 Original Article
Original Article

Affiliation:
1Institute of Molecular Sciences (IMS), Centro de Química Estrutural (CQE), Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisboa, Portugal
Email: luisfrija@tecnico.ulisboa.pt
ORCID: https://orcid.org/0000-0003-3252-3482
Affiliation:
2Center of Marine Sciences (CCMAR), Department of Chemistry and Pharmacy (FCT) Gambelas Campus, University of Algarve, P-8005-039 Faro, Portugal
Affiliation:
2Center of Marine Sciences (CCMAR), Department of Chemistry and Pharmacy (FCT) Gambelas Campus, University of Algarve, P-8005-039 Faro, Portugal
Affiliation:
3CBIOS - Research Center for Health Sciences & Technologies, ULusófona de Humanidades e Tecnologias, 1749-024 Lisboa, Portugal
4iMed.ULisboa - Research Institute for Medicines, Faculdade de Farmácia, Universidade de Lisboa, 1649-003 Lisboa, Portugal
Affiliation:
5LAQV/REQUIMTE, Laboratório de Microbiologia, Departamento de Ciências Biológicas, Faculdade de Farmácia, Universidade do Porto, 4050-313 Porto, Portugal
Affiliation:
6Faculty of Medicine, Clinic Academic Center of Coimbra (CACC), Coimbra Institute for Clinical and Biomedical Research (iCBR), University of Coimbra, 3000-548 Coimbra, Portugal
7Center for Innovative Biomedicine and Biotechnology (CIBB), University of Coimbra, 3000-548 Coimbra, Portugal
Affiliation:
6Faculty of Medicine, Clinic Academic Center of Coimbra (CACC), Coimbra Institute for Clinical and Biomedical Research (iCBR), University of Coimbra, 3000-548 Coimbra, Portugal
7Center for Innovative Biomedicine and Biotechnology (CIBB), University of Coimbra, 3000-548 Coimbra, Portugal
8PhD Programme in Experimental Biology and Biomedicine, Institute for Interdisciplinary Research (IIIUC), University of Coimbra, 3030-789 Coimbra, Portugal
9Center for Neuroscience and Cell Biology (CNC), University of Coimbra, 3004-504 Coimbra, Portugal
Affiliation:
6Faculty of Medicine, Clinic Academic Center of Coimbra (CACC), Coimbra Institute for Clinical and Biomedical Research (iCBR), University of Coimbra, 3000-548 Coimbra, Portugal
7Center for Innovative Biomedicine and Biotechnology (CIBB), University of Coimbra, 3000-548 Coimbra, Portugal
10Department of Life Sciences, Centre for Functional Ecology, University of Coimbra, 3000-456 Coimbra, Portugal
Affiliation:
2Center of Marine Sciences (CCMAR), Department of Chemistry and Pharmacy (FCT) Gambelas Campus, University of Algarve, P-8005-039 Faro, Portugal
ORCID: https://orcid.org/0000-0002-9447-2855
Affiliation:
3CBIOS - Research Center for Health Sciences & Technologies, ULusófona de Humanidades e Tecnologias, 1749-024 Lisboa, Portugal
4iMed.ULisboa - Research Institute for Medicines, Faculdade de Farmácia, Universidade de Lisboa, 1649-003 Lisboa, Portugal
Email: p1609@ulusofona.pt
ORCID: https://orcid.org/0000-0001-7992-8343
Explor Drug Sci. 2023;1:420–434 DOI: https://doi.org/10.37349/eds.2023.00028
Received: July 05, 2023 Accepted: September 06, 2023 Published: November 23, 2023
Academic Editor: Fernando Albericio, University of KwaZulu-Natal, South Africa; University of Barcelona, Spain
Aim: This study discloses the synthesis and the antimicrobial and anticancer activities of four molecules of structural basis saccharin-thiadiazolyl (4), saccharin-pyridyl (6, 8), and tetrazole-thiadiazolyl (11).
Methods: Antimicrobial properties of the molecules were evaluated by the well-diffusion method, against Gram-positive bacteria [Staphylococcus aureus American Type Culture Collection (ATCC) 25923, Staphylococcus epidermidis ATCC 12228, Mycobacterium smegmatis ATCC 607], Gram-negative bacteria (Pseudomonas aeruginosa ATCC 27853) and yeast (Saccharomyces cerevisiae ATCC 2601 and Candida albicans ATCC 10231) strains. The anticancer activity of the compounds was assessed through i) proliferation assays for HCT116, MCF-7, and A375 human cell lines [cells were treated with serial dilutions of compounds and the effect on cell propagation was evaluated by sulforhodamine B (SRB) assay]; ii) antiproliferative and cytotoxic assays for glioma-type cell lines A172 (glioblastoma), U87 (brain-likely glioblastoma), and H4 (neuroglioma; cells were treated with diverse concentrations and the cell viability was assessed using a modified Alamar blue® assay).
Results: Compound 11 exhibited significant inhibitory activity against S. aureus and S. epidermidis, with the further molecules demonstrating some inhibitory potential against all the tested Gram-positive, Gram-negative, and yeast strains. Similarly, derivative 11 showed an interesting antiproliferative activity against human colon adenocarcinoma (HCT116), human breast adenocarcinoma (MCF-7), and melanoma (A375) cells, with 50% growth inhibition (GI50) values varying from 3.55 µmol/L to 11.5 µmol/L, in the same order of magnitude of those shown by etoposide. Treatment of brain-like glioblastoma cells (U87) with 11, at the concentration of 100 µg/mL, induced a decrease in cell viability by 50% after 48 h and 72 h. Besides, results attained for A172 cells have shown that compound 11 only induces a significant decrease in cell viability upon treatment at 100 µg/mL for 72 h. A divergent observation was recorded for H4 cells, where the treatment with derivative 11 had promoted a significant decrease in cell viability (< 40–60%), even at concentrations as low as 0.39 µg/mL, after 24 h.
Conclusions: This investigation reveals the potential of distinct azole-based conjugates, in particular the tetrazole-thiadiazolyl (11) derivative, as scaffolds worth further investigations, in the frame of antimicrobial and antineoplastic chemotherapy.
The genesis and exploration of novel molecules with significant pharmacological activities remains a topic of intense research in medical sciences. The incessant search for new drugs is evident by a massive number of reported studies and is largely encouraged by the fact that various pathogens are able to improve processes of resistance to front-line drugs in their early life [1–6].
The amplified rate of bacterial resistance, associated with the slow development of new antibacterial agents, prefigures a crunch in this area. Actually, the global antibacterial clinical library is composed of many agents resulting from adjustments in primary classes of antibiotics [7]. Within the efforts by the scientific community to address the World Health Organization (WHO)’s Global Action Plan on Antimicrobial Resistance [8], principally in fighting Gram-negative bacteria, novel antibacterial drugs of greater specificity are urgently required [9].
E. faecium, S. aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species (ESKAPE) pathogens are the leading source of nosocomial infections worldwide [10, 11]. From these, the Gram-positive E. faecium and S. aureus may be tackled by glycopeptides of second-generation such as dalbavancin and oritavancin or the oxazolidinone linezolid, which have been described as antibacterial agents against infections by methicillin-resistant S. aureus (MRSA) [12, 13] and vancomycin (VAN) resistant enterococci (VRE) [11, 14]. The most recent successes concern the treatment of multidrug-resistant Gram-positive pathogens. However, even for these cases, a fast growth of resistance is expected, which makes the exploration of new antibacterial agents urgent [15–17]. Note, for example, that S. aureus, Enterococcus faecalis, E. faecium, and Staphylococcus epidermidis cause relevant nosocomial or healthcare-related infections, a major reason for mortality and morbidity worldwide [18].
Along with the need to develop new antibacterial agents that can circumscribe the growing resistance of bacteria to antibiotics, there is certainly an urgency in developing revolutionary treatments for cancer. Despite significant advances, the treatment of cancer remains a serious challenge, with novel and highly effective drugs and anticancer procedures still being pursued.
A tactical methodology exploring new drug candidates is the structure modification of existing drugs or other molecules with proven biological activity. In recent years, small molecules including five-membered heterocyclic moieties have become the subject of growing interest in designing antimicrobial and antitumor agents. Among the stated heterocyclic motifs are found, with some relevance, saccharin, tetrazole, and 1,3,4-thiadiazole derivatives (see Figure 1, for examples of drugs comprising such heterocycles in their structure).
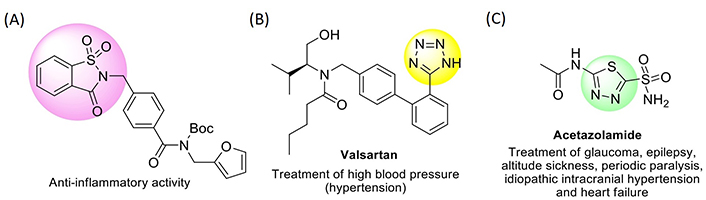
Structure representation of drugs containing saccharin (A), tetrazole (B), and 1,3,4-thiadiazole (C) frameworks
For more than a century, saccharin (1,1-dioxo-1,2-benzothiazol-3-one, 1; Figure 2) has been regularly used as a non-caloric artificial sweetener in the form of its water-soluble salts (mainly the sodium, ammonium, and calcium salts). The debate on its toxicity to humans lasted about thirty years since results on carcinogenicity in laboratory animals were published [19–21] and has not yet reached an authentic consensus. Nonetheless, various N-substituted saccharin analogues were evaluated for in vitro biological activity [22–25]. For example, structure-activity-relationship (SAR) studies have shown that the saccharin framework is an active unit for the development of inhibitors of human leukocyte elastase (HLE), cathepsin G (Cat G), and proteinase 3 (PR3), as well as anti-mycobacterial and central nervous system agents [26–28]. More recently, diverse saccharin-based antagonists for the interferon-signaling pathway were recognized, showing antitumor activity based on the inhibition of the cancer-related isoforms in humans [29].
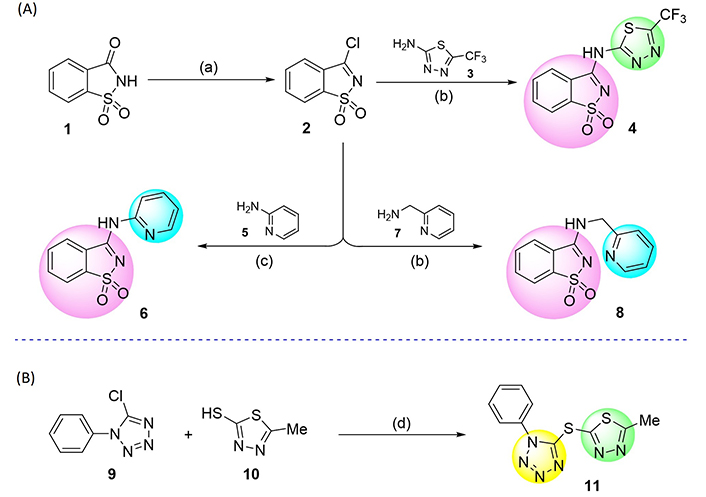
Approach to the synthesis of saccharin-thiadiazolyl (4), saccharin-pyridyl (6, 8), and tetrazole-thiadiazolyl (11) derivatives. Reaction conditions: (a) PCl5, 220°C, 4 h; (b) anhydrous tetrahydrofuran (THF), reflux, N2 atmosphere, 24 h; (c) 1,4-dioxane, 80°C, N2 atmosphere, 24 h; (d) NaH, anhydrous THF, room temperature, N2 atmosphere, 3 h
Likewise, tetrazole-based derivatives have attracted considerable attention due to their broad spectrum of biological activities. Several chemical, physical, physicochemical, and biological properties of tetrazoles are strictly correlated to their capability to act as acids and bases. In the tetrazole ring, the four nitrogen atoms connected in sequence, are able to be involved in proton-exchange processes, rendering this heterocycle both remarkable in the structure and distinctive in acid-base characteristics. The tetrazolic acid fragment, -CN4H, has comparable acidity to the carboxylic acid group, -CO2H, and is almost allosteric with it, but is metabolically more stable at the physiologic pH [30]. Consequently, approaches involving the substitution of -CO2H by -CN4H groups in molecules with pharmacological interest are of major significance [31], with the tetrazole moiety featuring, for example, in molecules with antibiotic, anti-hypertensive, anti-asthmatic, anti-tubercular, or anti-malarial activity [32–35]. Several tetrazole-based molecules have also shown promising activity as anticonvulsants, anticancer, and anti-human immunodeficiency virus (HIV)-1 [36–38].
1,3,4-Thiadiazoles are also an important class of heterocyclic compounds, with various biological activities and respective therapeutic applications. Usually, the bioactive properties of the 1,3,4-thiadiazole scaffold are ascribed to the fact that this ring is a bioisostere of pyrimidine, a motif present in the structure of three nucleic bases. Consequently, many 1,3,4-thiadiazole-based derivatives bear the capacity to disrupt processes related to DNA replication, with consequent inhibitory properties of both bacterial and cancer cell growth. Several 1,3,4-thiadiazole-based molecules were found to display a broad range of biological activities, such as antimicrobial [39], anticancer [40], antifungal [41], anti-inflammatory [42], antituberculosis [43], antioxidant [44], anticonvulsant [45], anti-depressant and anxiolytic [46], and anti-hypertensive [47]. The most notable thiadiazole-based drug so far is acetazolamide [N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide] (Figure 1), recognized as a carbonic anhydrase inhibitor, used for the treatment of high-altitude illness [48], epileptic seizures [49], glaucoma [50], idiopathic intracranial hypertension [51], hemiplegic migraine [52], or obstructive sleep apnea [53].
Following up on recent research aimed at the discovery of biologically active heterocyclic derivatives of the azole-type, strongly inspired by the conviction of Barry Sharpless on the conception of new molecules (“If chemists choose simple reactions, where there is a strong intrinsic drive for the molecules to bond together, they avoid many of the side reactions, with a minimal loss of material.” Barry Sharpless, Nobel prize in Chemistry 2001 and 2022), in the present work we disclose a preliminary biological survey covering distinct antineoplastic and antimicrobial activities of four hybrid-azoles, viz., saccharin-thiadiazolyl, saccharin-pyridyl, and tetrazole-thiadiazolyl conjugates.
All reagents for synthesis were acquired from commercial supplies and used without additional purification. Analytical thin layer chromatography (TLC) was accomplished using Merck (Darmstadt, Germany) TLC silica gel 60 F254 aluminium sheets and visualized under ultraviolet (UV) or by appropriate stain (p-anisaldehyde and potassium permanganate were the most used). 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded using a 400 MHz Bruker (Billerica, MA, USA) instrument or a 500 MHz JEOL system (Peabody, MA, USA) equipped with a Royal HFX probe, using dimethyl sulfoxide (DMSO)-d6 as deuterated solvent. All coupling constants are expressed in Hz and chemical shifts (δ) are described in parts per million (ppm), downfield from an internal standard of tetramethylsilane (TMS). Multiplicities are given as singlet (s), doublet (d), double doublet (dd), double-double-doublet (ddd), double triplet (dt), triplet (t), triple triplet (tt), and multiplet (m). Melting points (°C) were obtained on an SMP30 melting point apparatus and were uncorrected. High-resolution mass spectrometry (HRMS) was conducted on a Thermo Scientific HRMS (Waltham, MA, USA), model Orbitrap Elite, capable of MSn, n up to 10, available at the analytical services of Centre of Marine Sciences (CCMAR).
Saccharin (12 g, 56 mmol) and phosphorus pentachloride (16 g, 66 mmol) were added to a round bottom flash connected to a Graham condenser. The mixture was stirred at 220°C for 4 h, over a heating plate. Then, the reaction was allowed to cool to 180°C, and phosphorus oxychloride was distilled from the reaction medium under vacuum. Once the reaction mixture cooled to 100°C, toluene (1 mL) was first added, followed by the addition of chloroform (9 mL), when the temperature attained 70°C. The final product crystallised upon cooling the mixture slowly to room temperature. The recovered crystals were washed with cold toluene (30 mL) and chloroform (20 mL), giving pure 3-chloro-1,2-benzisothiazole 1,1-dioxide as colourless needles (4.8 g, 24 mmol, 36% yield). 1H NMR (400 MHz, DMSO-d6): δ 8.17 (d, J = 7.5 Hz, 1H), 8.00 (t, J = 8.0 Hz, 2H), and 7.93 (d, J = 7.3 Hz, 1H) ppm. 13C NMR (101 MHz, DMSO-d6): δ 160.61 (s), 139.17 (s), 135.72 (s), 134.89 (s), 127.32 (s), 124.95 (s), and 121.26 ppm (s). Found: C, 41.5%; H, 2.0%; N, 6.9%; calcd for C7H4NO2SCl: C, 41.7%; H, 2.0%; N, 7.0%. MS (EI, m/z): 201 [M]+. M.p. 143°–145°C.
3-Chloro-1,2-benzisothiazole 1,1-dioxide (1.00 g, 4.96 mmol) and 2-amino-5-trifluoromethyl-1,3,4-thiadiazole (0.923 g, 5.46 mmol) were dissolved in dry THF (30 mL). The mixture was stirred at 60°C for 24 h under a nitrogen atmosphere, then the solvent was evaporated under reduced pressure, distilled water (30 mL) was added, and the supernatant was extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered, and evaporated under reduced pressure. Recrystallization from ethanol gave compound 4 light green crystals (0.69 g, 2.07 mmol, 42% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.38–8.29 (m, 1H), 8.05 (dd, J = 6.0 Hz and 2.6 Hz, 1H), and 7.88–7.82 ppm (m, 2H). 13C{1H} NMR (126 MHz, DMSO-d6) δ 166.63, 159.67, 152.51, 142.30, 134.60, 134.31, 129.18, 124.79, 122.29, 121.55, and 119.39 ppm. HRMS (ES+, m/z) calcd for C10H6F3N4O2S2 (M + H)+: 334.98788; found: 334.98758. M.p. 320°–321°C (lit. [54]: 320°–321°C; see 1H, 13C{1H} NMR and mass spectra at Figures S1–3).
A mixture of 3-chloro-1,2-benzisothiazole 1,1-dioxide (1.00 g, 4.96 mmol) and pyridin-2-amine (0.515 g, 5.46 mmol) in 1,4-dioxane (30 mL) was stirred at 80°C for 24 h, under a nitrogen atmosphere. The solvent was evaporated under reduced pressure and the remaining solid residue was washed with distilled water (30 mL), filtered, and then dried under reduced pressure at room temperature. Recrystallization of the solid from acetone gave compound 6 yellow crystals (0.58 g, 2.24 mmol, 45% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.74 (s, 1H), 8.74–8.65 (m, 2H), 8.52 (ddd, J = 4.8, 1.8, and 0.8 Hz, 2H), 8.29 (d, J = 8.4 Hz, 2H), 8.14–8.07 (m, 1H), 8.01 (ddd, J = 8.4, 7.5, and 1.9 Hz, 1H), 7.93–7.86 (m, 2H), and 7.33 ppm (ddd, J = 7.4, 4.8, and 0.9 Hz, 1H). 13C{1H} NMR (126 MHz, DMSO-d6) δ 157.04, 150.85, 148.39, 140.53, 138.93, 134.01, 133.65, 128.06, 124.28, 121.64, 121.37, and 116.48 ppm. HRMS (ES+, m/z) calcd C12H10N3O2S (M + H)+: 260.04882; found: 260.0486 Diff: 0.85 ppm. M.p. 247°–249°C (lit. [55]: 247°–249°C; see 1H, 13C{1H} NMR, and mass spectra at Figures S4–6).
A solution of 3-chloro-1,2-benzisothiazole 1,1-dioxide (1.00 g, 4.96 mmol) and pyridin-2-ylmethanamine (0.564 mL, 5.46 mmol) in dry THF (30 mL) was stirred at 60°C for 24 h, under a nitrogen atmosphere. The solvent was evaporated under reduced pressure and the supernatant was washed with cold distilled water (30 mL) and ethyl acetate (3 × 20 mL). The organic phase was dried with anhydrous MgSO4, filtered, and finally dried under reduced pressure. Recrystallization from ethyl acetate gave compound 8 as yellow needles (0.68 g, 2.49 mmol, 50% yield). 1H NMR (500 MHz, DMSO-d6) δ 10.71 (t, J = 5.5 Hz, 1H), 8.84–8.80 (m, 1H), 8.45–8.41 (m, 1H), 8.37 (td, J = 7.8 Hz and 1.7 Hz, 1H), 8.02–7.98 (m, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.89–7.84 (m, 2H), 7.82 (ddd, J = 7.4, 5.6, and 1.3 Hz, 1H), and 5.01 ppm (d, J = 5.7 Hz, 2H). 13C{1H} (126 MHz, DMSO-d6) δ 159.77, 156.39, 149.26, 142.27, 137.07, 133.67, 133.27, 127.64, 123.14, 122.80, 121.91, 121.33, and 47.65 ppm. HRMS (ES+, m/z) calcd C13H12N3O2S (M + H)+: 274.06447; found: 274.0644 Diff: 0.26 mol/L. M.p. 257°–259°C (lit. [56]: 260°C; see 1H, 13C{1H} NMR, and mass spectra at Figures S7–9).
5-Methyl-1,3,4-thiadiazole-thiol (1.00 g, 7.56 mmol, 1 eq.) and NaH (0.74 g, 30.25 mmol, 4 eq.) were added to dry THF (20 mL) and the mixture was stirred for 3 h, at room temperature, under anhydrous conditions. Then, a solution of 5-chloro-1-phenyl-1H-tetrazole (1.37 g, 7.56 mmol, 1 eq.) in dry THF (20 mL) was gently added, using a syringe. The mixture was stirred for 24 h, at room temperature, under a nitrogen atmosphere. The solvent was evaporated under reduced pressure and the remaining solid was washed with freezing distilled water (30 mL) and extracted with dichloromethane DCM (3 × 20 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered, and evaporated under reduced pressure. The collected solid was dissolved in acetone and allowed to crystallise at room temperature, giving the final product pale yellow crystals (0.5 g, 1.81 mmol, 24% yield). 1H NMR (500 MHz, DMSO-d6) δ 7.76–7.72 (m, 2H), 7.67 (tt, J = 3.9 Hz and 2.9 Hz, 3H), and 2.72 ppm (s, 3H). 13C{1H} NMR (126 MHz, DMSO-d6) δ 169.90, 157.37, 150.03, 132.90, 131.15, 129.95, 125.28, and 15.45 ppm. HRMS (ES+, m/z) calcd C10H9N6S2 (M + H)+: 277.03246; found: 277.0320 Diff: 1.66 ppm. M.p. 97°–99°C (see 1H, 13C{1H} NMR, and mass spectra at Figures S10–12).
The antimicrobial properties of compounds 4, 6, 8, and 11 were evaluated by the well diffusion method [57], against Gram-positive bacteria [Staphylococcus aureus American Type Culture Collection (ATCC) 25923, Staphylococcus epidermidis ATCC 12228, and Mycobacterium smegmatis ATCC 607], Gram-negative bacteria (Pseudomonas aeruginosa ATCC 27853), and yeast (Saccharomyces cerevisiae ATCC 2601 and Candida albicans ATCC 10231) strains. DMSO solutions for each of the compounds, at the concentration of 10 mg/mL, were used in all the assays. Agar plates were inoculated with a standardized inoculum of the tested microorganism. Several wells (about 5 mm in diameter) were made into the solid medium to place the samples and the controls. The Petri dishes were incubated at 37°C for 24 h.
Human colon adenocarcinoma HCT116 cells were provided by Dr. Vogelstein (The Johns Hopkins Kimmel Cancer Center, Baltimore, USA); human breast adenocarcinoma MCF-7 and melanoma A375 were from ATCC (Rockville, MD, USA). Cells were cultured in RPMI-1640 with Ultra-Glutamine (Lonza, VWR, Portugal) with 10% fetal bovine serum (FBS; Gibco, Alfagene, Portugal), at 37°C with 5% CO2. Mycoplasma detection was routinely performed using the MycoAlertTM PLUS kit (Lonza).
Human cell lines were seeded in 96-well plates at a density of 5.0 × 103 cells/well (HCT116, MCF-7, and A375), and allowed to adhere for 24 h. Cells were treated with serial dilutions of compounds (ranging from 0.1 μmol/L to 50 μmol/L), for an additional 48 h. Effect on cell proliferation was measured by sulforhodamine B (SRB) assay, as described by Raimundo et al. [58] and the concentration of compound that induces 50% growth inhibition (GI50) was then determined for each cell line using the GraphPad Prism software version 7.0 (La Jolla, CA, USA).
U87 (brain-likely glioblastoma), A172 (glioblastoma), and H4 (neuroglioma) cell lines were a kind gift from Prof. C. Vitorino (University of Coimbra, Portugal). Cells were cultivated in Dulbecco’s modified eagle’s medium-high glucose (DMEM-HG; Biowest, Nuaillé, France) supplemented with 10% (v/v) of heat-inactivated FBS (Sigma, St. Louis, MO, USA) and 1% (v/v) of penicillin-streptomycin (Sigma, St. Louis, MO, USA), being maintained at 37°C with 5% CO2.
U87, A172, and H4 cell lines (1 × 104 cells/well) were seeded in 96-well plates. After 24 h, compounds 4, 6, 8, 11, and ArgKa extract were added to U87 cells, while compound 11 was administered to A172 and H4 cell lines. Cells were treated with the compounds at the same concentrations, ranging from 0.39 µg/mL to 100 µg/mL, or with the vehicle control (DMSO) and further incubated for 24, 48, and 72 h. Cell viability was assessed via a modified Alamar blue® assay [59]. Briefly, a solution of DMEM-HG medium with 10% (v/v) of a stock solution of resazurin salt dye at a concentration of 0.1 mg/mL was prepared and further added to each well after 24, 48, and 72 h. After incubation (4 h), the absorbance of the plate was read at 570 nm and 600 nm in a BioTeck (BioTek Instruments, Inc., Winooski, VT, USA). The absorbance results were obtained by the Gen5 program. Cell viability was calculated using the following equation:
Data were analyzed using GraphPad Prism v.7.04. All experiments were performed in triplicate and acquired results were expressed as mean ± standard deviation (SD). Statistical analysis was performed by one-way ANOVA, using the multiple comparison test Dunnett. A value of P < 0.05 was considered significant.
The synthesis of 3-chloro-1,2-benzisothiazole 1,1-dioxide (2), one of the key molecular scaffolds used for the production of the tested compounds, was initially carried out through halogenation of saccharin (1) by adapting the procedure followed by Ismael et al. [60]. Saccharin derivatives 4, 6, and 8 were synthesized by reacting the amino-thiadiazole 3 or amino-pyridines 5 and 7 with pseudo-saccharyl chloride (2). The building blocks were combined via nucleophilic displacement of the chloride anion by the amine functionality (Figure 2A).
Sequentially, molecule 11 was synthesized by coupling 5-chloro-1-phenyl-1H-tetrazole (9) with 5-methyl-1,3,4-thiadiazole-2-thiol (10; Figure 2B) via nucleophilic displacement of chloride anion by the thiolate functionality. All the reactions proceeded efficiently, adjusting equivalent experimental protocols earlier established by some of us [61–65], giving crystalline and chemically stable products in satisfactory yields.
The antimicrobial properties of molecules 4, 6, 8, and 11 were evaluated by using the agar well diffusion method, against Gram-positive (S. aureus, S. epidermidis, and M. smegmatis), Gram-negative (P. aeruginosa), and yeast (S. cerevisiae and C. albicans) strains, obtained from ATCC (Table 1).
Growth inhibition area of the compounds 4, 6, 8, and 11 at 10 mg/mL by the agar well diffusion method against Gram-positive, Gram-negative bacteria, and yeast strains
| Compound | Zone of inhibition/mm | |||||
|---|---|---|---|---|---|---|
| S. aureus | S. epidermidis | M. smegmatis | P. aeruginosa | S. cerevisiae | C. albicans | |
| 4 | n.a. | 7 | n.a. | n.a. | 7 | n.a. |
| 6 | n.a. | n.a. | n.a. | n.a. | 8 | n.a. |
| 8 | 6 | n.a. | n.a. | n.a. | 8 | n.a. |
| 11 | 15 | 12 | n.a. | n.a. | 9 | n.a. |
| Positive control | VAN 26 | VAN 27 | RIF 48 | NOR 33 | NYS 31 | NYS 29 |
n.a.: not active; NOR: norfloxacin; NYS: nystatin; RIF: rifampicin
At the concentration of 10 mg/mL (DMSO solution) all the tested molecules showed modest inhibitory potential against S. cerevisiae (inhibition area between 7 mm and 9 mm). Besides, derivatives 8 and 4 also showed slight inhibitory activity against S. aureus (6 mm) and S. epidermidis (7 mm) strains, respectively. Inversely, derivative 11 exhibited promising inhibition activity against S. aureus (15 mm) and S. epidermidis (12 mm; Table 1).
Subsequently, to the examination of antimicrobial activity, compounds 4, 6, 8, and 11 were submitted to anticancer evaluation against distinct types of cancer cells, namely: human colon adenocarcinoma (HCT116), human breast adenocarcinoma (MCF-7), and melanoma (A375).
Based on the GI50 values obtained by SRB assay [58], a promising antiproliferative activity against the aforementioned cancer cells has been clearly attested for compound 11. In fact, GI50 values for derivative 11 range from 3.55 µmol/L to 11.5 µmol/L on the tested cells, significantly lower than those observed for the remaining derivatives (Table 2).
Effect of compounds on the growth of human cancer cells
| Compound | GI50 (µmol/L) | ||
|---|---|---|---|
| HCT116 | MCF-7 | A375 | |
| 4 | > 50 | > 50 | > 50 |
| 6 | > 50 | 35.5 ± 1.5 | 23.8 ± 1.9 |
| 8 | > 50 | > 50 | > 50 |
| 11 | 3.55 ± 0.15 | 11.5 ± 0.5 | 9.6 ± 1.1 |
| Etoposide | 0.71 ± 0.06 | 2.70 ± 0.1 | 0.85 ± 0.09 |
GI50 values were determined by SRB assay after 48 h treatment (growth obtained with the vehicle set as 100%). Data are mean ± standard error of the mean (SEM) of four to six independent experiments. Etoposide was used as a positive control
Following prior cytostatic evaluation of molecules 4, 6, 8, and 11, the cytotoxic/antiproliferative potential of such molecular entities, together with ArgKa, was assessed using Alamar blue® assay [59]. Primarily, U87 cells (brain-likely glioblastoma) were treated for 24, 48, and 72 h with distinct concentrations (100, 50, 25, 6.25, 1.56, and 0.39 µg/mL) of the different compounds. Through data analysis, it was observed that treatment with the higher concentration of compound 11 (100 µg/mL) induced a decrease in U87 cell viability by 50% after 48 h and 72 h (**** P < 0.0001). For a concentration of 50 µg/mL of the same derivative, a considerable decline in cell viability (approx, 60%) was also observed after 72 h. On the other hand, the treatment of U87 cells using concentrations of 11 below 50 µg/mL did not produce a significant effect, once their proliferative capability did not drop below 80% relative to the control condition (untreated cells; see Figure 3). Precisely the same scenario was observed for the treatment with derivatives 4, 6, and 8, for all the tested concentrations (see Figure 3). From these results, it becomes evident that compound 11 is the one showing undeniable cytotoxic activity against brain tumor cells.
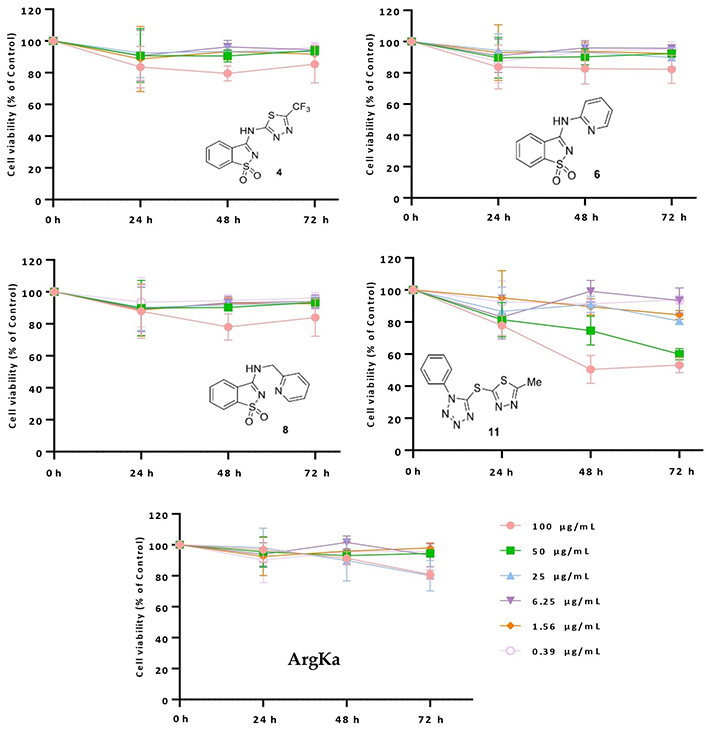
Cytotoxic/antiproliferative effect of derivatives 4, 6, 8, 11 and ArgKa against U87 cell line. Cells were treated with the same extract concentrations for 24, 48, and 72 h, and subsequently subjected to Alamar Blue® assay. Cell viability is represented by three independent experiments and is expressed as the percentage of survival compared to control (untreated cells)
Supported by the results obtained on U87 cells, the cytotoxic profile of 11 was also assessed in additional glioma cell lines [A172 (glioblastoma) and H4 (neuroglioma)] after 24, 48, and 72 h of treatment. According to the data obtained for A172 cells, only the treatment with 100 µg/mL of derivative 11 induces a significant decrease in cell viability after 72 h of treatment (**** P < 0.0001), which, by a first instance, would not be expected (Figure 4A). Nevertheless, a completely different situation was observed for H4 cells (Figure 4B), where treatment with all concentrations of 11 promoted a significant decrease in cell viability (< 40–60%; **** P < 0.0001) after 24 h of incubation (Figure 4B). A similar cytotoxic profile was observed after 48 h and 72 h of treatment with the higher concentrations of this molecule (25, 50, and 100 µg/mL; **** P < 0.0001).
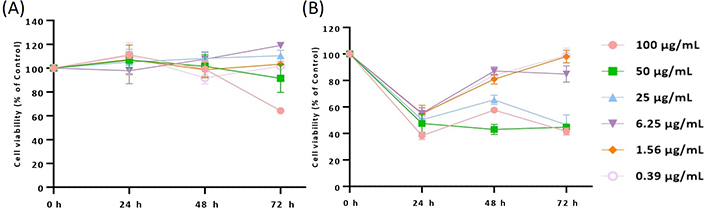
Cytotoxic/antiproliferative profile of derivative 11 in A172 (A) and H4 (B) cell lines. Cells were treated with the same extract concentrations for 24, 48, and 72 h, and subsequently subjected to Alamar Blue® assay. Cell viability is represented by three independent experiments and is expressed as the percentage of survival compared to control (untreated cells)
The present research work reveals the design, synthesis, and biological activity (antimicrobial and anticancer properties) of saccharin-thiadiazolyl (4), saccharin-pyridyl (6, 8), and tetrazole-thiadiazolyl (11) conjugates.
To the best of our knowledge, the derivatives 4 and 11 are completely new. Their design and synthesis have been developed by us through this investigation. Differently, molecules 6 and 8 have been previously synthetised [56, 66], although using dissimilar experimental protocols. Besides, these derivatives including pyridine moiety have not been earlier subjected to similar biological activities assessment to those reported, particularly as potential anticancer agents, justifying their selection.
The antimicrobial properties of the synthesized molecules were assessed against strains of Gram-positive (S. aureus, S. epidermidis, and M. smegmatis) and Gram-negative bacteria (P. aeruginosa), and also against strains of yeasts (S. cerevisiae and C. albicans). With the exception of derivative 11, which displayed promising inhibitory activity against S. aureus and S. epidermidis, all the other molecules exhibited modest inhibitory potential against the tested strains. It should be noted that from a structural point of view, compound 11 differs significantly from the other three amino-linked saccharyl-based conjugates under study. This molecule comprises two heterocycles, tetrazole, and thiadiazole, linked through a sulfur bridge instead of an amine moiety. The promising results shown by compound 11 with regard to its particular inhibitory activity against strains of Gram-positive bacteria prompts us to prepare new analogues based on the structural tetrazole-S-thiadiazole scaffold, altering the functional groups coupled to heterocyclic rings, to build a chemically diverse library for antimicrobial assessment and selection of leads and further development.
Concerning the studies of anticancer activity, treatment of human colon adenocarcinoma (HCT116), human breast adenocarcinoma (MCF-7), and melanoma (A375) cells with the compounds under scrutiny, exposed an encouraging antiproliferative activity of derivative 11 against these cancer cell lines, with GI50 values oscillating from 3.55 µmol/L to 11.5 µmol/L. The main highlight is the result obtained in the assay with cells of the HCT116 type, where a GI50 value of 3.55 µmol/L was observed, which is within the same order of magnitude of the value disclosed to the drug etoposide, used as a positive control. Such antiproliferative activity stimulated an expedited and ampler investigation using compound 11 as a cytostatic or chemotherapeutic agent for cancer through additional cell lines and divergent assays.
The treatment of brain-likely glioblastoma cells (U87) with compound 11 at the concentration of 100 µg/mL induced a decrease in cell viability by 50% after 48 h and 72 h, while data obtained for glioblastoma (A172) cells have shown that only the treatment with 100 µg/mL of the drug induces a significant decrease on cell viability after 72 h. On the other hand, treatment of neuroglioma (H4) cells with derivative 11 promoted a significant decrease on cell viability (< 40–60%) after 24 h.
Based on the cytotoxic effects achieved for compound 11 on brain-likely glioblastoma (U87) and neuroglioma (H4) cells, it seems worthy to explore their antitumor potential under additional investigation programs.
As previously discussed for antimicrobial assays, derivative 11 differs considerably from the other three amino-linked saccharyl-based conjugates, comprising tetrazole and thiadiazole units, linked through a sulfur bridge instead of an amine moiety. Likewise, the auspicious results shown by compound 11 in the anticancer tests prompt us to prepare different molecules based on the structural tetrazole-S-thiadiazole scaffold, altering the functional groups coupled to heterocyclic rings, to build a chemically diverse library for anticancer assessment. A detailed SAR analysis is difficult to do correctly at this point in the project. We intend to carry out a complete SAR analysis (including computational) when we achieve results with a larger number of compounds from this chemical family.
ATCC: American Type Culture Collection
d: doublet
ddd: double-double-doublet
DMSO: dimethyl sulfoxide
GI50: 50% growth inhibition
HRMS: high-resolution mass spectrometry
m: multiplet
NMR: nuclear magnetic resonance
ppm: parts per million
s: singlet
SAR: structure-activity-relationship
SRB: sulforhodamine B
t: triplet
THF: tetrahydrofuran
VAN: vancomycin
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/100828_sup_1.pdf.
LMTF: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing—original draft, Writing—review & editing. PR: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing—review & editing. MLSC: Supervision, Funding acquisition, Writing—review & editing. BECG and ICCC: Investigation, Methodology. VMSI: Methodology, Formal analysis. LS, BGN, MM, and CC: Investigation, Methodology, Validation, Formal analysis.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
This work was partially supported by the Foundation for Science and Technology (FCT), Portugal, via Strategic Projects [UIDB/00100/2020, UIDP/00100/2020 (CQE), LA/P/0056/2020 (IMS), UIDB/04326/2020, UIDP/04326/2020, LA/P/0101/2020 (CCMAR), UIDB/04539/2020, UIDP/04539/2020, LA/P/0058/2020 (CIBB), UIDB/04567/2020, UIDP/04567/2020 (CBIOS)]. Funds from operational programs CRESC Algarve 2020 and COMPETE 2020 through project EMBRC.PT [ALG-01-0145-FEDER-022121] are object of acknowledgement. LMTF expresses gratitude to FCT for the work contract nº IST-ID/115/2018. ICCC and MM thank FCT for the PhD grants [SFRH/BD/08242/2020, SFRH/BD/146441/2019], respectively. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

