H3N2 (A/Wisconsin/67/2005; EC50 of 83.1 μmol/L) and H7N9 (A/Anhui/1/2013; EC50 of 221 μmol/L). The inhibition was affected by resistance mutations Leu49 to Ile, Met403 to Thr, and particularly Glu447 to Lys, which are in the HA stalk region. At this point, docking calculations carried out using the structures of H1N1 (A/California/04/2009; PDB ID 3UBQ) and H5N1 (A/Vietnam/1194/2004; PDB ID 2IBX) HAs as templates supported the feasibility of binding around the HA stalk region defined by the CR6261 epitope residues.
Finally, following previous studies about the antiviral activity of oleanane acid triterpenes saponins [102–104], compound OA-10 (Figure 12F) was found to be effective against H5N1, H3N2, and H9N2 subtypes with EC50 values ranging from 6.7 μmol/L to 19.6 μmol/L [105]. Surface plasmon resonance (SPR) analysis using recombinant H5N1 (A/Vietnam/1203/2004) HA protein supported the interaction with HA (Kd estimated to be around 3.0 × 10−12 mol/L) [105]. By means of docking calculations, the interaction was proposed to occur at the HA1-HA2 interface in the HA stem region. The 3-O-β-chacotriosyl moiety was relevant for the binding, as the C2-OH and C3-OH groups of the L-rhamnose moiety (linked to C2-OH of D-glucose) formed hydrogen bonds with Gly202 and Val182, respectively, whereas the C3-OH of D-glucose and the C2-OH of the L-rhamnose moiety (linked to C4-OH of D-glucose) formed hydrogen bonds with Asp192 and Gln422, respectively. Moreover, numerous hydrophobic contacts were found between the oleanane moiety with His381, Trp212, Lys382, and Ile452.
As noted above, arbidol adopts a similar orientation in the binding pocket (site A) present in H3 (A/Hong Kong/1/1968; PDB ID 5T6N) and H7 (A/Shanghai/2/2013; PDB ID 5T6S). This binding mode is facilitated by the occurrence of conformational changes in the side chains of charged residues that establish salt bridges in the apo species of H7 HA (PDB ID 4LN6) [70]. However, the antiviral activity of arbidol against group 1 and 2 influenza A viruses is challenged by the differences observed upon comparison of the structural features present in the binding site of group 1 and 2 HAs, as can be noted in the comparison of the X-ray crystallographic structures for H1, H3, and H7 HA subtypes (Figure 14A and B) [70–73].
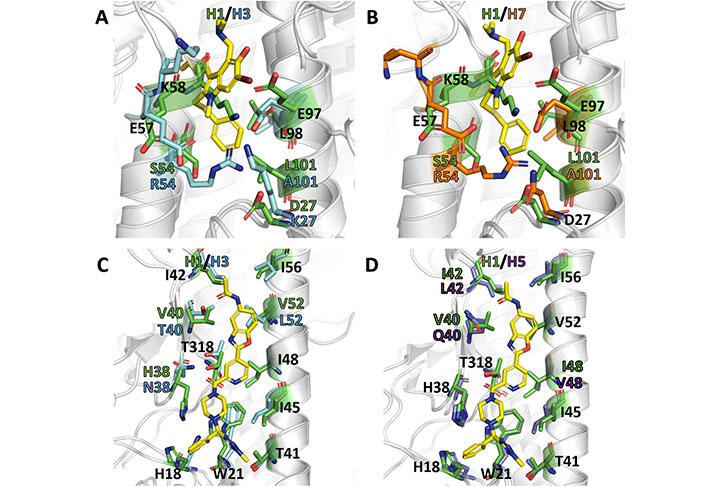
Comparative analysis of binding sites A and B in different X-ray crystallographic structures of HA. (A) Superposition of the site A in the group 1 (H1) HA (PDB ID 6CF7); (B) superposition of the and the arbidol-bound complex formed with group 2 (H3: PDB ID 5T6N; H7: PDB ID 5T6S) HAs; (C) superposition of the site B in the JNJ4796-bound complex formed with group 1 (H1) HA (PDB ID 6CF7) with the arbidol-bound complex formed with group 2 H3 (PDB ID 5T6N); (D) superposition of the site B in the JNJ4796-bound complex formed with group 1 (H1) HA (PDB ID 6CF7) with the arbidol-bound complex formed with group 1 H5 (PDB ID 6CFG) HAs. The ligands (arbidol and JNJ4796) are shown as yellow sticks
The most prominent change concerns the presence of the last helical turn (corresponding to residues 56–59 in 6CF7) in H1 HA, which is unfolded in the X-ray structures of the arbidol-bound complexes with H3 and H7 HAs. Furthermore, there is a shift of around 3.5 Å in the position of the HA2 helix relative the long central helices, one from the same protomer (helix C) and the other from the neighboring protomer (helix C’), that shape the walls of site A. Taken together, these changes enlarge the volume of the binding cavity and increase the accessibility of arbidol.
Can these structural differences be reconciled with the broad spectrum activity of arbidol? Although the antiviral activity of arbidol may result from the interaction at other sites in group 1 HAs, it may be speculated that the large structural remodeling required for the binding of arbidol to site A may be energetically more demanding due to the occurrence of stabilizing interactions formed between specific residues present in group 1 HAs. In particular, the disruption of the salt bridge formed by Lys58 and Glu97’ (helix C’) may be compensated by the electrostatic interaction formed between Arg54 and Glu97’ in H3 (5T6N) and H7 (5T6S) HAs, while the replacement of Arg54 by Ser in H1 (6CF7) HA would be less effective in providing such energetic compensation. Furthermore, it is also worth noting the substitution of Met59 (6CF7) by Thr in both H3 (5T6N) and H7 (5T6S) HAs, since the presence of Met may stabilize a hydrophobic cluster formed by Trp92 (helix C), Tyr94’ (helix C’), Pro293, Phe294, and the aliphatic chain of Lys307.
Comparison of the X-ray structures of JNJ4796 bound to group 1 H1 HA (6CF7) with the structures of group 2 H3 (5T6N) points out the lack of relevant rearrangements of the protein skeleton in site B (Figure 14C). However, it also reveals the existence of differences in specific residues of the binding site, such as the replacement of His381, Val401, and Val522 (6CF7) by Asn, Thr, and Leu (5T6N), respectively. The nature of Asn38 as a glycosylation site in group 2 HAs and the larger steric clash that may result from the Val522 to Leu substitution, which is close to the benzoxazole ring of JNJ4796, may explain the group 1 specificity of this compound. In this regard, the replacement of Val401 in H1 HA (6CF7) by Gln40 in H5 HA (6CFG; Figure 14D) may justify the approximately 25-fold decrease in potency of JNJ4796 to this latter subtype.
Overall, this demonstrates the relevant effect played by changes in the residues that shape the binding site on the antiviral activity of fusion inhibitors, making it necessary to pay attention to apparently minor differences in the design of novel compounds [95–97].
Starting from an in-silico diversity-driven screening of 495,000 molecules, compounds N-(1-(4-(1H-Imidazol-1-yl)phenyl)ethyl)-3,4-dimethylbenzenesulfonamide (Figure 15A) and 2-((5-fluoro-2-methylphenyl)amino)-2-oxoethylquinoline-2-carboxylate (Figure 15B) were found to be active against two H1N1 viral strains (A/RomaISS/02/08 and A/Parma/24/09) [106]. Recent studies have confirmed the inhibitory activity of these compounds against the H3N2 (A/Parma/05/06) subtype, with EC50 values ranging from 0.5 μmol/L to 0.098 μmol/L and from 0.062 μmol/L to 0.125 μmol/L, respectively [107]. The experimental assays pointed out the ability of these compounds to interfere with the recognition of SA by HA, and docking calculations showed that the 2-oxoethylquinoline-2-carboxylate derivative adopted a similar pose in the RBS of the H1 and H3 HA subtypes. Interestingly, these compounds were also active in hemolysis assays, suggesting the ability to alter the HA functional role though binding to the stem region given the chemical similarity to JNJ4796 and (S)-F0045. The results obtained from docking computations in conjunction with MD (50 ns) simulations supported the recognition and binding of the 2-oxoethylquinoline-2-carboxylate derivative to site B. Therefore, these results suggest the possibility to inhibit HA through a mechanism that involves a dual binding site, which might lead to a more effective inhibitory effect.
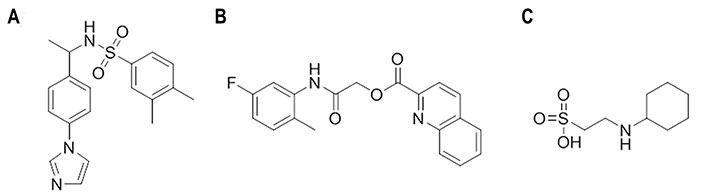
Chemical structures of small molecules presumably exerting the inhibitory effect through a dual binding site mechanism [106]. (A) N-(1-(4-(1H-Imidazol-1-yl)phenyl)ethyl)-3,4-dimethylbenzenesulfonamide; (B) 2-((5-fluoro-2-methylphenyl)amino)-2-oxoethylquinoline-2-carboxylate; (C) N-cyclohexyltaurine
These findings are aligned with the results reported for N-cyclohexyltaurine (Figure 15C) in previous studies [108]. Thus, X-ray crystallographic studies showed that this compound binds the RBS of H5N1 (A/Vietnam/1203/2004) and H3N2 (A/Hong Kong/1/1968) HAs (PDB IDs 6CF5 and 6CEX, respectively) [108]. Nevertheless, binding to the TBHQ/arbidol binding pocket was also suggested from the analysis of the electron density in the complex formed with H3 HA [108]. Although the ability to exhibit dual binding is facilitated by the small size and flexibility of N-cyclohexyltaurine, these results provide a basis to exploit the ability of certain chemical scaffolds for binding at multiple sites in HA.
The previous discussion highlights the identification of druggable sites in HA suitable for the identification of fusion inhibitors. However, given the structural complexity of HA and its conformational plasticity, the occurrence of alternative binding sites cannot be completely ruled out. Indeed, several studies have suggested other putative binding sites for a diverse set of compounds, which will be discussed here.
This compound (Figure 16A) was discovered through a SAR study of a series of pinanamine-based antivirals [109, 110]. In particular, M090 showed an EC50 of 0.3 μmol/L against influenza H1N1 (A/Guangzhou/GIRD/07/2009) virus-infected MDCK cells, and EC50 values of 0.34 μmol/L and 0.1 μmol/L in a cytopathic assay against several strains of H1N1 (EC50 of 1.5–6.9 μmol/L), H3N2 (EC50 of 4.6–5.4 μmol/L), H7N3 (EC50 of 4.6 μmol/L), and H9N2 (EC50 of 6.8 μmol/L). When M090 was administered at different time intervals since virus absorption, this compound exerted its inhibitory activity during the early steps of the virus life cycle (i.e., attachment, endocytosis, and fusion). Although the structural resemblance of M090 to inhibitors of the M2 proton channel supported originally its antiviral activity to the blockage of this protein, electrophysiology assays excluded this mechanism of action. Rather, resistance selection experiments revealed the occurrence of the Glu742 to Asp mutation, which is located at the top of the α-helix in the HA2 trimer. Accordingly, docking and subsequent MD (150 ns) refinement performed for the H1N1 (A/California/04/2009) HA (PDB ID 3AL4) suggested that M090 binds at the interface of the long helix (residues 82–93) and loop (residues 57–69; numbering started from the HA2 segment) through hydrophobic interactions: the thiophene ring forms contact with Ile892, Tyr3122, and Trp922, the pinane unit fills the region shaped by Ala652 and Val662, and the cyclopropyl group interacts with Phe882 and Pro3032 [110]. In addition, the N-H group is hydrogen bonded to Tyr3052. Finally, according to this binding mode, the Glu742 to Asp mutation could weaken the interactions formed by Glu74 and Arg76, destabilizing the local fold of HA2.
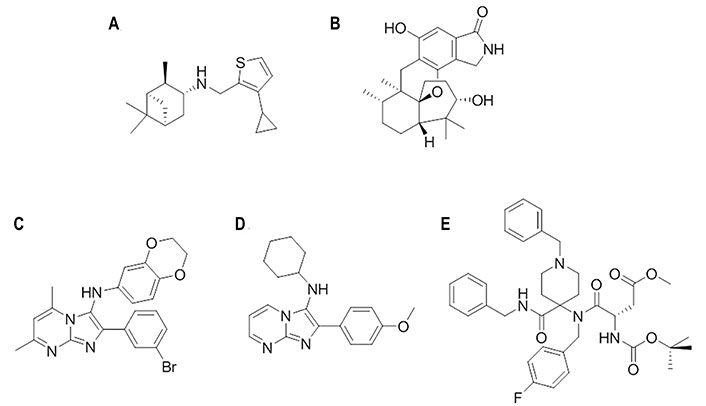
Chemical structures of small molecules presumably bound to other binding sites in HA [111]. (A) M090; (B) stachyflin; (C) CBS1194; (D) N-cyclohexyl-2-(4-methoxyphenyl)imidazo[1,2-a]pyrimidin-3-amine; (E) DICAM
The sesquiterpene stachyflin (Figure 16B) was found to inhibit the growth of H1, H2, H5, and H6 influenza viruses by preventing the fusion of the virus envelope with the cellular membrane [112]. The EC50 was generally comprised between 0.05 μmol/L against the viral replication of the H1N1 (A/WSN/1933) subtype and 4.7 μmol/L against the H5N1 (A/whooper swan/Mongolia/3/2005) strain. By means of docking calculations, stachyflin was proposed to bind a cavity on the HA2 region, which accommodates most amino acid substitutions found on the HA of stachyflin-resistant virus clones [112]. This pocket contained residues Asp372, Lys512, and Thr1072, which were substituted in the stachyflin-resistant strains. The lack of effect against the H3 virus subtype was attributed to the presence of 14 residue substitutions between the H1 and H3 HAs in the vicinity of the binding pocket.
A high-throughput screening campaign using two pseudotypes bearing H7 or H5 HAs led to the discovery of this compound (Figure 16C), which was active against group 2 during the early phase of viral infection, whereas no effect was observed against group 1. HA-mediated hemolysis was also inhibited, indicating that CBS1194 may target the HA stalk region [113]. Escape mutant analyses and docking calculations suggested that it might fit a pocket near the FP and residue Lys1172, surrounded by Lys392, Glu1142, Glu1202, and Lys1212. The steric hindrance generated upon binding might block the low pH-induced rearrangement of HA [113].
Subsequent SAR studies identified a series of structurally related compounds that retained the inhibitory activity (IC50 ≤ 200 nmol/L) against the H7N1 pseudovirus, whereas a weaker activity (IC50 ≥ 20 μmol/L) was measured against the H5N1 subtype [111]. The EC50 determined against the H3N2 (A/Hong Kong/11/1968) ranged from 30 nmol/L to 360 nmol/L, and a similar range of values was obtained for the oseltamivir-resistant H3N2 (A/Victoria/361/2011) strain (EC50 from 0.4 μmol/L to 1.5 μmol/L). Specifically, one of the most potent compounds (Figure 16D) had EC50 values of 0.03 μmol/L and 0.64 μmol/L, respectively. Despite the structural resemblance to CBS1194, docking computations suggested the TBHQ/arbidol pocket as the putative binding site, where it interacted with Tyr433, Phe303, Leu37, and Leu437. This binding mode was supported by the notable reduction in potency observed for selected mutations, such as the replacement of Tyr433 to His, which reduced the activity of the ligand by 1,400-fold, and the mutations of Leu37 to Ala and Val, which decreased the EC50 activity by 130-fold and 890-fold.
By exploiting the synthetic power of multicomponent reactions to generate molecular diversity in chemical libraries, a novel class of N-benzyl-4,4-disubstituted piperidines as influenza A virus fusion inhibitors with specific activity against the H1N1 subtype was discovered [114]. The lead compound, DICAM (Figure 16E), which showed an antiviral EC50 of 1.9 μmol/L against H1N1 (A/PR/8/34) virus, was found to act as an inhibitor of the low pH-induced HA-mediated membrane fusion process [114]. Computational studies suggested a putative binding site located at the bottom of the HA stem near the FP. Noteworthy, a π-stacking interaction between Phe92, which is located at the beginning of the FP, and the N-benzylpiperidine moiety of DICAM was found after MD (200 ns) refinement of the binding pose [114]. This direct interaction with the FP was assisted by an additional π-stacking interaction of the benzyl ring with Tyr1192, and a salt bridge between Glu1202 and the protonated piperidine nitrogen. To further confirm these interactions, methylation of this nitrogen, thus generating a permanent positive charge on this atom, led to a complete loss of the inhibitory potency, which was justified by the steric clash originated by the quaternary nitrogen for retaining the salt bridge with Glu1202. In addition, the insertion of two fluorine atoms in the benzyl ring was also detrimental for the activity, which was attributed to unfavorable repulsion with the lone pairs of carbonyl oxygens in Val1152 and Lys1162. This binding mode not only justified the SAR, but also provided a structural explanation for the H1N1-specific activity of these inhibitors, particularly regarding the seemingly conserved substitution of Lys1232 in A/PR/8/34 HA by Arg in A/Virginia/ATCC3/2009 HA due to the concomitant reduction in the volume of the binding pocket. Furthermore, the resistance-induced mutation Ser3261 to Val was close to the proposed new binding pocket.
Overall, these studies suggest the potential involvement of diverse binding sites in conjunction with novel mechanisms of action in HA, which might disclose new opportunities for antiviral drug design and development.
The challenging nature of HA to develop a “classical” structure-based drug design makes it necessary to open the scenario of alternative therapeutic approaches. They may be inspired by current drug discovery efforts undertaken for the treatment of diseases with undruggable targets, which are estimated to account for almost 90% of the human genome [115]. In this regard, TPD may open novel avenues for the design of antiviral agents that rely on PROTACs.
PROTACs permit to promote the degradation of a target protein by hijacking the ubiquitin-proteasome system. To this end, PROTACs are heterobifunctional molecules that contain three components: the ligand that acts as binding moiety to the protein of interest (POI), the E3 ubiquitin ligase ligand, and the linker that connects POI and E3 ligands, whose length and flexibility are pivotal for the effective formation and stabilization of the POI-PROTAC-E3 ternary complex. The absence of a linker connecting POI and E3 ligase ligands leads to another class of proteasome-driven degradative compounds named molecular glues (MGs). The formation of the ternary complex promotes POI (poly)-ubiquitination and finally degradation by the proteasome (Figure 17) [116, 117]. This strategy enables to fine tune disease-relevant biological pathways with potential downstream effects [118]. In addition, PROTACs catalyze the degradative process sub-stoichiometrically, as they can be recycled once released from the ternary complex [117, 119]. In fact, whereas generally classical inhibitors are required to have a high affinity for the target protein, PROTACs only need to bind tightly enough to induce proximity with E3 ligases, so that they can label POI targets for degradation.
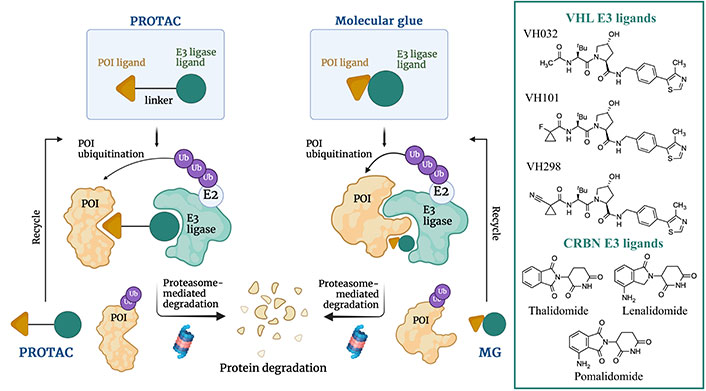
Schematic representation of the proteasome-induced protein target degradation mechanism promoted by MGs and PROTACs and chemical structures of the most common cereblon (CRBN) and von Hippel-Lindau (VHL) E3 ligases ligands successfully employed in TPD. Created with Biorender.com
CRBN and VHL proteins represent the most explored E3 ligases for the design of PROTACs. This obeys the fact that they are highly expressed in different cell lines, relatively safe, and well-characterized from a physicochemical, biological, biochemical, and pharmaceutical point of view [116, 117]. Typical CRBN E3 ligase ligands are represented by the immunomodulatory drugs as thalidomide, lenalidomide, and pomalidomide, whereas VHL E3 ligase ligands are VH032, VH101, and VH298 (Figure 17).
As noted above, viral proteins are prone to introduce mutations in their amino acid sequences, thus enabling to escape from classical antiviral strategies based on small molecules targeting known druggable binding sites. In this context, the application of TPD could help in circumventing drug resistance due to resistance mutations. The development of antiviral degraders is still in its infancy since very few compounds have been reported so far. Given the topic of this review, the discussion will be focused in influenza A antiviral degraders.
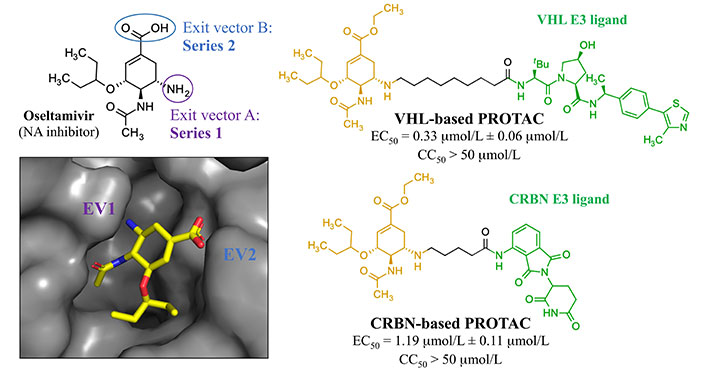
In 2022, Xu et al. [120] reported an antiviral PROTAC based on oseltamivir. Twenty-two PROTACs were designed by connecting this compound with CRBN and VHL E3 ligands (Figure 18). Two different exit vectors, one proceeding from the free amine and the other from the carboxylic moiety, were explored, and several linkers (as alkyl, PEGylated, piperazinyl, piperidyl, and triazolyl chains) were tested to connect to CRBN and VHL ligands. The SAR analysis suggested that a higher degree of flexibility in the linker was associated to a better antiviral activity. Among them, the VHL- and CRBN-based PROTAC were found to be the best-in-class antivirals (H1N1 EC50 of 0.33 μmol/L and 1.19 μmol/L, respectively). Dose-dependent degradative potency via ubiquitin-proteasome system, towards different wild-type and oseltamivir-resistant (H1N1 His274 to Tyr) HA strains, was also demonstrated for VHL-based PROTAC. The putative NA-VHL-based-VHL(E3) ternary complex derived by docking experiments is depicted in Figure 19.
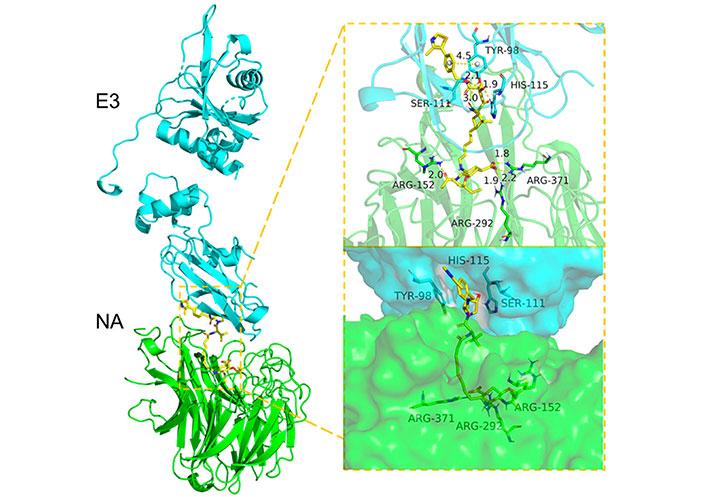
Docking-predicted NA-VHL-based-VHL(E3) ternary complex (NA, PDB ID 2HU0; VHL, PDB ID 5NW2)
Note. Reprinted from “Discovery of oseltamivir-based novel PROTACs as degraders targeting neuraminidase to combat H1N1 influenza virus,” by Xu Z, Liu X, Ma X, Zou W, Chen Q, Chen F, et al. Cell Insight. 2022;1:100030 (https://www.sciencedirect.com/science/article/pii/S277289272200027X?via%3Dihub). CC BY-NC-ND.
In 2022, Li et al. [121] reported two series of HA-targeting pentacyclic triterpenoid-based PROTACs. They were generated from the functionalization of an oleanolic OA derivative with CRBN (C1–C3 series) and VHL (V1–V6 series) E3 ligands via oligo (ethylene glycol) linkers (Figure 20A). Both series of PROTACs showed HA degradative effect but, unlike the NA-based PROTACs reported above, no antiviral effect was observed. Among the CRBN series, C3 (i.e., the one with the longest linker) exhibited the most competent degradation capacity (> 50% at 0.78 μmol/L). On the other hand, V1–V3 induced a concentration-dependent depletion of HA protein and possessed approximately 50% degradation capacity at a concentration of 33 μmol/L. Authors also reported that V3 (degradation concentration of 1.44 μmol/L) protected mice from influenza A virus-induced death and weight loss. The putative binding site of the OA was investigated by combination of docking and photo-crosslinking mass spectrometry (MS) experiments (Figure 20B) [121]. Crosslinking to the region defined by residues Asn15, Thr31, and Asn27 was suggested from the analysis of the HA complexes with the OA.
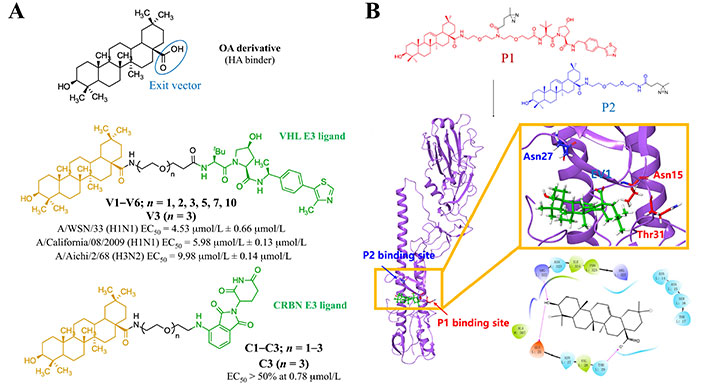
Strategy for the design of HA-targeting PROTAC compounds. (A) Available HA-based antiviral PROTACs; (B) putative HA-OA complex from photo-crosslinking MS and docking studies (PDB HA 1RVT)
Note. Adapted with permission from “Discovery of pentacyclic triterpenoid PROTACs as a class of effective hemagglutinin protein degraders,” by Li H, Wang S, Ma W, Cheng B, Yi Y, Ma X, et al. J Med Chem. 2022;65:7154–69 (https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c02013). © 2022 American Chemical Society.
In 2022, APL-16-5 (previously called asperphenalenone E), a microbial metabolite originally isolated from the plant endophytic fungus Aspergillus, was used to generate PROTACs targeting the PA protein of the viral polymerase [122]. APL-16-5 exerted a good antiviral activity with an EC50 of 0.28 μmol/L ± 0.01 μmol/L in HEK293T-Gluc cells infected with A/WSN/33. The chemical structure of the antiviral compound and its putative mechanism of action are shown in Figure 21.
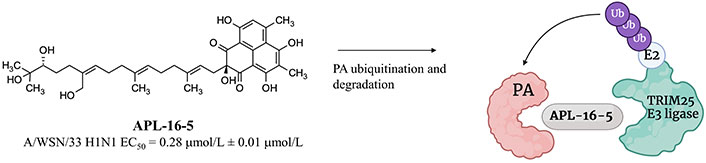
Chemical structure of APL-16-5 and its putative mechanism of action involving TRIM25 E3 ligase. Created with Biorender.com
APL-16-5 was reported to induce proteasome-dependent degradation of the PA protein by promoting proximity with the TRIM25 E3 ligase as suggested by SPR-liquid chromatography (LC)-MS/MS studies [122]. Influenza A viruses passaged in the presence of APL-16-5 showed that Asn228 to Lys and Ala704 to His mutants were resistant to APL-16-5, thus supporting the PA protein as the antiviral target for this compound [122]. APL-16-5 was able to reduce the level of this polymerase component, while remained unaffected those of other components, such as the polymerase basic proteins 1 and 2. As stated by Zhao et al. [122], APL-16-5 did not affect the expression of PA protein mRNA; hence, they speculated that the effect on RNA-dependent RNA polymerase levels might be related to the induction of protein instability in the PA component, which in turn promoted ubiquitination and degradation.
Over the past decades, the structural characterization of HA has played a crucial role not only in gaining deeper insight into its structure-function relationships, particularly regarding receptor binding function and fusion mechanism [123]. Another interesting issue concerns the complexity of the conformational plasticity of HA and its antigenicity. Altogether, this has facilitated the design and development of small molecule therapeutics and vaccines against influenza virus.
Most of the drug discovery efforts have been focused on the rather conserved stem domain, which encodes the fusion machinery. Up to now, X-ray crystallographic studies have disclosed the binding mode of five small molecule fusion inhibitors: TBHQ, arbidol, JNJ4796, (S)-F0045, and CBS1117 [68, 73, 97, 98]. These structures enabled the identification of the relevant chemical features that mediate the binding of these compounds to HA, providing thus useful information to explore the SAR for structurally related analogues. Furthermore, they have highlighted differences in the nature of the residues that may contribute to understanding the group-specific binding properties of these inhibitors [68, 73, 97, 98]. In addition, they have also provided insights about the molecular basis associated to the emergence of mutations that confer resistance to the inhibitory potency of these compounds.
The knowledge about the binding mode of these inhibitors can provide clues for guiding the design of optimized chemical scaffolds that may satisfy the requirement of binding affinity and broad spectrum of inhibitory activity. However, the success of structure-based methods is challenging for several reasons:
(ⅰ) The intrinsic conformational flexibility of HA may be responsible of small, but significant structural changes in the binding pocket, which may have a large impact on the expected potency of the designed compounds.
(ⅱ) The presence of glycosylated sites may affect the physicochemical properties of the binding pocket, such as the accessibility from the bulk solvent, the local flexibility around the binding cavity, and the structure of hydration at the protein surface.
(ⅲ) The ligand-promoted occurrence of resistance mutations is not always limited to the binding pocket or neighboring residues; sometimes they appear in regions located far from the site of action, suggesting that the stabilization of the pre-fusion state afforded by the fusion inhibitor may be counterbalanced by specific changes that could destabilize other regions implicated in the release of the FP, as reflected in changes in the acidic conditions to trigger membrane fusion.
In this context, in-silico drug discovery studies must be performed under a critical supervision, which must assess the suitability of the proposed binding mode. This is especially true when the putative binding site has not been resolved and its feasibility must be calibrated through a judicious assessment of the available experimental evidence. If properly executed, it can provide a rationale for the effect of chemical changes on the potency of the ligand, the group specificity, the mechanism of action in preventing the fusion membrane, and the impact of resistant-associated mutants. Clearly, a close synergy between structure-based computational studies and the biochemical and pharmacological information gained from antiviral assays is crucial for the identification of novel antiviral agents. In turn, validation of the mode of action of these compounds may open unexpected avenues to explore the development of novel and unique anti-influenza drugs.
In addition to the preceding discussion, it is also clear that the structural knowledge can be useful to make progress in the design of novel therapeutic strategies, such as PROTAC-based target degradation. Although still in an early stage, the results obtained up to now are encouraging and give support to the potential application of this approach, although more efforts are needed to effectively optimize both the efficiency in viral protein degradation and the pharmacokinetics profile of antiviral PROTACs. As for classical drug discovery campaigns, structural studies aiming at elucidating their mechanism of action are strongly encouraged since they can provide a solid basis for these necessary improvements.
CRBN: cereblon
EC50: median effective concentration
FP: fusion peptide
GH: globular head
HA: hemagglutinin
IC50: 50% inhibitory concentration
M2: matrix protein 2
MD: molecular dynamics
MDCK: Madin-Darby canine kidney
MGs: molecular glues
MS: mass spectrometry
NA: neuraminidase
NMR: nuclear magnetic resonance
OA: oleanolic acid
PA: polymerase acidic
PDB: protein data bank
POI: protein of interest
ProBiS: protein binding sites
PROTACs: proteolysis targeting chimeras
RBS: receptor binding site
SA: sialic acid
SAR: structure-activity relationships
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
SL: stem loop
TBHQ: tert-butylhydroquinone
TPD: targeted protein degradation
VHL: von Hippel-Lindau
vRNA: viral RNA
Dedicated to the memory of Dr. S. Velázquez. We are grateful for her enthusiasm, encouraging inspiration, and stimulating scientific discussions, which have been valuable for the preparation of this manuscript.
FJHP: Conceptualization, Investigation, Writing—original draft. AV: Investigation, Writing—original draft. MJC: Conceptualization, Validation, Writing—review & editing. TG: Conceptualization, Validation, Writing—review & editing, Supervision. FJL: Conceptualization, Investigation, Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The research was given financial support by the Spanish Ministerio de Ciencia e Innovación [PID2020-117646RB-I00 MCIN/AEI/10.13039/501100011033, PID2019-104070-RB-C21]; the Spanish Ministerio de Ciencia, Innovación y Universidades [PID2022-136307OB-C21/AEI/10.13039/501100011033/FEDER, UE]; Maria de Maetzu [CEX2021-001202-M]; Generalitat de Catalunya [2021SGR00671]; and Agencia Estatal Consejo Superior de Investigaciones Científicas (CSIC) [CSIC-PIE201980E100, CSIC-PIE201980E028, CSIC-PIE202380E095]. FJH has fellowship from the Spanish Ministerio de Ciencia e Innovación [PRE2021-100418]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.