 Review
Review

Affiliation:
1School of Life Sciences, Management Development Institute of Singapore, Singapore 148951, Singapore
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0004-3819-1401
Affiliation:
1School of Life Sciences, Management Development Institute of Singapore, Singapore 148951, Singapore
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0003-2705-7701
Affiliation:
2Department of Anatomy, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117594, Singapore
Email: dineshkumar@nus.edu.sg
ORCID: https://orcid.org/0000-0001-6535-5441
Explor Drug Sci. 2026;4:1008152 DOI: https://doi.org/10.37349/eds.2026.1008152
Received: November 09, 2025 Accepted: January 07, 2026 Published: February 26, 2026
Academic Editor: Fernando Albericio, University of KwaZulu-Natal, South Africa, Universidad de Barcelona, Spain
Ageing is a gradual, multifactorial process that leads to the deterioration of physical and mental health, increasing the risk of disease and eventually death. Indicators of ageing manifest at the molecular level, including genomic instability, telomere attrition, epigenetic alterations, mitochondrial dysfunction, loss of proteostasis, and dysregulation of key signalling pathways such as the mechanistic target of rapamycin (mTOR) and insulin signalling. These molecular hallmarks of ageing are interconnected, amplifying one another over time. The resulting cellular stress triggers apoptosis or drives cells into a pathological state known as cellular senescence, in which they secrete inflammatory, pro-ageing factors. Consequently, there is a progressive decline in tissue function and regenerative capacity, accompanied by atrophy and stem cell exhaustion under a chronically inflamed microenvironment. Although functional decline with age is irreversible, research indicates it can be delayed. In this review, we discuss the hallmarks of ageing, conventional pharmacological interventions with demonstrated anti-ageing effects in cellular and animal models, and emerging therapeutic strategies being explored as ageing becomes increasingly recognized as a major risk factor for disease development.
The world is confronted with an ageing population on an unprecedented scale. Global life expectancy has increased by approximately 58% from 1950 to 2025 and continues to rise [1]. In contrast, global fertility rates have declined sharply, from 4.9 children per woman in 1950 to 2.3 in 2021 [2, 3]. This combination of longer life expectancy and a decreasing birth rate has produced a demographic shift toward older populations. Projections suggest that the population aged over 80 will triple, and those over 65 will double within the next 30 years [4]. This demographic transition presents major challenges. Ageing is accompanied by a rising prevalence of chronic diseases and disabilities, contributing to escalating healthcare costs worldwide. Meeting this demand requires robust, integrated healthcare systems capable of managing both acute and chronic age-related conditions [1, 5]. Non-communicable diseases such as cardiovascular and neurodegenerative disorders now account for nearly one-quarter of the global healthcare burden among those over 60 [6, 7]. Consequently, global healthcare spending is projected to reach 3.5 trillion USD this year, with an annual growth rate of at least 10% [8, 9].
Beyond healthcare, ageing has profound socio-economic consequences. As the share of working-age adults declines, labour shortages, slower economic growth, and rising dependency ratios place additional fiscal and social strain on families and governments [4, 10]. Retirees face increasing medical costs paired with reduced income, forcing both individuals and economies to allocate more resources toward sustaining older populations [11]. This pressure underscores an urgent need for advances in anti-ageing and longevity research. With growing scientific insight and medical innovation, ageing is being studied as a multifactorial biological process influenced by genetic, cellular, and environmental mechanisms. This review explores current understanding of these mechanisms and highlights both established and emerging therapeutic approaches aimed at delaying this inevitable process.
Genomic instability refers to the accumulation of somatic mutations within the genome, which increases in frequency with age. These mutations include point mutations, deletions, chromosomal translocations, and DNA strand breaks. They can arise from endogenous sources, such as reactive oxygen species (ROS) generated during metabolism, or from exogenous sources, such as ultraviolet (UV) radiation. In normal cells, proteins such as DNA polymerases, glycosylases, nucleases, ligases, and protein complexes involved in DNA repair pathways, including base excision repair (BER) and nucleotide excision repair (NER), maintain genomic integrity [12]. As organisms age, the efficiency of DNA repair mechanisms declines due to downregulation of genes encoding these proteins, leading to the accumulation of unrepaired DNA damage in the form of mutations [6, 13]. Over time, when genetic damage reaches harmful levels, the cell cycle is halted, and apoptosis or cellular senescence is triggered [14].
Telomeres are protein–DNA structures located at the ends of linear chromosomes that act as protective caps, preventing chromosomal degradation and preserving genomic stability [15]. These repetitive non-coding sequences protect essential DNA from erosion during each round of replication, thereby addressing the end-replication problem [16]. Telomere attrition refers to the progressive shortening of telomeres over time due to both cell division and damage. The telomeric repeat sequence ‘TTAGGG’ is guanine-rich and particularly vulnerable to oxidative stress, especially from ROS [16, 17]. Oxidative damage accelerates telomere shortening beyond that caused by replication alone, and with age, cells exhibit a reduced capacity to counteract oxidative damage [18]. When telomere length reaches a critical threshold, cells undergo cell cycle arrest and are driven into cellular senescence or apoptosis [19].
Epigenetic alterations are heritable changes in gene activity that occur without modification of the underlying DNA sequence [6]. With age, four major types of epigenetic changes are typically observed: alterations in DNA methylation, histone protein dysregulation, non-coding RNA dysregulation, and changes in chromatin remodelling [20]. Ageing is associated with global hypomethylation, especially in repetitive regions, which can inappropriately expose normally silenced DNA to transcriptional activity and lead to aberrant gene expression. Conversely, hypermethylation may occur at promoters of protective genes, such as tumour suppressor genes, silencing their expression and reducing the cell’s capacity to prevent malignant transformation, thereby compromising genomic integrity [6].
Histone proteins, which regulate chromatin compaction and thereby control access to DNA, also undergo age-associated changes in abundance and structure. These changes can inappropriately activate normally silenced genes, such as oncogenes, or repress genes essential for genome maintenance and repair. Histone marks are crucial for recruiting DNA repair factors, and their dysregulation leads to impaired DNA repair [21]. Similarly, age-related dysregulation of non-coding RNAs, including microRNAs and long non-coding RNAs, disrupts normal post-transcriptional and transcriptional gene regulation, contributing to loss of homeostasis [22]. In addition, ageing is characterized by loss of constitutive heterochromatin and disruption of higher-order chromatin organization, due in part to altered chromatin-remodelling complexes, reduced sirtuin activity, and damage to the nuclear lamina [6]. These epigenetic and structural alterations, combined with genomic instability and inefficient repair, promote cellular stress and lead to apoptosis or cellular senescence, thereby contributing to ageing.
Proteostasis refers to the dynamic balance of protein synthesis, folding, quality control, and degradation necessary to maintain a functional proteome. With ageing, this balance becomes disrupted, leading to an accumulation of misfolded and aggregated proteins [23, 24]. Impaired protein degradation and reduced chaperone activity result in fewer correctly folded, functional proteins, while defects in protein trafficking further compromise cellular function. Aggregated proteins exert proteotoxic stress, interfering with normal cell processes and triggering cellular stress responses that may lead to apoptosis or cellular senescence. The accumulation of senescent cells and cell death associated with loss of proteostasis has been implicated in several age-related neurodegenerative disorders [25].
A decline in mitochondrial function and number is a common feature of ageing [26]. Mitochondria primarily generate adenosine triphosphate (ATP) through oxidative phosphorylation, which depends on the electron transport chain (ETC), comprising respiratory complexes I–IV and ATP synthase [27]. Ageing is associated with reduced activity of complexes I and IV, whereas changes in complexes II and III are generally less pronounced [28]. Reduced ETC efficiency promotes electron leakage, increasing ROS production and further damaging cellular components [29].
Mitochondrial DNA (mtDNA) accumulates mutations with age, similar to nuclear DNA, which further impairs mitochondrial function and exacerbates ROS generation. mtDNA is particularly vulnerable because it lacks protective histones and relies mainly on BER, with limited capacity for other repair pathways such as NER and mismatch repair (MMR) [29]. As a result, mtDNA damage accumulates, worsening mitochondrial dysfunction and contributing to cellular stress. In healthy cells, mitochondria undergo continuous fission and fusion to maintain quality control and enable the segregation or removal of damaged components through mitophagy [30]. With age, the expression of fission and fusion proteins becomes dysregulated, leading to either excessively fragmented or hyperfused mitochondria, both of which are functionally compromised [27, 30].
Mitochondrial dysfunction increases ROS production, amplifying macromolecular damage, while reduced oxidative phosphorylation leads to diminished ATP levels and an energy deficit that undermines cellular maintenance and repair, thereby heightening cellular stress [27]. The mitochondrial unfolded protein response (mtUPR), a stress pathway that maintains mitochondrial protein homeostasis, also declines with age. Under proteotoxic stress, the mtUPR activates transcription factors that translocate to the nucleus and upregulate genes encoding mitochondrial chaperones, co-chaperones, and proteases, promoting clearance of misfolded proteins and restoration of proteostasis [27, 31]. With ageing, reduced mtUPR efficiency impairs the removal of damaged proteins, destabilizes the ETC, increases ROS production, and promotes further mtDNA damage, ultimately driving mitochondrial dysfunction and, downstream, cellular senescence or apoptosis [31].
The mechanistic target of rapamycin (mTOR) is a serine/threonine protein kinase that regulates cellular metabolism, growth, survival, autophagy, and immune responses [32]. mTOR functions in two distinct complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [32, 33]. mTORC1 primarily senses nutrient and growth factor availability to regulate protein synthesis and metabolism, whereas mTORC2 is more closely associated with cell survival, proliferation, and cytoskeletal organization [34]. Together, these complexes help maintain the balance between anabolism and catabolism in response to cellular conditions [33].
With advancing age, mTOR activity is often found to be chronically elevated, promoting anabolic processes while inhibiting catabolic pathways such as autophagy, leading to loss of metabolic homeostasis [34, 35]. Although the precise mechanisms by which mTOR contributes to ageing are still being elucidated, hyperactivation of this pathway is known to increase oxidative and proteotoxic stress, reduce cellular stress resistance, and impair clearance of damaged macromolecules. These changes collectively accelerate functional decline and promote the onset of age-related pathologies. While the exact mechanism of action of this complex is still not fully understood, it is known to increase oxidative and proteotoxic stress within cells, decrease autophagy, cause mitochondrial dysfunction, and dampen the sensitivity and feedback control of nutrient-sensing pathways, thereby impairing the body’s ability to adapt to nutrient scarcity and hastening cellular senescence [34, 36]. The insulin/insulin-like growth factor (IGF)-1 signalling (IIS) pathway also activates mTOR, which can accelerate ageing when excessively stimulated and increases oxidative damage and ROS generation due to impaired mitochondrial activity [6].
Research shows that the body’s ability to respond to insulin deteriorates with age, although the precise pathophysiological mechanisms underlying this change remain unclear [37, 38]. Reduced insulin sensitivity results in hyperinsulinaemia, which has multiple downstream repercussions in cells and may be related to increased body fat and decreased lean mass [39–41]. Additionally, nutrient signalling through the IIS pathway, mediated by pancreatic beta cells during fasting–feeding cycles, is one of the primary pathways influencing ageing and becomes dysregulated with age [39, 42]. Insulin is secreted in response to glucose and promotes anabolic processes, while IGF-1 is a growth factor stimulated by hormones to support cell division and proliferation [43]. Under appropriate regulation, these mechanisms are beneficial, but with age they can become dysregulated and promote excessive anabolism, disrupting metabolic homeostasis and accelerating ageing [43, 44]. Specifically, the IIS pathway can drive ageing by suppressing the transcription factor forkhead box O (FOXO), which is essential for maintaining cellular quality control, particularly under stress and nutrient deprivation [44–46].
Among the nine hallmarks of ageing originally described by López-Otín et al. [6] (2013), which more recent work has expanded to fourteen, this review focuses on seven that most directly align with the therapeutic pathways discussed, to maintain a relevant scope [47]. These molecular indicators of ageing are not independent phenomena but are interconnected through overlapping pathways that exacerbate one another. Progression of dysfunction in one hallmark can precipitate decline in others. For example, epigenetic alterations can promote genomic instability and influence downstream factors such as telomere attrition and loss of proteostasis [6, 48]. Similarly, overactivation of mTOR increases the likelihood of mitochondrial dysfunction and loss of proteostasis, thereby promoting cellular senescence and further accelerating ageing [49, 50].
Cellular senescence usually occurs when damaged or stressed cells permanently stop dividing. Senescent cells secrete a collection of factors termed the senescence-associated secretory phenotype (SASP), which drives chronic inflammation and tissue damage [51]. The SASP includes pro-inflammatory cytokines such as interleukin (IL)-6, IL-8, IL-1β, tumour necrosis factor-alpha (TNF-α), monocyte chemoattractant protein-1 (MCP-1), growth factors, and matrix metalloproteinases (MMPs), which are persistently secreted, as described by Coppé et al. [52]. These factors recruit monocytes, macrophages, neutrophils, and other immune cells, leading to chronic immune activation, and can also induce senescence in neighbouring cells, amplifying the impact of ageing [52]. MMPs contribute to the degradation of the extracellular matrix, weakening tissue structural integrity [52]. Additionally, senescent cells activate senescent cell anti-apoptotic pathways (SCAPs), enabling them to resist apoptosis and persist, thereby amplifying their detrimental effects [53, 54]. When cells become senescent or undergo apoptosis in response to damage, the pool of functional cells within tissues declines. This loss is particularly critical in muscle, immune, and brain tissues, which already exhibit reduced regenerative potential with age. Senescence and apoptosis also deplete stem cell pools, as stem cells undergo repeated divisions to replace lost cells and eventually experience exhaustion, losing self-renewal and differentiation capacity despite the presence of telomerase [6, 55]. This stem cell exhaustion leads to impaired regeneration and tissue repair [56] (Figure 1).
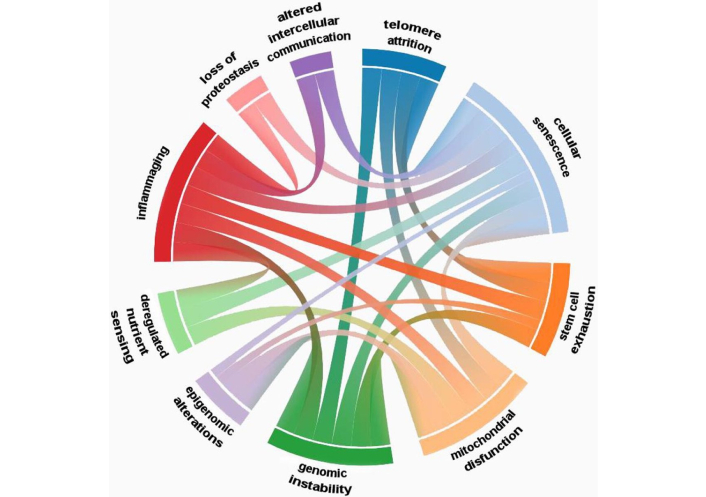
Interrelations between the hallmarks of aging. Reprinted from [56]. © 2023 The Authors. Licensed under CC-BY 4.0.
Although no drugs have been officially approved specifically for anti-ageing treatment, several therapies have attracted considerable interest for their mechanisms that promote anti-ageing and for their observed anti-ageing effects in individuals receiving them for other medical conditions.
Metformin is a small molecule widely noted for its potential anti-ageing effects in patients prescribed this drug as a first-line treatment for type II diabetes [57]. Multiple mechanisms of action have been proposed for metformin, many of which converge on improved geroprotection [58]. Primarily, metformin lowers blood glucose and lipid levels by activating AMP-activated protein kinase (AMPK), a central energy sensor. This activation promotes increased glucose uptake by myocytes, downregulates gluconeogenesis in hepatocytes, improves insulin sensitivity in both myocytes and hepatocytes, suppresses the expression of lipogenic enzymes, enhances fatty acid oxidation, and reduces serum triglyceride and low-density lipoprotein (LDL) levels [58–61]. These effects help maintain glycaemic control and confer cardioprotective benefits [62].
On a broader scale, AMPK activation indirectly suppresses mTOR signalling and shifts cellular metabolism away from excessive anabolism towards more catabolic and stress-resistant states, thereby mitigating pro-ageing mTOR activity [63, 64]. Metformin also reduces ROS production by partially inhibiting mitochondrial complex I and promotes mitochondrial biogenesis through the AMPK–peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) axis [65]. In addition, it exerts anti-inflammatory effects by inhibiting nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signalling and downregulating pro-inflammatory cytokines such as IL-6 and TNF-α, thereby attenuating the SASP [65]. These actions contribute to improved systemic homeostasis, reduced chronic inflammation, and preservation of genomic and cellular integrity. Furthermore, metformin influences epigenetic regulation by modulating DNA methylation and microRNA expression patterns associated with longevity [66]. Collectively, these multifaceted effects position metformin as a promising gerotherapeutic agent [67, 68]. Although metformin is not currently prescribed solely as an anti-ageing therapy, the landmark Targeting Ageing with Metformin (TAME) trial is in the preparatory phase and is designed to test whether metformin can delay the onset of multiple age-related diseases simultaneously [69] (Figure 2).
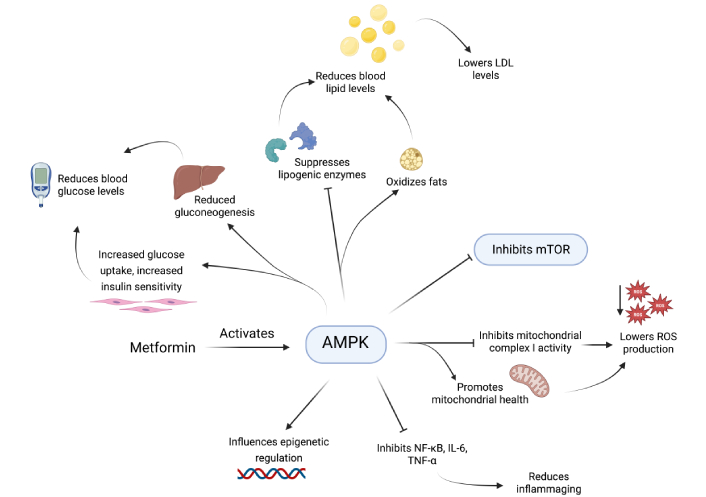
Mode of action of metformin via AMPK activation. Metformin activates adenosine monophosphate (AMP)-activated protein kinase (AMPK), which leads to enhanced glucose uptake and reduced hepatic gluconeogenesis, causing reduced blood glucose levels, suppresses lipogenic enzymes and oxidizes fats, reducing blood lipid levels, inhibits mechanistic target of rapamycin (mTOR) signalling, improves mitochondrial health and inhibits mitochondrial complex I activity, effectively reducing ROS production, inhibits nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), interleukin-6 (IL-6) and tumour necrosis factor-alpha (TNF-α) which reduces inflammaging, and allows for increased epigenetic regulation. LDL: low-density lipoprotein; ROS: reactive oxygen species. Created in BioRender. Dhinakaran, M. (2026) https://BioRender.com/gycdzpa.
Dasatinib and quercetin are agents that have received increasing attention in anti-ageing research. Dasatinib was initially developed as an anticancer drug but was later found to be efficacious when combined with quercetin, forming a potentially powerful senolytic regimen for age-related disorders [70–72]. The dasatinib–quercetin (DQ) combination can selectively eliminate senescent cells, which accumulate with age and contribute to chronic inflammation and tissue dysfunction [73]. Dasatinib inhibits multiple tyrosine kinases that are often overexpressed in senescent cells, selectively targeting senescent preadipocytes, haematopoietic progenitors, chondrogenic stem cells, subsets of fibroblasts, and endothelial cells [70, 71, 74, 75]. It also interferes with SCAPs and induces apoptosis in these pathogenic cells [76]. Quercetin, a flavonoid, helps reduce fat accumulation and senescence markers [75].
Several translational studies have shown that DQ treatment significantly reduces senescent cell burden and improves various health parameters, including bone health, metabolic function, inflammation, and cardiac performance [75, 77, 78]. DQ has also shown promise in aesthetic applications, such as skin rejuvenation, by eliminating senescent dermal fibroblasts and enhancing collagen density [79]. Currently, dasatinib is clinically prescribed for certain forms of leukaemia, whereas quercetin is available over the counter mainly for its antioxidant and anti-inflammatory properties. This combination is not yet approved by the U.S. Food and Drug Administration (FDA) for any indication [79, 80]. However, multiple clinical trials are underway to evaluate its potential to mitigate ageing, including studies on reversing osteoporosis in elderly women, improving physical function in individuals with idiopathic pulmonary fibrosis, enhancing gait and cognitive performance in older adults, slowing the progression of Alzheimer’s disease, and assessing its impact on biological ageing [81–83].
Monoclonal antibodies offer high specificity in targeting ageing-associated pro-inflammatory cytokines such as IL-6 and were initially developed for conditions like autoimmune diseases and cancer [84–86]. More recently, they have been explored as potential agents to reduce chronic low-grade inflammation in ageing-associated disorders [87–89]. IL-6, a widely produced cytokine, exerts pleiotropic effects in tissues surrounding senescent cells [90]. It is a major driver of inflammation and participates in a positive feedback loop with mitochondrial dysfunction [86]. Moreover, elevated IL-6 has been implicated in skewing haematopoietic stem cells towards myeloid differentiation, increasing mutational risk, and adversely affecting cardiac tissue [91, 92].
By suppressing IL-6 signalling, these adverse effects may be mitigated and the pace of ageing-related decline potentially reduced [86]. By neutralising specific SASP components, monoclonal antibody therapies aim to restore tissue homeostasis and function. The humanised monoclonal antibody tocilizumab and the fully human antibody sarilumab bind to IL-6 receptors and prevent IL-6 from engaging them, thereby blocking downstream signalling cascades initiated by this cytokine [93, 94]. Inhibiting IL-6-mediated pathways has shown potential to improve tissue integrity and function [86, 91]. Ongoing clinical trials are investigating whether IL-6 inhibition can slow aspects of biological ageing and improve outcomes in patients with inflammatory disorders such as rheumatoid arthritis, polymyalgia rheumatica, and chronic kidney disease [95].
Stem cell-based therapies seek to replenish or rejuvenate aged tissues. Mesenchymal stem cells (MSCs) and induced pluripotent stem cells (iPSCs) show considerable promise for treating degenerative conditions of the brain, heart, and musculoskeletal system [96–98]. These cells can repair age-related tissue damage, modulate immune responses, and differentiate into multiple lineages, making them attractive candidates for regenerative applications [96–98]. Ageing, however, impairs the regenerative capacity of endogenous stem cells through mechanisms such as accumulated DNA damage, epigenetic alterations, mitochondrial dysfunction, and telomere shortening, underscoring the need for strategies to enhance stem cell efficacy [98].
At present, stem cell therapy is approved for certain indications, including graft-versus-host disease and specific haematological and immune disorders [99, 100]. Numerous clinical trials are ongoing to evaluate its utility in alleviating ageing-related degenerative conditions, such as Parkinson’s disease, osteoporosis, and ischaemic heart disease [74, 101, 102].
Beyond therapies that have shown benefit in other diseases and may be repurposed to slow ageing, several novel approaches are emerging as important areas of investigation. Although still largely experimental, these strategies are underpinned by a strong mechanistic rationale.
Clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 is a versatile genome-editing platform that enables precise modification of DNA to silence or alter specific genes [103, 104]. Guided by a 20-nucleotide single-guide RNA (sgRNA), Cas9 introduces double-strand breaks that are repaired by cellular DNA repair pathways, typically non-homologous end joining (NHEJ), resulting in gene knockouts, or by homology-directed repair (HDR) when a repair template is provided [103, 104] (Figure 3). This technology has markedly shortened the time required to generate genetically engineered mice, from over a year to a matter of months [105, 106].
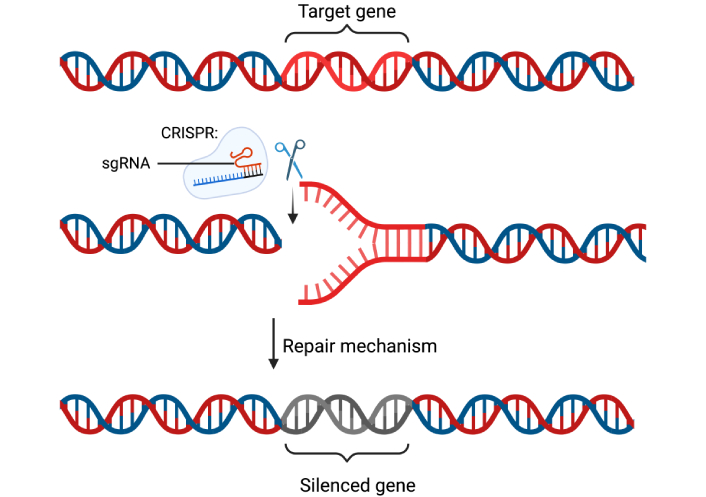
CRISPR-Cas9 mediated gene silencing mechanism. The clustered regularly interspaced short palindromic repeats-CRISPR-associated protein 9 (CRISPR-Cas9) mechanism uses sgRNA to direct the Cas9 nuclease to a specific DNA sequence, where it introduces a double-strand break that disrupts gene expression, leading to the silencing of a gene. sgRNA: single-guide RNA. Created in BioRender. Dhinakaran, M. (2026) https://BioRender.com/dqtxiea.
In mouse zygote microinjection or electroporation experiments, high-efficiency editing has been reported, with up to 100% NHEJ and 27% HDR in ten-eleven translocation methylcytosine dioxygenase 2 (Tet2)-targeted embryos [107]. These studies demonstrate that CRISPR-Cas9 can efficiently produce knockouts, knock-ins, large deletions, conditional alleles, and multiplexed gene alterations in a single step [105, 108, 109]. Despite concerns regarding large deletions at target sites, mosaicism, and off-target effects, the speed, scalability, and cost-effectiveness of CRISPR-Cas9 have made it indispensable for functional genomics, disease modelling, and preclinical therapeutic ageing research [105, 108]. For example, CRISPR-Cas9 has been used in mouse models to reduce senescence in hepatocytes and attenuate liver ageing by inactivating the KAT7 gene, which codes for a histone acetyltransferase, thereby extending lifespan [110]. In another study, epigenetic reactivation of telomerase reverse transcriptase (TERT) using CRISPR-Cas9 extended the lifespan of T cells in vitro and delayed their senescence [111]. With a growing number of studies applying CRISPR-Cas9 to interrogate molecular drivers of ageing, CRISPR-based rejuvenation strategies are now actively being explored.
Sirtuin 6 (SIRT6), with its first functional characterisation in 2006, has gained increasing attention as a key molecular target in ageing research due to its multifaceted roles, which have been shown to extend lifespan in preclinical models [112, 113]. SIRT6 is a nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase involved in critical metabolic signalling pathways. Quercetin derivatives have also been shown to modulate SIRT6 activity, with some acting as activators [114]. SIRT6 promotes longevity by enhancing DNA repair, maintaining chromosomal integrity, regulating energy metabolism, and suppressing SASP production [115–119].
SIRT6 protects telomeres by deacetylating histone H3 in telomeric regions, compacting chromatin to shield DNA from damage [115]. It also promotes BER, NHEJ, and homologous recombination (HR) by activating BER enzymes and poly(ADP-ribose) polymerase 1 (PARP1), thereby enhancing cellular resistance to DNA damage and supporting longevity [116, 117]. This protective effect was demonstrated in a study showing SIRT6-mediated repair of UV-induced DNA damage [116]. In metabolism, SIRT6 represses glycolysis through histone deacetylation, redirecting glucose towards efficient mitochondrial respiration [119]. SIRT6-deficient mice exhibit increased glycolytic flux, leading to dysregulated glucose uptake and hypoglycaemia [119]. SIRT6 also inhibits gluconeogenesis, modulates β-cell insulin secretion, and improves insulin sensitivity [119]. In lipid metabolism, SIRT6 suppresses proprotein convertase subtilisin/kexin type 9 (PCSK9) expression via histone acetylation, preserving LDL receptor levels and reducing cardiovascular risk [120, 121].
Furthermore, SIRT6 attenuates chronic inflammation by deacetylating histones at NF-κB gene promoters, suppressing inflammatory cytokine production [122]. It interacts with nuclear factor erythroid 2-related factor 2 (NRF2) to upregulate antioxidant genes, protecting against ROS [123]. Early human clinical trials are now underway to evaluate SIRT6 upregulation using modulators and activators, with initial data indicating promising safety and effective target engagement [48, 82, 124].
A 2023 University College London study identified the coatomer protein complex I (COPI) pathway as a novel target for senescent cell elimination using RNA interference (RNAi) screens [125]. In normal cells, COPI mediates retrograde vesicle transport between the endoplasmic reticulum and Golgi apparatus, involving cargo selection, ADP-ribosylation factor 1 (ARF1) activation, coat assembly, vesicle budding and scission, uncoating, targeting, and fusion [126]. In senescent cells, ARF1 activity is upregulated [127]. ARF1 requires N-terminal myristoylation for activation; N-myristoyltransferase inhibitors (NMTi) suppress this, thereby repressing COPI function [126].
Pharmacological or genetic COPI suppression induces Golgi apparatus dispersal, impaired autophagy, and apoptosis via the unfolded protein response. While NMTi affect both normal and senescent cells, Golgi apparatus disruption and reduced secretion occur selectively in the latter [125]. This inhibits SASP factors trafficking (e.g., IL-6, IL-8), preventing secretion despite ongoing translation [125]. Although target cells remain senescent, this approach reduces inflammation, limits paracrine senescence spread, protects stem cells, and slows tissue degeneration [126]. Preclinical data are promising, and clinical trials may follow soon [128].
RNAi uses short hairpin RNA (shRNA) or small interfering RNA (siRNA) to silence genes by degrading target messenger RNA (mRNA) [129]. Double-stranded RNA is processed by Dicer into siRNA, which loads into the RNA-induced silencing complex (RISC). RISC binds complementary mRNA, cleaving it for degradation [130, 131]. In anti-ageing research, RNAi targets senescent cells or SASP to restore tissue function [132, 133].
Clinical trials are testing RNAi for age-related conditions. For age-related macular degeneration (AMD), targeting myeloid differentiation primary response 88 (MYD88) mRNA reduces inflammation and tissue damage [134, 135]. In amyloidosis, a precursor to atherosclerosis and dementia, transthyretin (TTR) mRNA targeting reduces amyloid deposition [136–138]. For fibrosis, common in the elderly and impairing organ repair, perilipin 2 (PLIN2) targeting alleviates liver fibrosis [139]. These applications highlight RNAi’s potential in age-related diseases.
Therapeutic strategies, both repurposed and emerging, show strong preclinical promise. However, clinical trials for ageing interventions face challenges, particularly in identifying validated biomarkers as surrogate endpoints [140]. Epigenetic clocks, telomere length, and inflammatory profiles are common, but their predictive value requires further validation [140, 141]. Composite biomarkers may offer greater accuracy, though standardised frameworks are lacking [142, 143]. Regulatory hurdles persist, as agencies like the FDA and European Medicines Agency (EMA) prioritise disease-specific approvals over broad geroprotection [132, 133].
Ethical concerns centre on balancing lifespan extension with healthspan. Interventions must enhance functional wellbeing, not just longevity, to avoid prolonged frailty [144]. Access inequities loom large, with ageing research often excluding disadvantaged groups, exacerbating disparities [145, 146]. Low- and middle-income countries face barriers like poor infrastructure and financial constraints [147]. Advanced therapies risk creating a “longevity divide” favouring the affluent population [148]. Solutions include global infrastructure investment, and policies ensuring affordability and equity.
Ageing remains an inevitable biological process that affects every individual through interconnected molecular hallmarks, including genomic instability, telomere attrition, epigenetic alterations, proteostasis loss, mitochondrial dysfunction, mTOR dysregulation, and cellular senescence. This review has analysed these hallmarks and highlighted how repurposed therapies—such as metformin, DQ combinations, monoclonal antibodies, and stem cell interventions—demonstrate translational promise by targeting these pathways to improve metabolic homeostasis, reduce inflammation, and enhance cellular repair. Emerging strategies, including SIRT6 upregulation, COPI pathway inhibition, RNAi therapeutics, and CRISPR-Cas9 gene editing, offer innovative opportunities to address ageing at its biological roots, with preclinical data showing lifespan extension and tissue rejuvenation in model organisms.
Despite these advances, significant challenges persist. Reliable biomarkers, such as epigenetic clocks and composite inflammatory profiles, require further validation for use as surrogate endpoints in clinical trials. Regulatory frameworks from agencies like the FDA and EMA prioritise disease-specific approvals, complicating direct testing of geroprotective agents. Ethical considerations are paramount, emphasising the need to extend healthspan alongside lifespan to avoid prolonged frailty, while addressing access inequities that could create a “longevity divide” favouring affluent populations.
Future progress demands multidisciplinary integration: rigorous mechanistic studies, inclusive clinical trials, standardised biomarker frameworks, and policy reforms for equitable access. By combining lifestyle interventions with pharmacological and biotechnological innovations, anti-ageing research holds transformative potential to compress morbidity, enhance quality of life, and extend human longevity in the decades ahead.
AMPK: AMP-activated protein kinase
ARF1: ADP-ribosylation factor 1
ATP: adenosine triphosphate
BER: base excision repair
Cas9: clustered regularly interspaced short palindromic repeats-associated protein 9
COPI: coatomer protein complex I
CRISPR: clustered regularly interspaced short palindromic repeats
DQ: dasatinib–quercetin
EMA: European Medicines Agency
ETC: electron transport chain
FDA: Food and Drug Administration
HDR: homology-directed repair
IGF: insulin-like growth factor
IIS: insulin/insulin-like growth factor-1 signalling
IL: interleukin
LDL: low-density lipoprotein
MMPs: matrix metalloproteinases
mRNA: messenger RNA
mtDNA: mitochondrial DNA
mTOR: mechanistic target of rapamycin
mTORC1: mechanistic target of rapamycin complex 1
mTORC2: mechanistic target of rapamycin complex 2
mtUPR: mitochondrial unfolded protein response
NER: nucleotide excision repair
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
NHEJ: non-homologous end joining
NMTi: N-myristoyltransferase inhibitors
RISC: RNA-induced silencing complex
RNAi: RNA interference
ROS: reactive oxygen species
SASP: senescence-associated secretory phenotype
SCAPs: senescent cell anti-apoptotic pathways
siRNA: small interfering RNA
SIRT6: sirtuin 6
TNF-α: tumour necrosis factor-alpha
UV: ultraviolet
MLD: Investigation, Writing—original draft, Writing—review & editing, Visualization. SM: Investigation, Writing—original draft, Writing—review & editing, Visualization. DKS: Conceptualization, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 6501
Download: 51
Times Cited: 0
